Pirfect 200 Mg Sert Kapsül Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIPIRFECT 200 mg Sert Kapsül 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir sert kapsül, 200 mg pirfenidon içerir. Yardımcı maddeler:Yardımcı maddeler için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMSert Kapsül Beyaz-beyazımsı toz içeren, Size No:2 Mor opak gövde, mor opak kapak sert jelatin kapsül (Sığır jelatini) 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarPİRFECT, yetişkinlerde hafif ve orta derecede İdiyopatik Pulmoner Fibrozis'in tedavisinde endikedir. 4.2. Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:PIRFECT tedavisi, IPF tanı ve tedavisinde uzman hekimler tarafından yürütülmelidir.
PIRFECT için önerilen başlangıç dozu, toplam doz günde 800 mg/gün olacak şekilde yemeklerle birlikte alınan günde 4 defa 200 mg/sert kapsüldür. Ayrıca doz, semptomlara bağlıolarak uygun şekilde artırılabilir. 1 Sert kapsüller oral yolla bütün halinde, bir miktar su ile alınmalıdır. Bulantı, sersemlik gibi yan etkilerin oluşma riskinin azaltılması için mutlaka yemeklerden sonra kullanılmalıdır. 2400 mg/gün üzerindeki dozlar hiçbir hastada kullanılmamalıdır. 14 ardışık gün veya daha fazla süreyle pirfenidon tedavisi almayan hastalar, başlangıç dozundan başlayarak, kademeli olarak ve gözlem altında, önerilen günlük doza kadar süren tedaviyeyeniden başlamalıdır. 14 ardışık günden daha az bir zaman için tedavi kesilirse, titrasyon yapılmaksızın önceden önerilen günlük dozdan tedaviye devam edilebilir. Doz ayarlaması ve güvenli kullanım için öneriler Gastrointestinal olaylar.Gastrointestinal yan etkilerden dolayı tedaviye intolerans gösteren hastalara, ilacı yiyeceklerle birlikte alması gerektiği hatırlatılmalıdır. Eğer semptomlar sürerse,PIRFECT dozu 1 -2 sert kapsül azaltılabilir ve günde 4 defa yemeklerle birlikte tolere edilebilenönerilen günlük doza yeniden artırılabilir. Eğer semptomlar devam ederse, hasta semptomlarıgidermek adına tedaviye 1-2 hafta ara verme konusunda bilgilendirilmelidir.Fotosensitivite reaksiyonu veya kızarıklık.Hafif veya orta derecede fotosensitivite reaksiyonu veya kızarıklık/döküntü yaşayan hastalara güneş koruyucu krem kullanımı önerilmeli ve güneşemaruziyeti önleme bilgisi verilmelidir. PIRFECT dozu günde 800 mg'a azaltılabilir (günde 4defa 200 mg'lık 1 sert kapsül). Eğer kızarıklık 7 gün sonra da devam ederse, PIRFECT 15 güniçin kullanılmamalı, doz artırma periyoduyla aynı olacak şekilde önerilen günlük doza yenidenartırılmalıdır.Ağır fotosensitivite reaksiyonu veya kızarıklık gözlenen hastalar tedaviyi kesmek ve medikal destek alma konusunda bilgilendirilmelidirler. Kızarıklık geçtiğinde, PIRFECT yenidenbaşlanabilir ve hekimin yönlendirmesine göre önerilen günlük doza yeniden artırılabilir. Uygulama şekli:PIRFECT oral uygulama içindir. PIRFECT bir bardak su ile yutulmalıdır, bulantıyı önlemek için yiyeceklerle birlikte alınmalıdır. Özel popülasyonlara ilişkin ek bilgiler:Karaciğer yetmezliği:Hafif ve orta derecede hepatik bozukluğu (Child-Pugh Sınıf A ve B) olan hastalarda doz ayarlaması gerekmemektedir. Bununla birlikte, hafif ve orta derecede hepatik bozukluğu olanbazı bireylerde pirfenidonun plazma seviyesi artabileceğinden bu popülasyonda PIRFECTdikkatli kullanılmalıdır. Hastalar eğer pirfenidon ile birlikte bir CYP1A2 inhibitörü dekullanıyorlarsa, toksisite belirtilerine karşı sıklıkla monitörize edilmelidir. Pirfenidon, ağırhepatik bozukluğu olan hastalarda veya son dönem karaciğer hastalığı olan hastalardaçalışılmamıştır ve bu hastalarda kullanılmamalıdır. Tedavi sırasında karaciğer fonksiyonlarınınkontrol edilmesi ve artış durumunda doz ayarlaması yapılması gerekebilir. Böbrek yetmezliği:Hafif veya orta dereceli renal bozukluğu olan hastalarda doz ayarlaması gerekmemektedir. Pirfenidon şiddetli renal yetmezliği olan hastalarda (CrCl<30 mL/dk) veya diyaliz gerektirenson dönem renal bozukluğu olan hastalarda kontrendikedir. 