Betafact 500 Iu/10 Ml Iv Enjeksiyon İçin Liyofilize Toz İçeren Flakon Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADIBETAFACT 500 IU/ 10 mL IV enjeksiyonluk çözelti hazırlamak için liyofilize toz ve çözücü 2. KALİTATİF VE KANTİTATİF BİLEŞİMSterilEtkin madde:İnsan koagülasyon faktörü IX 50 IU/ 1 mL Yardımcı maddeler:Heparin sodyum (Domuz bağırsağı mukozasından5 IU elde edilen heparin) Yardımcı maddeler için 6.1'e bakınız. BETAFACT'ın spesifik aktivitesi yaklaşık 110 IU /mg proteindir. İnsan plazmasından üretilmektedir. 3. FARMASÖTİK FORMIV kullanım için liyofilize toz ve çözücü çözelti Beyaz toz Hazırlama sonrası elde edilen çözelti renksiz ya da biraz opaktır. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarHemofili B (konjenital faktör IX eksikliği) hastalarında kanamaların tedavisinde ve önlenmesinde endikedir. 4.2. Pozoloji ve uygulama şekliTedaviye, hemofili tedavisinde tecrübeli bir doktorun gözetimi altında başlanmalıdır. Pozoloji/ uygulama sıklığı ve süresi:Yerine koyma tedavisinin süresi ve dozu, koagülasyon bozukluğunun ve kanamanın şiddetine, yerine ve hastanın klinik durumuna da bağlıdır. Uygulanan faktör IX miktarı, Dünya Sağlık Örgütü'nün (WHO) faktör IX ürünleri için belirlediği internasyonel Ünite (IU) olarak ifade edilmiştir. Plazmadaki faktör IX aktivitesi ya yüzde (normal insan plazmasına göre) veya İnternasyonel Ünite (Plazma faktör IX için Uluslararası Standartlara göre) olarak ifade edilir. 1/11 1 IU faktör IX aktivitesi, 1 mL normal insan plazmasında bulunan faktör IX miktarına eşdeğerdir (%100). Faktör IX dozunun hesaplanması, vücut ağırlığının her kg'ı için 1 IU BETAFACT'ın plazmadaki faktör IX aktivitesini normalin %1,08'i oranında arttırmasına dayanmaktadır.Gerekli doz, aşağıdaki formül ile belirlenir: Gerekli IU = vücut ağırlığı (kg) X faktör IX düzeyinde (normale göre %) sayısıistenen artış (IU/dL) X 0,93
2/11 Profilaksi Ağır hemofili B hastalarında kanamalara karşı uzun süreli profilaksi için genellikle kullanılan doz 3-4 gün aralıklarla kg başına 20-40 IU'dir ( 20-40 IU/kg ). Bazı durumlarda, özelliklegenç hastalarda daha kısa doz aralıkları veya daha yüksek dozlar gerekli olabilir. Tedavi sırasında faktör IX seviyelerinin uygun şekilde belirlenmesi uygulanacak doz ve tekrarlanan infüzyonların sıklığını belirlemek için önerilmektedir. Özellikle majörameliyatlarda, koagülasyon analizi (plazma faktör IX aktivitesi) ile yerine koyma tedavisininizlenmesi gereklidir. Yarı ömür ve gerikazanımda farklılık göstermelerine bağlı olarakhastaların faktör IX'a cevapları farklı olabilmektedir. Önceden Tedavi Almamış HastalarGüncel bilgiler bölüm 4.8'de belirtilmiştir, fakat özel bir doz önerisi yoktur. Hastalar, faktör IX inhibitör gelişimi için izlenmelidirler. Eğer beklenen faktö r IX aktivite plazma düzeyleri elde edilmez ise veya uygun bir doz ile kanama kontrol edilemez ise, faktörIX inhibitörünün bulunup bulunmadığı belirlenmelidir. mL'de 10 Bethesda Ünitesinde (BU)daha az inhibitör var ise, ek olarak faktör IX uygulanması, inhibitörü nötralize edebilir.