Haemate P 500 IU I.V. Enjeksiyon/İnfüzyon İçin Toz İçeren Flakon Kısa Ürün BilgisiKan ve Kan Yapıcı Organlar » Kanama Durdurucu İlaçlar » K Vitamini ve Diğer Hemostatikler » Kan Koagülasyon (pıhtılaşma) Faktörleri » Anti hemofilik faktor viii KISA URUNBILGISI1.BEŞERI TIBBI URUNUN ADIHAEMATE P 500 lU I.V. enjeksiyon/infüzyon için toz içeren flakon2.KALITATIF VE KANTİTATIF BİLEŞİMEtkin maddeler:İnsan Koagülasyon Faktör VIII (FVIII: C- Faktör VIII Aktivitesi) 500 IU (50 IU/ mL) Von Willebrand Faktör (VWF: RCo- Ristosetin Kofaktör Aktivitesi) 1200 IU (120 IU/ mL)HAEMATE P 500' de; Von Willebrand Faktör için spesifik aktivite yaklaşık 3 -17 VWF: RCo/ mg protein, Faktör VIII için spesifik aktivite 2- 6 IU FVIII/ mg protein' dir.* * Faktör VIII' in potens değeri (IU) Avrupa Farmakopesi Kromojenik Analiz Yöntemi' ne göre hesaplanır. Yardımcı maddeler:Sodyum klorür 40-80 mgSodyum sitrat70-140 mg Sodyum hidroksit (eser miktarda pH ayarlamak için) Yardımcı maddeler için 6.1' e bakınız. 3.FARMASÖTIK FORMEnjeksiyon ya da infüzyonluk çözelti hazırlamak için toz ve çözücüsü Hazırlanmış çözelti renksiz, berrak ila hafif opalesan bir çözelti.4.KLINIK ÖZELLIKLER4.1. Terapötik endikasyonlarVon Willebrand hastalığında (VWH) hemoraji ya da cerrahi kanamaların tedavisinde ve profilaksisinde, tek başına desmopressin (DDAVP) tedavisinin yetersiz kaldığı ya da kontrendike olduğu durumlarda kullanılır.Hemofili A (konjenital Faktör VIII eksikliğinde) hastalığında kanamaların profilaksisi ve tedavisinde kullanılır. HAEMATE P sonradan kazanılmış Faktör VIII eksikliğinde ve Faktör VIII' e karşı antikor geliştiren hastaların tedavisinde kullanılabilir. 4.2. Pozoloji ve uygulama şekliUygulanacak doz; hastanın klinik durumuna, kanamanın şiddetine, ölçülen VWF ve FVIII eksikliğinin derecesine göre, bu konuda uzman doktorlar tarafından belirlenmelidir.Pozoloji/Uygulama sıklığı ve süresi:Von Willebrand hastalığının tedavisinde;Genellikle, 1 IU /kg VWF: RCo uygulanması halinde; VWF: RCo sirkülasyon seviyesi 0.02 IU/ mL (% 2) oranında artar. VWF: RCo seviyesi 0.6 IU/mL' den (%60) büyük ve FVIII: C seviyesi 0.4 IU/mL' den (%40) büyük olmalıdır. Kanamanın durması için vücut ağırlığına göre; 40 - 80 IU/ kg VWF ve 20 - 40 IU FVIII/ kg kullanılması tavsiye edilir. Başlangıç dozu için gereken miktar 80 IU/ kg VWF' dür. Özellikle Tip III Von Willebrand hastalarında, gerekli seviyenin korunabilmesi için diğer tiplere göre daha fazla doz uygulanması gerekebilir. Cerrahi müdahalelerde ve şiddetli travmalarda hemorajinin önlenmesinde; Aşırı kanamadan korunmak için kanama sırasında ya da sonrasında cerrahi müdahalelerden 1 ya da 2 saat önce enjekte edilmelidir. Uygun doz her 12 - 24 saat arasında tekrarlanmalıdır. İçeriğinde VWF bulunduran FVIII preparatlarıyla yapılan uzun süreli tedavilerde, hastada FVIII seviyesinde aşırı artış görülebileceği dikkate alınmalıdır. Tedaviden 24- 48 saat sonra verilen FVIII dozunun azaltılması veya doz intervalinin genişletilmesi kontrolsüz FVIII artışını engelleyecektir. Hemofili A hastalığının tedavisinde; Uygulanacak FVIII seviyesi miktarı uluslararası birim (IU) olarak belirlenir ve FVIII ürünleri için güncel WHO standardı ile hesaplanır. Plazmadaki FVIII aktivitesi (FVIII-C) ya yüzde olarak (normal insan plazması) ile ilgili olarak ya da IU (plazmadaki FVIII için belirlenen uluslararası standarda göre) ifade edilir. 