2 Pediyatrik popülasyon:Pediyatrik popülasyonda IPF tedavisinde pirfenidon kullanımına ilişkin veri bulunmamaktadır. Geriyatrik popülasyon:65 yaş ve üzeri hastalarda doz ayarlaması gerekmemektedir. 4.3. Kontrendikasyonlar Etkin maddeye veya yardımcı maddelerden herhangi birine aşırı duyarlılık Pirfenidon ile anjiyoödem hikayesi (bkz. Bölüm 4.4). Fluvoksamin ile eş zamanlı kullanımda (bkz. Bölüm 4.5), Şiddetli karaciğer yetmezliği veya terminal dönem karaciğer hastalığı olanlarda (bkz.Bölüm 4.2 ve 4.4), Şiddetli böbrek yetmezliği (CrCl<30 ml / dak) veya diyalizi gerektiren son evre böbrekhastalığı (bkz. Bölüm 4.2 ve 5.2). 4.4. Özel kullanım uyarıları ve önlemleriPirfenidon kullanımının, fotogenotoksisite testlerinde ışığa maruz kalma sonucunda anormal bir kromozom yapısına neden olduğu gösterilmiştir; bu nedenle hastaya, ilacınışığa maruz kalma üzerine ciltte kanser oluşumuna neden olabilme potansiyelininaçıklanması ve anladığının sözlü onayı alındıktan sonra uygulanması önemlidir.Karaciğer fonksiyonuPirfenidon tedavisine başlamadan önce karaciğer fonksiyon testleri (ALT, AST ve bilirubin) yapılmalıdır. Pirfenidon tedavisi boyunca ALT, AST ve bilirubin düzeylerinde değişiklikmeydana gelebilir. Bu nedenle tedavinin ilk 6 aylık döneminde ayda 1 ve daha sonra her üçayda bir karaciğer fonksiyon testleri yapılmalıdır (bkz. Bölüm 4.8). Hepatik bozuklukOrta derecede hepatik bozukluk olan kişilerde (örn. Child-Pugh Sınıf B), Pirfenidon maruziyeti %60 artmıştır. Önceden hafif ve orta derecede hepatik bozukluğu bulunan hastalarda (örn.Child-Pugh Sınıf A ve B) artan maruziyet ihtimaline karşı pirfenidon dikkatli kullanılmalıdır.Hastalar toksisite belirtileri için, özellikle pirfenidon ile birlikte CYP1A2 inhibitörü bir ilaçalıyorlarsa, sıklıkla monitörize edilmelidir. Pirfenidon, şiddetli hepatik bozukluğu olanbireylerde çalışılmamıştır ve şiddetli hepatik bozukluğu olan hastalarda kullanılmamalıdır. Fotosensitivite reaksiyonu ve ciltte kızarıklıkPIRFECT tedavisi sırasında direkt gün ışığına maruziyetten (güneş lambaları dahil) kaçınılmalıdır veya en aza indirilmelidir. Hastalar, günlük güneş koruyucu kullanımı, güneşemaruziyeti engelleyecek kıyafetler giyme ve fotosensitiviteye neden olacak diğer tıbbiürünlerin kullanımı konusunda bilgilendirilmelidir. Hastalar fotosensitivite veya cilttekızarıklık semptomlarını hekimlerine bildirmeleri konusunda uyarılmalıdır. Ağır fotosensitivitereaksiyonları yaygın değildir. Hafiften ağıra fotosensitivite reaksiyonu veya ciltte kızarıklıkdurumunda doz ayarlaması veya geçici süreyle tedaviye ara verme gerekebilir. AnjivoödemNefes almada güçlük veya hırıltılı solunum ile birlikte görülebilen yüzde, dudaklarda ve/veya dilde şişme gibi anjiyoödem raporları (bazıları ciddi) pazarlama sonrasında pirfenidon 3 kullanımıyla birlikte görülmüştür. Bu sebeple, PIRFECT tedavisi uygulanan hastalarda anjiyoödem belirtileri görülmesi durumunda tedavi hemen kesilmelidir. Anjiyoödem geçirenhastaların tedavisi standart bakım prosedürüne göre gerçekleştirilmelidir. Pirfenidondan dolayıanjiyoödem geçmişi olan hastalarda PİRFECT kullanımı kontrendikedir (Bkz. Bölüm 4.3). SersemlikPirfenidon kullanan hastalarda sersemlik raporlanmıştır. Bu sebeple, dikkat ve koordinasyon gerektiren aktivitelerden önce, hastalar bu