İnhibitör titreleri 10 BU'dan fazla olan veya yüksek anamnestik cevap veren hastalarda,(aktif) protrombin kompleks konsantresi (aPCC) veya aktif faktör VII (FVIIa) kullanımı gözönünde bulundurulmalıdır. Bu tedaviler, hemofili hastalarının tedavisinde deneyimlidoktorlar tarafından yürütülmelidir. Uygulama şekli:İntravenöz yoldan uygulanmalıdır. Faktör IX'un, 4 mL/dk'dan daha yüksek hızlarda uygulanması önerilme mektedir. Ürünün uygulamadan önce hazırlanışı için Bölüm 6.6.'ya bakınız. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karaciğer yetmezliği:Böbrek ve karaciğer yetmezliği olan hastalarda minimum dozda ve pratik olarak mümkün olan en yavaş infüzyon hızıyla uygulanmalıdır. Pediyatrik popülasyon:Çocuk ( 6 yaş altında olanlarda dahil ) ve erişkinlerde pozoloji benzerdir. (bölüm 5.1'e bakınız)Geriyatrik popülasyon:65 yaşının üzerindeki hastalarda doz ayarlaması ve minimum infüzyon hızıyla uygulama gereklidir. 4.3. Kontrendikasyonlar Preparatın içindeki aktif madde veya Bölüm 6.1'de belirtilen maddelerden herhangibirine, özellikle heparin (düşük molekül ağırlıklı heparin dahil) veya türevleri karşı alerjikolduğu bilinen hastalarda, Ağır tip II heparinle indüklenmiş trombositopeni (HIT) öyküsü olanlardakullanılmamalıdır. adresinden kontrol edilebilir. Güvenli elektronik imza aslı ile aynıdır. Dokümanın doğrulama kodu : !YnUyZW56ZlAxQ3NRaklURG83SHY33/11 4.4. Özel kullanım uyarıları ve önlemleri_Virüs güvenliğiBETAFACT, insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibiçeşitli hastalıklara yol açabilen enfeksiyon yapıcı ajanlar içerebilirler.BETAFACT'da Varyant Creutzfeldt-Jacob hastalığının bulaşma riski teorik olarakminimumken, klasik Creutzfeldt-Jacob hastalığının bulaşma riski hiçbir kanıtladesteklenmez. Alınan önlemlere rağmen, bu tür ürünler halen potansiyel olarakhastalık bulaştırabilir. Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıp kalmadığının izlenmesi, belirli virüsenfeksiyonlarının halihazırda varlığının test edilmesi ve belirli virüslerin yokedilmesi ve/veya inaktivasyonu ile azaltılmıştır. Bütün bu önlemlere rağmen, buürünler hala potansiyel olarak hastalık bulaştırabilirler. Ayrıca, henüz bilinmeyenenfeksiyon yapıcı ajanların bu ürünlerin içerisinde bulunma ihtimali mevcuttur. HIV, HBV, HCV gibi zarflı virüsler ve HAV gibi zarflı olmayan virüslerin etkisi için önlemlerin alınmasına dikkat edilmelidir. Parvovirüs B19 gibi zarflı olmayanvirüslere karşı alınan tedbirler sınırlı sayıda olabilir. Parvovirüs B19 e nfeksiyonu,gebelikte (fetal enfeksiyon) ve immün yetmezlik ya da kırmızı kan hücreüretiminde artış olan hastalarda tehlikeli olabilir (hemolitik anemi gibi). Doktor, bu ilacı hastaya reçete etmeden veya uygulamadan önce hastası ile risk ve yararlarını tartışmalıdır._Düzenli olarak koagülasyon faktörü alan hastaların, hepatit A ve hepatit B'ye karşı aşılanması önerilmektedir. Hastalar açısından BETAFACT her uygulandığında, hastayla ürünün