1IU FVIII-C normal insan plazmasındaki 1 mL FVIII miktarına eşdeğerdir. Gerekli FVIII miktarı hesaplanırken deneysel bulgular temel alınır. Vücut ağırlığının her 1 kg için 1 IU FVIII plazmadaki FVIII aktivitesini normal aktivitenin % 2oranında artırır (2 IU/ kg). Gerekli miktar aşağıdaki formüle göre hesaplanır: Vücut Ağırlığı (kg) X İstenen FVIII miktarı (% ya da IU/ dL) x 0.5 Uygulama miktarı ve uygulama sıklığı her bir bireyin klinik etkinlik düzeyine göre belirlenir. Aşağıdaki hemorajik olgularda faktör VIII düzeyi o döneme ait plazma aktivitesi düzeyinin (normalin %' si ya da IU/dL olarak) altına düşmemelidir. Aşağıdaki tablo ameliyatlardaki ve kanamalı olgularda kullanılacak dozun miktarını belirleyebilmek için kullanılabilir:
Ciddi hemofili A hastalığı olanlarda kanamaya karşı uzun süreli profilaksi için 2 ila 3 günlük aralıklarla, vücut ağırlığının her kg. için 20 ila 40 IU faktör VIII verilmelidir. Eğer gerekiyorsa başka tedavi önlemleri de alınmalıdır. Bazı durumlarda özellikle genç hastalarda uygulanacak doz aralıklarım kısaltmak yada yüksek dozlarda uygulamak gerekebilir. Hastalar, faktör VIII inhibitörlerinin gelişimi açısından izlenmelidir. Eğer beklenen faktör VIII aktivite plazma seviyesi elde edilemezse ya da uygun doz ile kanama kontrol altına alınmazsa faktör VIII inhibitörlerinin varlığına karar vermek için analiz yürütülmelidir. Hastada yüksek seviyede inhibitör varlığında, faktör VIII tedavisi etkisiz olabilir ve diğer tedavi yöntemleri göz önünde bulundurulmalıdır. Bazı hastaların tedavisi hemofili ile ilgili deneyimleri fazla olan doktorlar tarafından yürütülmelidir. Ayrıca bakınız 4.4.Özel kullanım uyarıları ve önlemleri Uygulama şekli:HAEMATE P intravenöz yolla uygulanır.Ürünü hazırlandığında uygulamadan önceki sıcaklığı oda yada vücut sıcaklığında olmalıdır. Hastanın rahat edebileceği bir yerde intravenöz yolla yavaş bir şekilde uygulanır. Ürün şırınga içine çekilir çekilmez hemen kullanılmalıdır. Yüksek dozda faktör uygulanması gerekli olan durumlarda bu uygulama infüzyon olarak yapılabilir. Bunun için kullanıma hazır ürün uygun bir infüzyon sistemine transfer edilir. Enjeksiyon ya da infüzyon dakikada 4 mL'yi geçmemelidir. Ani değişmeler için hasta gözlenmelidir. Eğer HAEMATE P kullanımı ile ilgili herhangi bir değişiklik meydana gelirse infüzyon oranı düşürülebilir ya da uygulama kesilebilir ve hastanın klinik koşulları belirlenmelidir.