tıbbi ürünün yol açabileceği sersemlik ile ilgilibilgilendirilmelidirler. Klinik çalışmalarda, sersemlik yaşayan hastaların çoğu tek bir olaytecrübe etmiş ve 22 günlük ortalama devam süresinde birçok olay da iyileşmiştir. Eğersersemlik iyileşmiyorsa veya ağırlaşıyorsa, doz ayarlaması veya PIRFECT tedavisininkesilmesi gereklidir. HalsizlikPirfenidon alan hastalarda halsizlik raporlanmıştır. Bu yüzden, dikkat ve koordinasyon gerektiren aktivitelerden önce, hastalar bu tıbbi ürünün yol açabileceği halsizlik ile ilgilibilgilendirilmelidirler. Kilo kaybıPirfenidon alan hastalarda kilo kaybı rapor edilmiştir. Hekimler hastanın ağırlığını takip etmeli ve kilo kaybı klinik anlamlılık gösterdiğinde hastanın uygun kalori alımını sağlamalıdır. PİRFECT sunset yellow (E110) içermesi sebebiyle alerjik reaksiyonlara sebep olabilir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriPirfenidonun yaklaşık %70-80'i, CYP2C9, 2C19, 2D6 ve 2E1'i içeren diğer CYP izoenzimlerinin minör katkılarıyla, CYP1A2 tarafından metabolize edilmektedir. Greyfurt suyu tüketimi CYP1A2'nin inhibisyonu ile ilişkilidir bu nedenle pirfenidon ile tedavi sırasında greyfurt suyu tüketiminden kaçınılmalıdır. Fluvoksamin ve CYP1A2 inhibitörleriFaz 1 çalışmasında, pirfenidon ve fluvoksaminin [diğer CYP izoenzimleri (CYP2C9, 2C19 ve 2D6) üzerinde inhibitör etkilere sahip CYP1A2'nin güçlü bir inhibitörü] birlikte kullanılması,sigara içmeyen hastaların pirfenidon maruziyetinde 4-kat artışla sonuçlanmıştır. Eş zamanlı fluvoksamin kullanan hastalarda pirfenidon kontrendikedir (bkz. Bölüm 4.3). Pirfenidon tedavisine başlamadan önce fluvoksamin tedavisi kesilmeli ve pirfenidon tedavisisırasında pirfenidonun azalmış klerensi nedeniyle fluvoksamin kullanımından kaçınılmalıdır.Pirfenidon tedavisi sırasında hem CYP1A2'nin hem de pirfenidon metabolizmasında yer alanbir veya daha fazla diğer CYP izoenzimi (örn. CYP2C9, 2C19 ve 2D6)'nin inhibitörü olan diğertedavilerden kaçınılmalıdır. İn vitroin vivoekstrapolasyonlar, CYP1A2'nin güçlü ve seçici inhibitörlerinin (örneğin, enoxacin), pirfenidona maruziyeti yaklaşık 2 ila 4 kat artırma potansiyeline sahip olduğunugöstermektedir. Eğer pirfenidonun güçlü ve seçici CYP1A2 inhibitörü ile birlikte kullanılmasıönlenemezse, pirfenidonun dozu günde 800 mg'a azaltılmalıdır (günde dört kez 200 mg).Pirfenidon tedavisiyle ilişkili advers reaksiyonlar açısından hastalar yakından izlenmelidir.Gerekirse pirfenidon tedavisi durdurulmalıdır (bkz. Bölüm 4.2 ve 4.4).4 Pirfenidon ve 750 mg siprofloksasinin birlikte uygulanması (orta derecede CYP1A2 inhibitörü) pirfenidona maruz kalmayı %81 oranında artırır. Günde iki kez 750 mg dozunda siprofloksasinalımı gerekli ise, pirfenidonun dozu günde 1600 mg'a düşürülmelidir (günde dört kez 400 mg).Pirfenidon, siprofloksasin günde bir veya iki kez 250 mg veya 500 mg'lık bir dozdakullanıldığında dikkatli kullanılmalıdır. Pirfenidon, diğer orta derecede CYP1A2 inhibitörleri (örn., amiodaron, propafenon) ile tedavi edilen hastalarda dikkatli kullanılmalıdır. CYP2C9 (örn. amiodaron, flukonazol), 2C19 (örneğin kloramfenikol) ve 2D6 (örn. fluoksetin paroksetin) gibi pirfenidonun metabolizmasında yer alan bir veya daha fazla CYP izoenziminingüçlü inhibitörleriyle CYP1A2 inhibitörleri eş zamanlı olarak kullanıldığında özel dikkatgösterilmelidir. CYP1A2 indükleyicileriOrta derecede CYP1A2 indükleyicileri (örneğin omeprazol), eşzamanlı kullanıldığında teorik olarak pirfenidon plazma seviyelerinin düşmesine neden olabilir. Hem CYP1A2 indükleyicilerinin hem de pirfenidonun metabolizmasında rol oynayan diğer CYP izoenzimlerinin güçlü