seri numarası arasındaki bağlantının korunabilmesi için, ürünün adı ve seri numarası kaydedilmelidir. Hipersensitivite Diğer intravenöz protein ürünlerinde olduğu gibi, alerjik tip aşırı duyarlık reaksiyonları mümkündür. Ürün faktör IX dışında insan proteinlerinin izlerini taşır. Bu semptomlar oluşurise ürünün kullanımına derhal son verilmeli ve doktoruna başvurulmalıdır. Hastalar, ürtiker, göğüste sıkışma, hırlama, hipotansiyon ve anafilaksi gibi aşırı duyarlık reaksiyonlarının erken belirtileri hakkında bilgilendirilmelidirler. Şok durumunda, standartşok tedavisi yöntemleri uygulanmalıdır. InhibitörTekrarlayan faktör IX tedavisi sonrası uygun biyolojik test kullanılarak Bethesda Ünitesi (BÜ) olarak titre edilen inhibitör (nötralize edici antikor) bakımından hastalar izlenmelidir. Literatürde faktör IX inhibitörünün oluşumu ve alerjik reaksiyonlar arasında korelasyon gösteren raporlar bulunmaktadır. Bu nedenle, alerjik reaksiyonlarla karşılaşan hastalara, 4/11 faktör IX inhibitörü için test yapılması gerekir. Faktör IX inhibitörü geliştiren hastaların, Faktör IX kullanımı ile anafilaksi riskinin arttığı unutulmamalıdır. Faktör IX konsantreleri ile oluşabilen alerjik reaksiyon riski nedeniyle, Faktör IX'un ilk uygulamaları, alerjik reaksiyonlar için uygun tıbbi bakımın sağlanabileceği tıbbi gözetimaltında yapılmalıdır. Daha önce tedavi edilmemiş, klinik deneyimin sınırlı olduğu hastalarda ve 6 yaşından küçük çocuklarda ürün dikkatle kullanılmalıdır. Listelenen uyarılar ve önlemler hem yetişkinlerdehem de çocuklarda geçerlidir. TromboemboliTrombotik komplikasyonların potansiyel riski nedeniyle, bu ürün karaciğer hastalığı olan kişilere, ameliyat sonrasında hastalara, yenidoğanlara veya trombotik pnömoni ve dissemineintravasküler koagülasyon (DIC) riski olan hastalara uygulandığında uygun biyolojik testyapılarak trombotik ve tüketim koagülopatisinin erken belirtileri klinik olarak izlenmelidir. Bu durumların her birinde, BETAFACT ile tedavinin yararı, bu komplikasyonların riskine karşı değerlendirilmelidir. Bu tıbbi ürünün içerisinde heparin bulunmaktadır. Alerjik reaksiyonlara ve kanın pıhtılaşma sistemini etkileyebilecek olan kan hücre sayısında düşmelere neden olabilir. Geçmişindeheparinle indüklenmiş alerjik reaksiyonu olan hastaların heparin ihtiva eden ilaçlarıkullanmaktan kaçınmaları gerekir. Bu tıbbi ürün her flakonunda 26 mg sodyum ihtiva eder. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriBugüne kadar BETAFACT ile başka ilaç etkileşimi bildirilmemiştir. Buna rağmen, diğer ilaçlarla karıştırılması önerilmez. Özel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyonEtkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/ Doğum kontrolü (Kontrasepsiyon)BETAFACT'ın çocuk doğurma potansiyeli bulunan kadınlarda kullanımına ilişkin özel bir öneri veya, tedavi sırasında veya sonrasında doğum kontrolünün gerekli olduğuna dairherhangi bir bilgi söz konusu değildir. Gebelik dönemiHayvanlar üzerinde yapılan çalışmalar, gebelik/ ve-veya/ embriyonal/fetal gelişim/ ve-veya / doğum/ ve-veya doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. BU BELGE SOKİSıME5ki)niinsarila¥aı,yö'ffeiiik; potansiyel riska^li,nme1mekt1ettirebs titck gov tr/Basvura/EImza/Kontro1adresinden kontrol edilebilir. Güvenli elektronik imza aslrile aynıdır. Dokümanın doğrulama kodu : !YnUyZW56ZlAxQ3NRaklURG83SHY35/11 Dolayısıyla, mutlak gerekmedikçe BETAFACT gebelerde kullanılmamalıdır. Laktasyon dönemiBETAFACT ile hayvanlar üzerinde hiçbir laktasyon çalışması yapılmamıştır. Dolayısıyla, mutlak gerekmedikçe BETAFACT laktasyon döneminde kullanılmamalıdır. Üreme yeteneği /FertiliteHayvan üreme çalışmaları yürütülmemiştir. İnsanlardaki üreme yeteneği/ fertiliteyi etkileyip etkilemediği bilinmemektedir. 4.7. Araç ve makine kullanımı üzerindeki etkilerBETAFACT'ın araç ve makine kullanımı üzerine etkisi yoktur. 4.8. İstenmeyen etkilerFaktör IX uygulama sırasında oluşan etkiler güvenlik profili özetinde belirtilmiştir. Hipersensitivite ve alerjik reaksiyonlar (anjiyoödem, infüzyon yerinde yanma ve batma, üşüme, kızarıklık, yaygın ürtiker, baş ağrısı, hipotansiyon, letarji, bulantı, huzursuzluk,taşikardi, göğüste sıkışma, karıncalanma, kusma, hışıltılı solunum içeren) seyrek görülebilirve bazı vakalarda ciddi anaflaksiye (şok dahil) progresyon gösterebilir. Alerjik reaksiyon öyküsü olan ve faktör IX inhibitörlü hemofili B hastalarında immün tolerans indüksiyon tedavisi denenmesi sonrası nefrotik sendrom rapor edilmiştir. Hemofili B hastaları faktör IX'a karşı inhibitör geliştirebilir. Eğer inhibitör oluşursa, bu yetersiz yanıt olarak değerlendirilir ve böyle vakalarda uzmanlaşmış hemofili merkezleri ileiletişime geçilmesi önerilir. Düşük saflıktaki ürünlerde yüksek riskli olarak, faktör IX uygulaması sonrası tromboembolik olaylar gelişme riski mevcuttur. Miyokard enfarktüsü, dissemineintravasküler koagülasyon, venöz tromboz ve pulmoner emboli oluşumu düşük saflıktafaktör IX ürünü kullanmakla ilişkilendirilebilir. Yüksek saflıkta faktör IX ürünü ile bu tür yan etkilerin görülmesi seyrektir. Bulaşıcı ajanlarla ilgili güvenlik bilgileri için bölüm 4.4'e bakınız. Yan etkiler Tablosu Yapılan çalışmada BETAFACT 50 IU/mL ve BETAFACT 100 IU/mL ile toplamda 8054 tedavi gününde ilacı kullanan 8/109 (%7.3) hastada 17 yan etki raporlanmıştır. Sıklıklar şu şekilde tanımlanmıştır: Çok yaygın (>1/10), yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000),bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubu içinde, yanetkiler azalan şiddete göre sınıflandırılırlar. 6/11
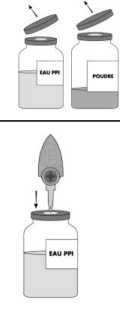