(Ayrıca bakınız Bölüm 4.4.) Genel talimatlar: Çözelti berrak veya hafif opak olmalıdır. Filtre edildikten/çekildikten sonra (aşağıya bakınız) hazırlanmış ürün uygulanma öncesi çökeltiye veya renk bozulmasına karşı göz ile incelenmelidir. Artık içeren (kalıntı/parçacık) veya berrak olmayan çözeltileri kullanmayınız. Mix2Vial setini içeren filtreler artık içeren kalıntı ya da parçacıkları tamamiyle söker. Filtrasyon dozaj hesaplarını etkilemez. Çözeltiyi, filtrasyondan sonra da toz parçacıkları yada partiküller içeriyorsa kullanmayınız. Hazırlanma ve enjektöre çekilme aseptik şartlarda gerçekleştirilmelidir. Uygulama sonrası kullanılmayan ürün veya atık madde, yerel gerekliliklere uygun şekilde bertaraf edilmelidir. Hazırlanması: Çözücüyü oda sıcaklığına getiriniz. Toz ve çözücü flakonlarının kapaklarının çıkarıldığından ve stoperlerin aseptik bir solüsyonla silinerek Mix2Vial paketinin açılmasından önce kendiliğinden kuruduğundan emin olunuz. l.Kapağı soyarak Mix2Vial paketini açınız. Mix2Vial' i blister paketinden çıkarmayınız. 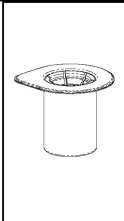 l
l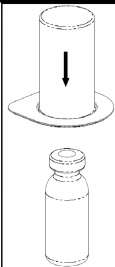 r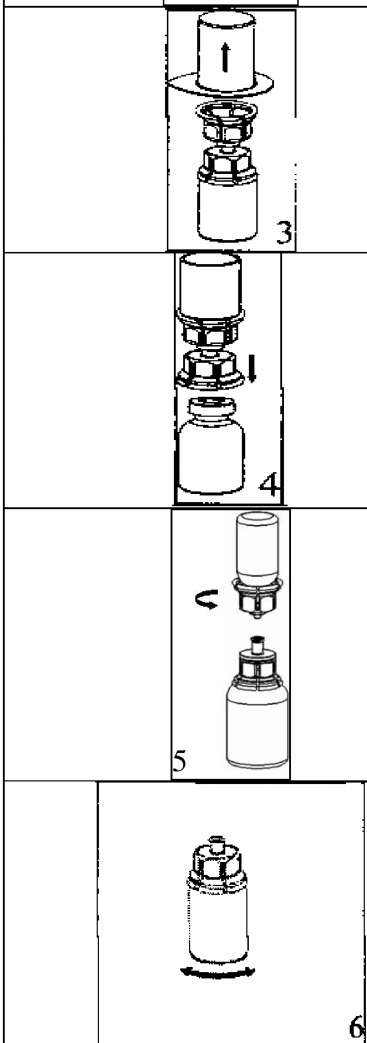 3. Kenarından tutarak ve dikey şekilde yukarıya doğru çekerek, blister paketini Mix2Vial setinden dikkatlice çıkarınız. Sadece blister paketini çekerek çıkardığınızdan ve Mix2Vial setini çıkarmadığınızdan emin olun. 4. Toz flakonunu düz ve sağlam bir zemin üzerine yerleştiriniz. Çözücü flakonunu takılı olan Mix2Vial setiyle birlikte ters çevirin ve saydam adaptör ucunun tepe noktasını, toz flakonun stoperinden aşağıya doğru bastırınız. Çözücü otomatik olarak toz flakonunun içine akacaktır. 5. Bir elinizle Mix2Vial setinin ürün tarafını, diğer elinizle de çözücü tarafını tutunuz ve ürünün çözülmesi sırasında aşırı köpük oluşmasını önlemek için seti dikkatli bir şekilde iki parçaya ayırınız. Çözücü flakonunu mavi Mix2Vial adaptörü takılı şekilde atınız. 6. Tozun tamamen çözündüğünden emin oluncaya kadar ürün flakonunu saydam adaptör takılı şekilde hafifçe sağa sola çeviriniz. Çalkalamayınız. 7. Boş, steril bir şırıngaya hava çekiniz. Ürün flakonu yukarıya doğru iken, şırıngayı Mix2Vial'ın Luer Lock bağlantı parçasına bağlayınız. Ürün flakonuna hava enjekte ediniz. 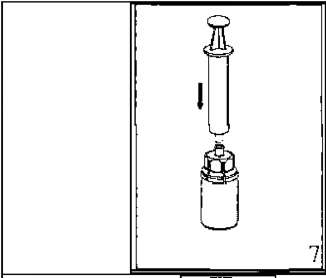 8 9. Çözelti şırıngaya aktarıldıktan sonra, şırınganın gövdesini sıkıca tutunuz (şırınganın sapını aşağıya bakacak şekilde tutarak) ve saydam Mix2Vial adaptörünü şırıngadan ayırınız. Çözeltiyi hemen yavaş intravenöz enjeksiyonla veya infüzyonla uygulayınız Ve ürün dolu şırangaya kanın girmediğinden emin olunuz. Özel popülasyonlara ilişkin ek bilgiler: Böbrek/Karaciğer yetmezliği:HAEMATE P' nin böbrek/karaciğer yetmezliği olan hastalarda kullanımı ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle HAEMATE P' nin bu hastalarda kullanımında tedbirli olunmalı ve hasta açısından yarar/ zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır.Pediyatrik popülasyon:Hemofili A hastalığına sahip çocuklarda HAEMATE P' nin dozajı ile ilgili herhangi bir klinik çalışma bulunmamaktadır.Bu nedenle çocuk vücut ağırlığı (kg'da) başına dozun ayarlanmasıyla kullanılabilir. İnfüzyon hızı düşük tutulmalıdır. HAEMATE P' nin çocuklarda kullanımında tedbirli olunmalı ve çocuk açısından yarar/ zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır. 