indükleyicileri olarak etki gösteren tıbbi ürünlerin birlikteuygulanması, pirfenidon plazma seviyelerinin önemli ölçüde düşmesine neden olabilir.Mümkünse bu tıbbi ürünlerin kullanımından kaçınılmalıdır. Özel popülasyonlara ilişkin ek bilgilerPediyatrik popülasyonPirfenidonun, düşük doğum ağırlığına sahip bebeklerde, yeni doğan bebeklerde, emzirilen yenidoğanlarda, bebeklerde veya çocuklarda güvenliliği belirlenmemiştir. Geriyatrik popülasyonYaşlı hastalarda fizyolojik fonksiyon genellikle gerilemiştir; bu nedenle pirfenidon dikkatle uygulanmalıdır. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon)Pirfenidonun çocuk doğurma potansiyeli bulunan kadınlar ve doğum kontrolü üzerine olan etkisi bilinmemektedir. Çocuk doğurma potansiyeline sahip kadın hastalara tedavi sırasındaetkili bir doğum kontrol yöntemi kullanmaları önerilmelidir. Gebelik dönemiHamilelerde pirfenidon kullanımına dair bir veri bulunmamaktadır. Hayvanlarda pirfenidonun ve/veya metabolitlerinin plasentadan transferi, pirfenidonun ve/veya metabolitlerinin amniyotik sıvıda birikmesi potansiyeli ile meydana gelir. 5 Yüksek dozlarda (>1000 mg/kg/gün) sıçanlarda gebelik süresinde uzama ve fötal yaşam şansında azalma görülmüştür. Laktasyon dönemiPirfenidonun veya metabolitlerinin insan sütüne geçip geçmediği bilinmemektedir. Hayvanlardan elde edilen farmakokinetik veriler, pirfenidonun ve/veya metabolitlerinin sütegeçmesi ve beraberinde sütte birikimi potansiyelini göstermektedir. Risk göz ardı edilemez. Emzirmenin çocuğa faydası ve pirfenidon terapisinin anneye faydası dikkate alınarak, emzirmenin kesilmesi veya pirfenidon terapisinin kesilmesi yönünde bir karar alınmalıdır. Üreme yeteneği/FertiliteKlinik-öncesi çalışmalarda, fertilite üzerinde advers etkiler gözlemlenmemiştir. 4.7. Araç ve makine kullanımı üzerindeki etkilerPIRFECT sersemlik ve halsizliğe sebep olabilir, bu da araç ve makine kullanımını etkileyebilir. 4.8. İstenmeyen etkilerPirfenidonun güvenliliği 1650 gönüllü ve hasta ile klinik çalışma ile değerlendirilmiştir. 170'ten fazla hasta beş yıldan fazla süre ve bir kısmı da on yıla kadar, açık çalışmalarda araştırılmıştır. Pirfenidon ile 2403 mg/gün dozda, plasebo karşılaştırmalı klinik çalışma esnasında en yaygın raporlanan advers reaksiyonlar sırasıyla aşağıdaki gibidir:
Ciddi advers reaksiyonlar 2403 mg/gün pirfenidon ile tedavi edilen hastalar ve plasebo alanlar arasında benzer sıklıklarda kaydedilmiştir. Üç pivotal Faz 3 çalışmasında önerilen doz olan 2403 mg/gün pirfenidon kullanan 623 hastada >%2 sıklığında raporlanan advers reaksiyonlar Tablo 1'de gösterilmiştir. Pazarlama sonrasıçalışmalardan elde edilen advers reaksiyonlar da Tablo 1'de listelenmiştir. Advers reaksiyonlarSistem Organ Sınıfı ve sıklık gruplandırmasına göre listelenmiştir [Çok yaygın (>1/10), yaygın(>1/100 ila <1/10), yaygın olmayan (>1/1.000 ila <1/100), seyrek >1/10.000 ila <1/1.000), çokseyrek (< 1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor)] adversreaksiyonlar azalan ciddiyetlerine göre sıralanmıştır. 6
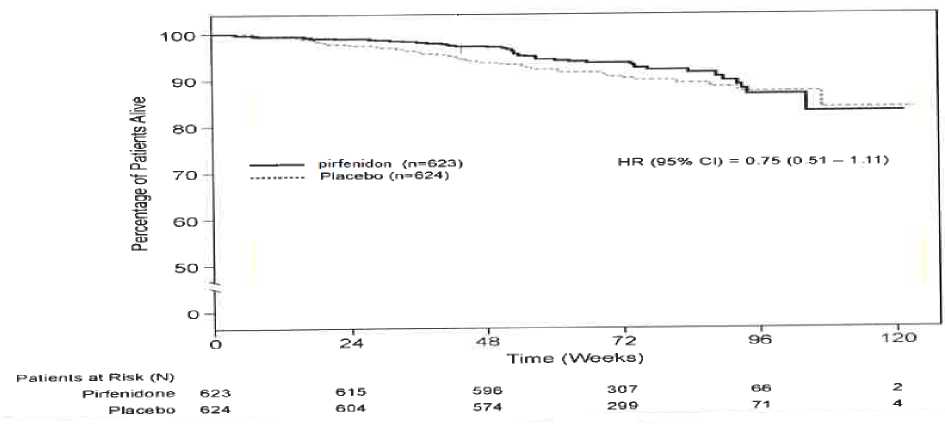
Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr;e- posta:[email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99)7 4.9. Doz aşımı ve tedavisiDoz aşımı konusunda yetersiz klinik bilgi mevcuttur. Pirfenidonun günde 4806 mg'a kadar olan çoklu dozları, günde üç defa altı adet 267 mg'lık sert kapsül olarak sağlıklı erişkin gönüllülere12-günlük doz artırma periyodundan sonra uygulanmıştır. Advers reaksiyonlar hafif, geçici vepirfenidon için en sık rapor edilen advers reaksiyonlar ile tutarlıdır. Şüphelenilen bir doz aşımı halinde, yaşamsal işaretlerin izlenmesi ve hastanın klinik durumunun yakından izlenmesi dahil destekleyici tıbbi bakım sağlanmalıdır. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Diğer immünosupresanlar ATC kodu: L04AX05 Pirfenidonun etki mekanizması tam olarak belirlenmemiştir. Bununla birlikte, çeşitli in vitro sistemlerde ve pulmoner fibröz hayvan modellerinde (bleomisin- ve transplant-indüklenmişfibröz) veriler pirfenidonun hem antifibrotik hem de antiinflamatuvar özellikler ortayakoyduğunu gösterir. İdiyopatik pulmoner fibrozis (İPF), tümör nekroz faktörü-alfa (TNF-a) ve interlökin-1-beta (IL-1P) dahil proenflamatuvar sitokinlerin sentezi ve salımı tarafından etkilenen bir kronik fibrotik ve enflamatuvar pulmoner hastalıktır ve pirfenidonun çeşitli uyaranlara yanıt olarakinflamatuvar hücrelerin birikmesini azalttığı gösterilmiştir. Pirfenidon, fibroblast proliferasyonunu fibrozis ile ilişkili proteinlerin ve sitokinlerin üretimini ve dönüştürücü büyüme faktörü-beta (TGF-P) ve trombosit kaynaklı büyüme faktörü (PDGF)gibi sitokin büyüme faktörlerine yanıt olarak hücre-dışı matrisin biyosentezindeki vebirikmesindeki artışı azaltır. SağkalımPirfenidonun plaseboya kıyasla sağkalımı, Çalışma 1, 2 ve 3'te primer son noktayı desteklemek amacıyla keşifsel analiz şeklinde hesaplanmıştır. Tüm nedenlere bağlı mortalite, çalışmasüresince ve mevcut takip periyodunda, ölümün sebebine ve hastanın tedaviye devam edipetmediğine bakmaksızın değerlendirilmiştir. Tüm nedenlere bağlı mortalite, istatistiksel olarakanlamlı bir farklılık göstermemiştir (bkz. Grafik 1). 8
Klinik etkililikPirfenidonun klinik etkililiği İPF'li hastalarda dört Faz 3, çok merkezli, randomize, çift-kör, plasebo-kontrollü çalışılmıştır. Faz 3 çalışmalarının üçü (PIPF-004, PIPF-006 ve PIPF-016)çok-uluslu, biri (SP-3) Japonya'da yürütülmüştür. PIPF-004 ve PIPF-006, pirfenidon 2.403 mg/gün tedavisi ile plasebonun karşılaştırılmasıdır. Çalışmaların tasarımı, PIPF-004'te ara doz grubunu (1197 mg/gün) içeren birkaç istisna dışındaneredeyse aynıdır. İki çalışmada da, tedavi en az 72 hafta boyunca günde üç defa uygulanmıştır.İki çalışmada da primer varış noktası, başlangıçtan 72. haftaya kadar, yüzde beklenen ZorluVital Kapasite (ZVK)'nin değişimidir. PIPF-004 çalışmasında, pirfenidon alan hastalarda (N=174), plasebo alan hastalara kıyasla (N=174, p=0,001) başlangıçtan tedavinin 72. haftasına kadar, beklenen ZVK düşüşü yüzdesibelirgin bir şekilde azalmıştır. Pirfenidon tedavisi başlangıçtan 24. (p=0,014), 36. (p<0,001),48. (p<0,001) ve 60. (p<0,001) haftalara kadar da, beklenen ZVK düşüşü yüzdesini belirgin birşekilde azaltmıştır. 72. haftada, beklenen ZVK yüzdesinde başlangıçtan düşüş >%10 (İPFmortalite riskinin indikatör eşiği) Pirfenidon alan hastalarda %20, plasebo alan hastalarda %35olarak gözlenmiştir (Tablo 2).
Pirfenidon alan hastalarda plasebo alan hastalara kıyasla rank ANCOVA ile önceden belirlenmiş altı dakika yürüyüş testi (6MWT) sırasında başlangıçtan 72. haftaya kadar bir farkolmasa da, rastgele bir analizde Pirfenidon alan hastaların %37'sinde, PIPF-004'teki plaseboalan hastaların %47'sine kıyasla 6MWT'de >50 m azalma gözlenmiştir. 9 PIPF-006 çalışmasında, Pirfenidon tedavisi (N=171) başlangıçtan 72. haftaya kadar, plasebo ile karşılaştırıldığında (N=173; p=0,501) yüzde beklenen ZVK düşüşünü azaltmamıştır.Bununla beraber, pirfenidon tedavisi başlangıçtan 24. (p<0,001), 36. (p=0,011) ve 48.(p=0,005) haftaya kadar yüzde beklenen ZVK düşüşünü azaltmıştır. 72. Haftada, ZVK'de>%10 düşüş pirfenidon alan hastaların %23'ünde ve plasebo alan hastaların %27'sindegörülmüştür (Tablo 3).