Önceden tedavi almamış hastalar Bir klinik çalışmada önceden tedavi almamış 11 hasta BETAFACT ile tedavi edilmiştir. Total 662 uygulama günü BETAFACT uygulanmış ve inhibitör rapor edilmemiştir. Bütün klinik çalışmalarda, BETAFACT ile tedavi edilmiş önceden tedavi almamış 6 hastanın dahil olduğu 14 ağır hemofili B ( FIX < %1) hastanın hiçbirinde faktör IXinhibitörü gelişmemiştir.En az 8 aylık izlemin olduğu son vizitte ortalama uygulama günü 63 (aralık 5-205) gündür. Pazarlama sonrası BETAFACT tedavisinde 1'i önceden tedavi almamış,1'i önceden tedavi almış 2 hastada inhibitör gelişmiştir. Pediyatrik popülasyon 109 hastanın dahil olduğu güvenlik analizinde, ilk enjeksiyon yapıldığında 12 yaş altı olan 44 hastanın 24'ü 6 yaş altındaydı. Çocuklardaki yan etkilerin sıklığı, tipi ve ciddiyeti erişkinlerden farklı değildi. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir ( BETAFACT ile doz aşımı vakası bildirilmemiştir. 7/11 Farmakoterapötik grup: Antihemorajik: Koagülasyon faktörü ATC kodu: B02BD04Etki mekanizması: Faktör IX, molekül ağırlığı yaklaşık 68.000 Da. olan tek zincirli bir glikoproteindir. Faktör IX, K vitaminine bağlı olarak karaciğerde sentezlenen bir koagülasyon faktörüdür. Faktör IX, intrinsik koagülasyon yolunda faktör XIa ile ve ekstrinsik koagülasyon yolunda faktör VII/doku faktör kompleksi ile aktive edilir. Aktive faktör VIII ile beraber aktive faktörIX, faktör X'u aktive eder. Aktive olan faktör X, protrombini trombine dönüştürür. Trombin,fibrinojeni fibrine dönüştürerek pıhtı oluşumunu sağlar. Hemofili B, doğuştan, cinsiyet kromozomuna bağlı geçiş gösteren faktör IX düzeylerinin düşük olması ile cerrahi travma veya kaza sonucu veya kendiliğinden olan eklem, kas ve içorganlarda kanamaya neden olan kan pıhtılaşma bozukluğu hastalığıdır. Yerine koymatedavisi ile faktör IX plazma seviyesi yükseltilir, böylece faktör eksikliği ve kanamaeğilimleri geçici olarak düzeltilir. Klinik çalışmada 6 yaş altı (10'u önceden tedavi almamış) 13 hasta BETAFACT ile tedavi edilmiştir. Her uygulama gününde minör kanama epizodları ve profilaksi için uygulananortalama doz 37 ve 39 IU /kg'dır. Pazarlama sonrası çalışmada 6 - 12 yaş arasındaki 11çocuk hastaya, 12- 18 yaş arasındaki 4 adölesan ve 18-65 yaş arası 27 erişkin hasta ilebenzer dozlarda BETAFACT uygulanmıştır. BETAFACT'ın plazma doruk düzeyleri, enjeksiyondan genellikle 15 - 30 dakika sonra gözlenir. İntravenöz uygulama sonrası, uygulanan faktör IX miktarının tamamı dolaşımdatespit edilebilir. İntravenöz uygulama sonrası absorbsiyon tam ve hızlıdır. Geri kazanma 1.08 ± 0.21 IU/dL/IU/kg'dır. Eğri altındaki alan 1888 ± 387 IU. h/dL'ye eşdeğerdir. Ortalama kalma süresi 44.2 ± 4.9 saattir. BETAFACT'ın yarı ömrü 33 ± 4 saattir. 8/11 Eliminasyonu dozla doğru orantılıdır. BETAFACT, insan plazmasının normal bir bileşenidir ve endojen faktör IX ile aynı etkiye sahiptir. Hayvanlarda üreme çalışmaları yapılmamıştır. Klinik öncesi çalışmalarda (Amestesti) BETAFACT'ın mutajenik potansiyeline ilişkin herhangi bir bulgu elde edilmemiştir.Tavşanlarda yapılan lokal bir tolerans çalışması, insan koagülasyon faktörü IX'un intravenözuygulama ile iyi tolere edildiğini ve kazara peri-venöz veya intra-arteriyel uygulamadurumunda bile tolere edildiğini göstermiştir. Heparin sodyum (Domuz bağırsak mukozasından elde edilen heparin) Sodyum klorür Lizin hidroklorür Arjinin Sodyum sitrat Sterilize enjeksiyonluk su Bu ilaç başka hiç bir ilaçla karıştırılmamalıdır. Sadece propilenden yapılmış enjeksiyon / infüzyon seti kullanılabilir, çünkü bazı infüzyon ekipmanlarının iç yüzeyinde insan plazması kökenli pıhtılaşma faktörlerinin absorbsiyonutedavinin başarısız olmasına yol açabilir. 