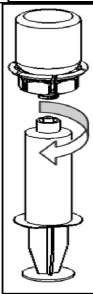 Geriyatrik popülasyon:HAEMATE P' nin 65 yaş üzerindeki yaşlı hastalarda kullanımı üzerine ve tedaviye cevapta yaşlılar ve gençler arasında farklılık olmadığını gösterebilecek klinik çalışmalar yeterli değildir.Bu nedenle HAEMATE P' nin yaşlılarda kullanımında tedbirli olunmalı ve yaşlı kişiler açısından yarar/ zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır. 4.3.KontrendikasyonlarHAEMATE P' nin bileşimindeki maddelerden herhangi birine karşı aşırı duyarlılığı bilinen hastalarda kontrendikedir.4.4.Özel kullanım uyarıları ve önlemleri Virüs Güvenilirliği:Insan kanı veya plazmasından hazırlanan tıbbi ürünlerin kullanımından kaynaklanan enfeksiyonların önüne geçmek için alınan standart önlemler, vericilerin seçilmesini, münferit bağışların ve plazma havuzlarının belli enfeksiyon belirteçleri için izlenmesini ve virüslerin etkisizleştirilmesi/uzaklaştırılması için etkili üretim basamaklarını kapsamaktadır. Buna karşın, insan kanı veya plazmasından hazırlanan ürünler uygulandığında, enfeksiyona neden olacak ajanların geçişi olasılığı bertaraf edilememektedir. Bu durum bilinmeyen veya gelişmekte olan virüsler ve diğer patojenler için de geçerlidir.Bu ilacın üretiminde kullanılan plazmalar Creutzfeldt Jacob (deli dana) ve new variant Creutzfeldt Jacob hastalıklarına karşı teorik enfeksiyon riskini minimize edebilmek için hazırlanmış prosedüre uygun olarak seçilen donörlerden alınmıştır. Yine de insan kanı ve plazmasından elde edilmiş ürünlerde enfeksiyon etkenlerinin bulaşma riski kesin olarak dışlanamaz. Bu durum henüz bilinmeyen patojenler için de geçerlidir.Alınan önlemlerin HIV, HBV ve HCV gibi kapalı virüsler için etkili olduğu düşünülmektedir.Alınan önlemler, HAV ve parvovirus B19 gibi kapalı olmayan virüsler için kısmi koruyuculuğa sahiptir. Parvovirus B19 enfeksiyonu, gebelerde (fetusun enfeksiyonu) ve bağışıklık sistemi yetersiz veya eritropoiesis artışı görülen hastalarda (örn. hemolitik anemi) ciddi seyredebilmektedir. Düzenli/tekrarlanan plazma kaynaklı ürünleri alan hastalar için uygun aşılama düşünülmelidir (Hepatit A ve Hepatit B). Hasta ile ürün serisi arasındaki bağıntıyı koruyabilmek amacı ile, hastaya her HAEMATE P uygulandığında ürünün ismi ve seri numarasının kayıt edilmesi önemle tavsiye edilmektedir. İntravenöz protein ürünlerinin kullanımında, alerjik tipi aşırı duyarlılık reaksiyonları görülebilir. Hastalar aşırı duyarlılık reaksiyonlarının neden olacağı ürtiker, genel ürtiker, göğüste sıkışma, göğüste hırıltı, hipotansiyon ve anafilaksi için bilgilendirilmelidir. Bu semptomlar görüldüğü takdirde hastaya ilacın kullanımını derhal kesmesi ve doktoruna başvurması gerektiği söylenmelidir. Hasta şoka girerse; şok tedavisinde geçerli olan tıbbi standart tedaviler uygulanmalıdır. Von Willebrand Hastalığı Özellikle klinik ya da laboratuar risk faktörleri bilinen hastalarda trombotik vakaların oluşma riski bulunmaktadır. Bu nedenle risk altındaki hastalar, olası trombozis belirtilerinin erken safhada teşhis edilebilmesi için yakinen takip edilmelidirler. VWF ürünleri kullanıldığında, hastada Faktör VIII aktivitesi (FVIII: C) seviyesinin aşırı ve kontrolsüz yükselmesini