Başlangıçtan 72. haftaya kadar 6MWT uzaklığındaki düşüş PIPF-006 (p<0,001, rank ANCOVA) çalışmasındaki plasebo ile kıyaslandığında belirgin bir şekilde azalmıştır. Ekolarak, bir adhocanalizde, pirfenidon alan hastaların %33'ü, 6MWT mesafesinde >50 m'lik birdüşüş gösterdi, bu da PIPF-006'da plasebo alan hastaların %47'sine karşılık gelmektedir.PIPF-004 ve PIPF-006'dan elde edilen havuzlanmış hayatta kalma analizinde, ölüm oranı pirfenidon 2403 mg/gün grubunda %7,8 iken, plasebo alan grupta %9,8 olarak hesaplanmıştır(HR 0,77 [%95 CI, 0,47-1,28]). PIPF-016, 2403 mg/gün Pirfenidonun plaseboya karşı karşılaştırılmalı çalışmasıdır. Tedavi günde üç defa 52 hafta boyunca uygulanmıştır. Primer varış noktası başlangıçtan 52. haftayakadar yüzde beklenen ZVK değişimidir. 555 hastanın tamamında, ortalama başlangıç yüzdebeklenen ZVK ve %DL COCObaşlangıçta %35'in altındadır.PIPF-016 çalışmasında, başlangıçtan tedavinin 52. haftasına kadar yüzde beklenen ZVK düşüşü, Pirfenidon alan hastalarda (N=278), plasebo alan hastalarla (N=277; p<0,000001, rankANCOVA) kıyaslandığında, belirgin bir şekilde azalmıştır. Pirfenidon ile tedavi başlangıçtantedavinin 13. (p<0,000001), 26. (p<0,000001) ve 39. (p=0,000002) haftasına kadar yüzdebeklenen ZVK düşüşü belirgin bir şekilde azalmıştır. 52. haftada, ZVK'de >%10 düşüşPirfenidon alan hastaların %17'ünde ve plasebo alan hastaların %32'sinde görülmüştür (Tablo4).
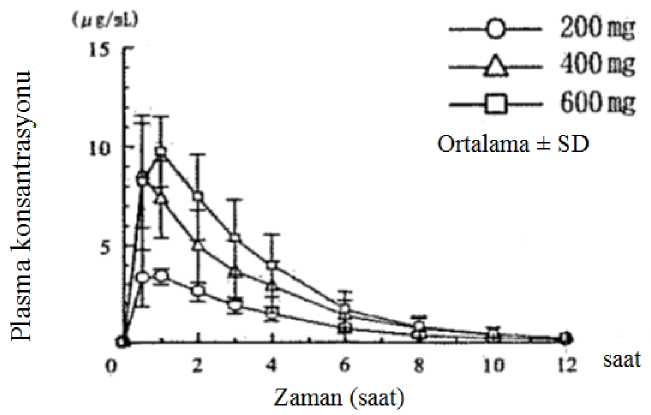
10 Başlangıçtan 52. haftaya kadar 6MWT uzaklığındaki düşüş PIPF-016 (p<0,036, mevkii ANCOVA) çalışmasındaki plasebo ile kıyaslandığında belirgin bir şekilde azalmıştır;Pirfenidon alan hastaların %26'sı, plasebo alan hastaların %36'sı ile karşılaştırıldığında,6MWT uzaklığında >50 m düşüş göstermiştir. PIPF-016, PIPF-004 ve PIPF-006 çalışmalarının 12. ayda önceden belirlenmiş toplam analizinde, tüm sebep-olunmuş mortalite, Pirfenidon 2403 mg/gün grubunda (%3,5, 623hastanın 22'si) plasebo alan hastalarla (%6,7, 624 hastanın 42'si) kıyaslandığında mortalitebelirgin bir şekilde azdır, sonuçlanan %48'inde ilk 12 ayda tüm sebep olunan mortalite riskindeazalma gözlenmiştir (HR 0,52 [%95 Cl, 0,31-0,87], p=0,0107, log-rank test). Japon hastalarda yapılan çalışmada (SP3), pirfenidon 1800 mg/gün (ABD'de ve PIPF-004/006'nın Avrupa popülasyonunda, 2403 mg/gün ile ağırlık-normalize edilmiş olarak karşılaştırılabilir) plasebo ile karşılaştırılmıştır (sırasıyla N = 110, N = 109). Pirfenidon iletedavi vital kapasitede (VC) ortalama düşüşü 52. Haftada (Primer varış noktası) plasebo ilekıyaslandığında belirgin bir şekilde azaltmıştır (-0,09±0,02 I karşı -0,16±0,02 I sırasıyla,p=0,042). Pediyatrik popülasyonAvrupa İlaç Ajansı, İPF'li pediyatrik popülasyonun tüm alt kümelerinde pirfenidon ile çalışmaların sonuçlarının yayınlanması yükümlülüğünden vazgeçmiştir. 5.2. Farmakokinetik özelliklerGenel özelliklerEmilim:Pirfenidon sert kapsüllerin gıda ile verilmesi, Cmax'ta büyük bir azalma (%50 oranında) ve EAA'da açlık durumuna kıyasla daha küçük bir etki ile sonuçlanır. Tokluk durumunda sağlıklıyaşlı yetişkin gönüllülere (50-66 yaş) tek bir doz 801 mg oral yoldan uygulandıktan sonra,pirfenidon emilim hızı yavaşlarken tokluk durumdaki EAA değeri açlık durumundakinin yaklaşık%80-85'i olarak hesaplanmıştır. Açlık durumunda 801 mg tabletin üç 267 mg'lık sert kapsül ilekarşılaştırıldığı çalışmada biyoeşdeğerlik gösterilmiştir. Tokluk durumda, 801 mg tablet, sertkapsüllere kıyasla EAA ölçümlerine dayanan biyoeşdeğerlik kriterlerini karşılarken, Cmax için%90 güven aralığı (%108,26 - %125,60), standart biyoeşdeğerlik sınırının) üst sınırını birazaşmıştır (%90 CI: %80,00 - %125,00). Gıdaların oral pirfenidon EAA üzerindeki etkisi, tablet vesert kapsül formülasyonları arasında tutarlıydı. Açlık durumuna kıyasla, her iki formülasyonun da gıda ile uygulanması pirfenidon Cmax değerini azaltırken, pirfenidon tabletin Cmax değeri pirfenidon sert kapsüllere göre biraz daha az (%50)azaltmıştır (%40'a karşılık %50). Aç karna ilaç alan gruba kıyasla, tok karna ilaç alan bireylerde, düşük yan etki insidansı (bulantı ve sersemlik) gözlenmiştir. Bu nedenle, pirfenidonun bulantı ve sersemlik insidansını azaltmakiçin gıda ile birlikte uygulanması tavsiye edilir. İnsanlarda pirfenidonun mutlak biyoyararlılığı belirlenmemiştir. Plazma Konsantrasyonları11 1. Aç karnına alınan tek dozAltı sağlıklı erkek yetişkine, aç karnına tek bir oral uygulama ile verilen 200 mg, 400 mg ve 600 mg pirfenidonun plazma konsantrasyonları ve farmakokinetik parametreleri Şekil 1'de veTablo 1'de gösterilmektedir. Hem Cmaks hem de EAA, doz ile yaklaşık olarak karşılaştırıldığında artmıştır.