30 ay İlaç hazırlandıktan sonra hemen kullanılmalıdır (çözelti hazırlandıktan sonra 25°C'de 3 saat stabildir). 2-8°C arasında buzdolabında saklanmalıdır. Dondurulmamalıdır. Donmuş ürünü çözüp kullanmayınız. Enjeksiyondan arta kalan ilaç olursa, uygun şekilde atılmalıdır. Bir kutuda: Tıpalı (bromobutil) flakon içerisinde (Tip I cam) enjeksiyonluk çözelti için liyofilize toz (500 IU); tıpalı (bromobutil) flakon içerisinde (Tip II cam) çözücü (10 mL), ayrıca birtransfer sistemi ve bir filtre iğnesi bulunur. 9/11 Her iki flakonu da (toz ve çözücü) oda sıcaklığına gelmesi için bekletiniz. Çözücü ve toz flakonunun koruyucu kapaklarını çıkartınız. Kauçuk tıpaların yüzeyini alkollü bir pamukla silerek dezenfekteediniz. Transfer sisteminin buzlu cam görünümlü koruyucu başlığını çıkartıp, çözücü flakonunun tıpasından içeri döndürerek batırınız. Transfer sisteminin diğer ucundaki ikinci koruyucu kapağı daçıkartınız. İki flakonu da yatay konuma getiriniz ve iğnenin serbest ucunutoz flakonunun kapağının ortasına hızla batırınız. Çözücüflakonunun içindeki iğnenin sürekli çözücü içinde kalmasınadikkat ediniz. Transfer iğnesi takılı durumda iken, her iki flakonu da dikeykonuma getiriniz. Çözücü toza gidecek şekilde, çözücü flakonu tozflakonunun üstünde olmalıdır. Transfer sırasında çözücü toz yüzeyinin her yanınapüskürtülmelidir. Çözücünün tamamının gitmiş olduğundan eminolun. Boş flakonu (çözücü) ve transfer sistemini çıkartınız. Toz tamamen çözünene kadar ve köpürtmeden flakonu hafifçedöndürerek sallayınız. Elde edilen çözelti renksiz ya da biraz opaktır. Bulanık ya da çökeltili çözeltiler enjekte edilmemelidir. Hazırlandıktan hemen sonra bir kerede ve intravenöz olarak uygulanmalıdır. Şırıngaya, bir intravenöz iğne veya epikraniyal iğne takınız, şırınganın havasınıçıkarınız ve deriyi dezenfekte ettikten sonra vene enjekte ediniz. Hazırlandıktan hemen sonra tek bir intravenöz doz olarak, enjeksiyon hızı 4mL/dakikayı geçmeyecek şekilde, yavaşça verilmelidir. Hazırlandıktan hemen sonra kullanılmalıdır (hazırlanan çözeltinin stabilitesi 25oC'de 3saattir). Kullanılmamış olan ürünler ya da atık materyaller 'Tıbbi Atıkların Kontrolü Yönetmeliği' ve 'Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelikleri'ne uygun olarak atılmalıdır. Er-Kim İlaç Sanayi ve Ticaret A.Ş. Zorlu Center, Levazım Mah. Koru Sk. No:2 D-Blok 342-345 34340, Beşiktaş-İstanbul Tel: (0212) 275 39 69 Faks: (0212) 211 29 77 137/34 26.12.2013 11/11 |
İlaç BilgileriBetafact 500 Iu/10 Ml Iv Enjeksiyon İçin Liyofilize Toz İçeren FlakonEtken Maddesi: İnsan Koagülasyon Faktörü Ix Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |


Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.