önlemek için tedavi devam ettiği sürece hastalar uzman bir doktor tarafından sürekli gözlenmelidir. İçeriğinde FVIII bulunduran VWF ürünleri alan hastalar, plazmadaki FVIII: C seviyesinin sürekli artışını önlemek için takip edilmelidir. FVIII: C seviyesinin aşırı yükselmesi trombotik vakaların oluşma riskini artırır, bu nedenle anti-trombotik önlemler göz önünde bulundurulmalıdır. Von Willebrand olan hastalarında başta Tip III hastaları olmak üzere VWF' ye karşı doğal olarak antikor (inhibitör) gelişebilir. Eğer uygulanan doz ile plazmada istenen FVIII: C seviyesine ulaşılamaz veya kanama kontrol altına alınamaz ise, uygulanacak doz için VWF inhibitörü performansı tekrar hesaplamalıdır. İnhibitör seviyesi yüksek olan hastalarda tedavinin etkin olmadığı ve diğer terapötik olasılıklar düşünülmelidir. Hemofili A Faktör VIII' e karşı vücutta antikor (inhibitör) oluşumu hemofili A hastalarının tedavisinde rastlanan, bilinen bir komplikasyondur. Bu inhibitörler genellikle Faktör VIII prokoagülan aktivitesine karşı gelişen immunoglobulinlerdir ve miktarı, plazmanın her bir mL' sinde bulunan BU (Bethesda Unitsj cinsinden hesaplanır. Antikor gelişme riski antihemofilik Faktör VIII' e maruz kalındığında olur ve bu riskin oluşma ihtimali maruz kalınan ilk 20 gün içinde yüksektir. Nadiren maruz kalınan ilk 100 gün sonrasında inhibitör gelişimi görülür. İnsan koagülasyon Faktörü VIII ile tedavi gören hastalarda inhibitör gelişimi uygun klinik araştırmalar ve laboratuar testleriyle dikkatli bir şekilde gözlenmelidir. İnhibitör seviyesi yüksek olan hastalarda tedavi etkin değil ise diğer tedavi seçenekleri düşünülmelidir (bakınız: Bölüm 4.8. İstenmeyen Etkiler) HAEMATE P her 500 IU da 1,521 mmol sodyum içerir. Sodyum diyeti alan hastalarda göz önünde bulundurulmalıdır. 4.5.Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriHAEMATE P diğer tıbbi ürünlerle karıştırılmamalıdır ve ilacın infüzyonu ayrı bir damardan yapılmalıdır.Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin hiçbir etkileşim çalışması yapılmamıştır.Pediyatrik popülasyonPediyatrik popülasyona ilişkin hiçbir etkileşim çalışması yapılmamıştır.4.6.Gebelik ve laktasyon Genel tavsiyeGebelik kategorisi: CÇocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü ( Kontrasepsiyon)HAEMATE P' nin çocuk doğurma potansiyeli bulunan kadınlarda kullanıldığında üreme kapasitesini etkileyip etkilemediğine ilişkin yeterli veri mevcut değildir. Bu nedenle çocuk doğurma potansiyeli bulunan kadınlarda planlanmış bir gebelikten önce uygun bir alternatif tedaviye geçilmelidir.Gebelik dönemiHAEMATE P' nin gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir. İnsanlara yönelik potansiyel risk bilinmemektedir. HAEMATE P gebelik döneminde gerekli olmadıkça kullanılmamalıdır.Bu durum Von Willebrand Hastalığı için farklıdır. Çünkü bu hastalık otozomal kalıtsal bir hastalıktır. Kadınlar erkeklere göre daha fazla etkilenir, çünkü kanama riski daha fazladır (örneğin menstürasyon, hamilelik, doğum ve jinekolojik komplikasyonlar gibi). Pazarlama deneyimlerine dayanılarak Von Willebrand