2. Tekrar kullanımBenzer şekilde, 12 sağlıklı yetişkin erkekte sırasıyla 200 mg, 400 mg ve 600 mg tekrarlı dozlarının ve dozajların kademeli olarak 6 gün süreyle her öğün sonrasında sabah, öğlen veakşam olarak günde üç defaya çıkarılmasının (ilk günde ve altıncı günde doz uygulaması, sabahve öğleden sonra olmak üzere günde iki defadır) ardından (toplam 18 gün) elde edilen plazmakonsantrasyonları Tablo 2'de bildirilmiştir. İlk gündeki ve altıncı gündeki her doz açısından, plazma konsantrasyonları benzer bir değişim eğilimi göstermiştir. İlk gün uygulamanın ardından hem C makshem de EAA, dozaj miktarındakiorantılı artışa uygun şekilde artmıştır.
12
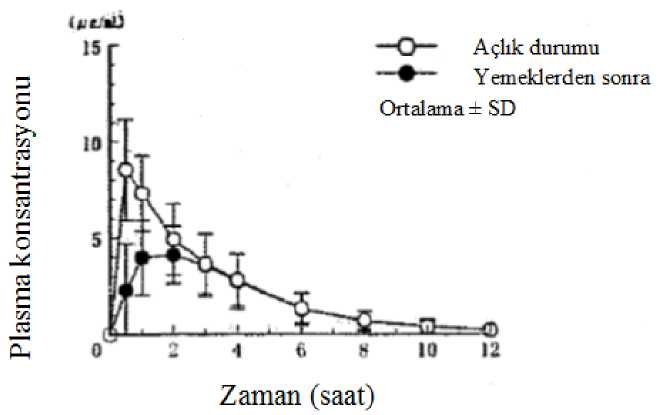
Besinlerin Etkisi400 mg'lık tek bir oral dozun yemeklerden sonra ve aç karınla uygulamasının ardından 6 sağlıklı yetişkin erkekteki plazma konsantrasyonları ve farmakokinetik parametreler Şekil 2'de veTablo 3'de gösterilmiştir. Yemek sonrası, Cmaks ve EAA değerleri anlamlı olarak azalmıştır veTmaks anlamlı olarak gecikmiştir.
Tok deneklerde, aç karnına olan grup ile karşılaştırıldığında, advers olayların (bulantı) insidansında bir azalma gözlemlenmiştir. Bu nedenle, bulantı insidansını azaltmak amacıylapirfenidonun yemek ile birlikte uygulanması önerilmektedir. 48 sağlıklı gönüllüde tokluk koşullarında yapılan biyoeşdeğerlik çalışmasında, PIRFECT 200 13 mg Film Tablet C maksmaks0-247.912,084±2.286,202 ng.sa/mL, Pirespa 200 mg Tablet için ise7.783,808±2.610,796 ng.sa/mL bulunmuştur.Dağılım:Pirfenidon, başta serum albumini olmak üzere insan plazma proteinlerine bağlanır. Genel ortalama bağlanma oranı klinik çalışmalarda gözlenen konsantrasyonlarda %50 ila %58 arasında değişmektedir (1 ila 100 mikrogram/ml). Ortalama oral kararlı durum dağılımı hacmiyaklaşık 70 litre olup, bu da pirfenidonun dokulara dağılımının orta derecede olduğunugöstermektedir. Biyotransformasyon:Pirfenidonun yaklaşık %70-80'i CYP1A2 ile daha az oranda da CYP2C9, 2C19, 2D6 ve 2E1 dahil olmak üzere diğer CYP izoenzimleri ile metabolize edilir. İn vitro veriler, IPF hastalarındatepe plazma konsantrasyonlarının üstündeki konsantrasyonlarda ana metabolitin (5-karboksi-pirfenidon) farmakolojik olarak etkili olduğunu göstermiştir. 5-karboksi-pirfenidona plazmamaruziyetinin arttığı orta derecede böbrek yetmezliği olan hastalarda bu klinik ile alakalıolabilir. Eliminasyon:6 sağlıklı yetişkin erkeğe 200 mg, 400 mg ve 600 mg, aç karnına tek doz oral yolla uygulandığında, her doz miktarında 48 saate kadar idrar içinde atılım oranı; değişmemiş halde%1den az, 5 -karboksi-pirfenidon olarak ise (ana metabolit) yaklaşık %90'dır. 48 sağlıklı gönüllüde tokluk koşullarında yapılan biyoeşdeğerlik çalışmasında, PIRFECT 200 mg Film Tablet t ı/2ı/2'si ise2,096±0,447 saat olarak bulunmuştur.Pirfenidonun oral klerensi orta derecede doyurulabilir özelliktedir. Sağlıklı yaşlı yetişkinlerde yapılan çoklu dozlu, doz-aralığı çalışmasında, günde üç kez 267 mg ila 1335 mg arasındadeğişen dozlar uygulandı, ortalama klerensi günde üç kez 801 mg'lık dozun üzerindeki