Faktör; akut kanamaların önlenmesinde ve tedavisinde önerilmektedir. Bununla birlikte hamile veya emziren kadınlarda Von Willebrand Faktör kullanımı ile ilgili herhangi bir klinik çalışma bulunmamaktadır. Bu nedenle, VWF ve FVIII preparatları gebelik döneminde gerekli olmadıkça kullanılmamalıdır. Laktasyon dönemiEtkin maddenin insan sütüyle atılıp atılmadığı bilinmemektedir. Etkin maddenin süt ile atılımı hayvanlar üzerinde araştırılmamıştır. Emzirmenin durdurulup durdurulmayacağına yada HAEMATE P tedavisinin durdurulup durdurulmayacağına/tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verilirken emzirmenin çocuk açısından faydası ve HAEMATE P tedavisinin emziren anne açısından faydası dikkate alınmalıdır.Üreme yeteneği / FertiliteHAEMATE P' nin fertilite üzerine doğrudan veya dolaylı olarak zararlı etkilerinin olduğu bilinmemektedir.4.7.Araç ve makine kullanımı üzerindeki etkilerAraç ve makine kullanmaya etkisi yoktur.4.8.İstenmeyen etkilerKlinik denemeler ve pazarlama deneyimlerinde elde edilen advers etkiler aşağıdaki sıklık derecelerine göre verilmiştir.Çok yaygın (> 1/10); yaygın (>1/100 ila < 1/10); yaygın olmayan (>1/1.000 ila < 1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) Kan ve lenf sistemi hastalıklarıÇok seyrek:Hipervolemi (çok fazla miktarda veya sık uygulama gerektiren durumlarda veya cerrahi olaylar öncesinde, sonrasında ya da cerrahi müdahaleler sırasında inhibitör oluşan tüm hastalar hipervolemi açısından gözlenmelidir). İntravasküler hemoliz, hematokrit değerinin düşmesi açısından A, B ve AB kan gruplu hastalar kontrol altında tutulmalıdır. Bağışıklık sistemi hastalıklarıÇok seyrek:Anjioödemin de dahil olduğu alerjik reaksiyon Anafilaktik reaksiyon Anafilaktik şok VWF ve FVIII' e karşı inhibitör gelişimi ve sonucunda yetersiz klinik cevap. Sinir sistemi hastalıklarıÇok seyrek:Baş ağrısı Uyuşukluk Huzursuzluk Sersemlik ve yorgunluk Kardiyak hastalıklarıÇok seyrek:Hipotansiyon Taşikardi Vasküler hastalıklarıÇok seyrek:Trombozis Solunum, göğüs bozuklukları ve mediastinal hastalıklarıÇok seyrek:Dispne Hışırtı Gastrointestinal hastalıklarıÇok seyrek:Mide bulantısı Kusma Deri ve deri altı doku hastalıklarıÇok seyrek:Ürtiker Kaşıntı Genel bozukluklar ve uygulama bölgesine ilişkin hastalıkları :Çok seyrek:Hipersensitivite reaksiyonlar Ateş 4.9. Doz aşımı ve tedavisiBugüne kadar VWF ve FVIII' in yüksek dozda ve aşırı kullanımından kaynaklanan herhangi bir yan etki raporu bildirilmemiştir. Bununla birlikte, yüksek dozda FVIII ile VWF içeren ürünlerin kullanımında trombozis riski unutulmamalıdır.5.FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antihemorajikler: Koagülasyon Faktörleri, Von Willebrand Faktör ve Koagülasyon Faktör VIII kombinasyonu ATC Kodu: B02BD06Etki Mekanizması: Von Willebrand Faktör HAEMATE P insan vücudunda doğal olarak bulunan VWF ile aynı şekilde etki eder. VWF eksikliği olan hastalarda hemostatik anormalliklerin düzeltilmesi için uygulanan VWF iki şekilde görev yapar: 1.VWF, vasküler zedelenmenin olduğu vasküler subendotelyumda trombosit adezyonunu sağlar (hem vasküler subendotelyuma bağlanarak hem de trombosit yüzeyine bağlanarak), primer hemostaz sağlayarak kanama zamanını kısaltır. İçerikte bulunan yüksek miktardaki büyük moleküler ağırlıklı VWF-multimerler sebebiyle bu etki çok kısa zamanda oluşur. 2.VWF içeren ürünler FVIII eksikliğinin düzeltilmesinde daha geç etki gösterirler. İntravenöz uygulamada VWF endojen FVIII' e (hasta tarafından normal olarak üretilen) bağlanır ve böylece bu faktörün stabilizasyonunu sağlayarak hızlı bir şekilde indirgenmesini engeller. Bundan dolayı, saf VWF' ün (çok düşük miktarda FVIII içeren VWF ürünü) FVIII seviyesini normale getirmesi ikincil bir etkidir ve ilk infüzyondan bir müddet sonra gerçekleşir. FVIII içeren VWF preparatları ise ilk infüzyondan sonra hemen FVIII: C seviyesini yeniden yapılandırarak normal seviyesine getirir. Faktör VIII HAEMATE P, insan vücudunda bulunan endojen FVIII ile aynı etkileri gösterir. Hemofilik hastalara infüzyon yapıldığı zaman FVIII hastanın dolaşımındaki VWF' e bağlanır. Aktive edilmiş Faktör VIII, aktive edilmiş Faktör VIII kofaktörü gibi davranarak, Faktör X' un aktive edilmiş Faktör X' a dönüşmesini hızlandırır. Aktive olan Faktör X, protrombini trombin haline çevirir. Daha sonra trombin fibrinojeni fibrine dönüştürür ve pıhtılaşma gerçekleştirilir. Hemofili A, cinsiyet ile bağlantılı, FVIII seviyesinin kanda düşmesi sonucu oluşan kan koagülasyon bozukluğunun görüldüğü kalıtsal bir hastalıktır. Kendi kendine, bir kaza sonucu veya bir cerrahi travma sonucu eklem, kas ve iç organlar içine fazla kanama oluşması ile sonuçlanır. Faktör VIII ile yerine koyma tedavisi ile hastada FVIII seviyesi yükselir ve böylece kanama bozukluğunun kısa süreli olarak düzeltilmesi sağlanır. 5.2. Farmakokinetik özelliklerVon Willebrand FaktörHAEMATE P' nin farmakokinetiği kanama olmayan durumdaki 28 VWF hastası (Tip I n=10; Tip IIA n=10; Tip IIM n=1; Tip III n=7) üzerinde değerlendirilerek aşağıdaki sonuçlar bulunmuştur. Emilim:Uygulama yeri açısından (intravenöz) ilaç direkt kana karışır. VWF: RCo (Ristosetin kofaktör aktivitesi) doruk plazma düzeyine genellikle enjeksiyondan sonraki 50 dakika içinde erişilir. Dağılım:Ortalama Tutulma Süresi (MRT) 13.7 saat ( 3.0 - 44.6 saat) Plazma Konsantrasyon-Zaman Eğrisinin Altında Kalan Alan (EAA): 1664 IU/ dL X saat (aralık: 142-3846 IU/ dL X saat) Biyotransformasyon:VWF: RCo aktivitesi için in vivo geri kazanım 1.9 {(IU/ dL)/ (IU/ kg)}' dir (aralık: 0.6-4.5 {(IU/ dL)/(IU/ kg)} Eliminasyon:VWF: RCo' nin ortalama terminal yarılanma zamanı (iki kompartımanlı model için) 9.9 saattir (2.8 - 51.1 saat). Başlangıcındaki ortalama yarılanma ömrü 1.47 saattir (0.28-13.86 saat) Ortalama klerens 4.81 mL/ kg/ saat (aralık: 2.08-53.0 mL/ kg/ saat). Doğrusallık/ Doğrusal olmayan durum:Doz cevap ilişkisi doğrusaldır.Faktör VIII Emilim:Uygulama yeri açısından (intravenöz) ilaç direkt kana karışır. İntravenöz enjeksiyon sonrasında, plazmadaki Faktör VIII aktivitesi (FVIII: C) hızla yükselir bunu hızlı bir düşüş izler ve sonradan aktivitedeki düşme yavaş olarak azalan bir ivme gösterir. FVIII doruk seviyesi enjeksiyondan 1 ile 1.5 saat içinde erişir. Dağılım:Ortalama Tutulma Suresi (MRT) 19.0 saattir (14.8 - 40.0 saat) Plazma Konsantrasyon-Zaman Eğrisinin Altında Kalan Alan (EAA): 36.1 {% X saat}/ {IU/ kg} (14.8 - 72.4 {% X saat}/ {IU/ kg}) Biyotransformasyon:Faktör VIII için in vivo geri kazanım ortalama her IU/ kg için: 1.73 IU/ dL' dir (her IU/ kg için 0.5- 4.13 IU/ dL) Eliminasyon:Hemofili A hastalarında ortalama yarılanma zamanı 12.6 saat (5.0- 27.7 saat) Ortalama klerens 2.8 mL /saat/ kg (1.4 - 6.7 mL/ saat/ kg) Doğrusallık/ Doğrusal olmayan durum:Doz cevap ilişkisi doğrusaldır.5.3. Klinik öncesi güvenlik verileriHAEMATE P etken madde olarak insan plazmasından elde edilen Faktör VIII ve Von Willebrand Faktör içerir. HAEMATE P' nin tek doz uygulanması çeşitli hayvan türlerinde toksik etki göstermemiştir. Heterolog insan proteini uygulanmasıyla izlenen antikor gelişimi nedeniyle sıradan hayvan kobaylarında tekrarlanan dozlarla (kronik toksisite, kanserojenik, mutajenik) yapılan ön klinik çalışmalar uygun bir performans göstermez.6.FARMASÖTIK ÖZELLIKLER6.1. Yardımcı maddelerin listesiİnsan albumin Aminoasetik asit Sodyum klorür Sodyum sitratSodyum hidroksit ya da hidroklorik asit (az miktarda pH ayarlayıcı) Enjeksiyonluk su 10 mL 6.2.GeçimsizliklerBu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır, sıvısı ve çözücüsü bu bölüm 6.1.' deki durumdan hariç tutulmalıdır.6.3.Raf ömrü36 aydır.6.4.Saklamaya yönelik özel tedbirlerHAEMATE P' yi 25 °C' nin altındaki oda sıcaklığında saklayınız. Dondurmayınız. Donmuş ürünü çözüp kullanmayınız.Kullanıma hazırlandıktan sonraki fizikokimyasal stabilite oda sıcaklığında 48 saat olarak ispatlanmıştır (maksimum 25°C). Mikrobiyolojik olarak bakıldığında HAEMATE P koruyucu içermez, sulandırılarak kullanıma hazırlanan ürünler hemen kullanılmalıdır. Eğer hemen kullanılmayacak ise oda sıcaklığında en çok 8 saat bekletilebilir. 6.5.Ambalajın niteliği ve içeriğiPlastik disk ve alimünyum kapaktan oluşan lastik infüzyon tıpası ile mühürlenmiş Tip II camdan (Avr. Far) yapılmış renksiz enjeksiyon flakonları.Çözücü flakon (enjeksiyonluk su): Plastik disk ve alimünyum kapaktan oluşan lastik infüzyon tıpası ile mühürlenmiş renksiz Tip I (Avr. Far) camdan yapılmış enjeksiyon flakonları. HAEMATE P 500 lU içeriği: 1 adet toz içeren flakon 1adet 10 mL enjeksiyonluk su içeren flakon 2adet alkollü bez 1 filtre transfer aracı 20/20 (Mix2Vial) 6.6.Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller ''Tıbbi Atıkların Kontrolü Yönetmeliği'' ve ''Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelik'' lerine uygun olarak imha edilmelidir.7.RUHSAT SAHİBİFarma-Tek İlaç San. ve Tic. Ltd. Şti. Şerifali Mah. Bayraktar Bulvarı. Beyan Sok. No:12 Ümraniye/İstanbul8.RUHSAT NUMARASI699.İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 26.05.2011 Ruhsat yenileme tarihi10.KÜB' ÜN YENİLENME TARİHİ |
İlaç BilgileriHaemate P 500 IU I.V. Enjeksiyon/İnfüzyon İçin Toz İçeren FlakonEtken Maddesi: Insan Koagulasyon Faktoru Viii Atc Kodu: B02BD06 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.