dozlardayaklaşık %25 azaldı. Sağlıklı yaşlı erişkinlerde tek dozlu pirfenidon uygulamasının ardından,ortalama terminal eliminasyon yarı ömrü yaklaşık 2,4 saat idi. Oral olarak uygulanan pirfenidondozunun yaklaşık %80'i, dozlamadan itibaren 24 saat içinde idrarla atılmıştır. Pirfenidonunönemli bir kısmı, 5-karboksi-pirfenidon metaboliti olarak ve %1'den az pirfenidon ise, idrardadeğişmeden atılır. Özel popülasyonlarKaraciğer yetmezliğiPirfenidon ve 5-karboksi-pirfenidon metabolitinin farmakokinetiği, orta derecede karaciğer yetmezliği olan (Child-Pugh B Sınıfı) ve normal karaciğer fonksiyonuna sahip olan kişilerdekarşılaştırılmıştır. Sonuçlar orta derecede karaciğer yetmezliği olan hastalarda tek doz 801 mgpirfenidon (3 x 267 mg sert kapsül) sonrasında pirfenidon maruziyetinde ortalama %60'lık birartış olduğunu gösterdi. Pirfenidon hafif ve orta şiddette karaciğer yetmezliği olan hastalardadikkatle kullanılmalı ve özellikle de bilinen bir CYP1A2 inhibitörü kullanmaları durumunda,hastalar toksisite belirtileri açısından yakından izlenmelidir. Pirfenidon, ağır hepatik bozukluğu olan hastalarda veya son dönem karaciğer hastalığı olan hastalarda çalışılmamıştır ve bu hastalarda kullanımı önerilmez. 14 Böbrek yetmezliğiNormal böbrek fonksiyonu olan hastalarla karşılaştırıldığında hafif ve şiddetli böbrek yetmezliği olan kişilerde, pirfenidonun farmakokinetiğinde klinik açıdan anlamlı bir farkgözlenmedi. Pirfenidon büyük oranda 5-karboksi-pirfenidona metabolize edilir. 5-karboksi-pirfenidonun ortalama (SD) AUC0-ro değeri, orta derecede (p = 0,009) ve ağır (p<0,0001)böbrek yetmezliği gruplarında, normal renal fonksiyona sahip gruptan anlamlı olarak dahayüksekti; 28,7 (4,99) mg*saat/L'ye karşılık sırasıyla 100 (26,3) mg*saat/L ve 168 (67,4)mg*saat/L.
AUC0-® = 0 anından sonsuza kadar konsantrasyon-zaman eğrisinin altındaki alan. a Normale karşı p-değeri = 1,00 (Bonferroni ile çift-karşılaştırma)b Normale karşı p değeri = 0,009 (Bonferroni ile çift-karşılaştırma)c Normale karşı p değeri < 0,0001 (Bonferroni ile çift-karşılaştırma) Orta derecede böbrek yetmezliği olan hastalarda 5-karboksi-pirfenidona maruz kalma 3,5 kat veya daha fazla artar. Orta derecede böbrek yetmezliği olan hastalarda metabolitin klinik olarakilgili farmakodinamik aktivitesi göz ardı edilemez. Pirfenidon alan hafif böbrek yetmezliği olanhastalarda doz ayarlaması gerekmez. Orta derecede böbrek yetmezliği olan hastalardapirfenidon dikkatli kullanılmalıdır. Şiddetli böbrek yetmezliği (CrCl<30ml / dk) veya diyalizigerektiren son dönem böbrek hastalığı olan hastalarda pirfenidon kullanımı kontrendikedir(bkz. Bölüm 4.2 ve 4.3). Sağlıklı bireylerde veya böbrek yetmezliği olan kişilerde yapılan 4 çalışmadan elde edilen popülasyon farmakokinetik analizleri ve İPF hastalarında yapılan bir çalışmada, pirfenidonunfarmakokinetiği üzerinde yaş, cinsiyet veya vücut ağırlığının klinik olarak anlamlı bir etkisibulunmamıştır. 5.3. Klinik öncesi güvenlilik verileriGüvenlik farmakolojisi, tekrarlanan doz toksisitesi, genotoksisite ve karsinojenik potansiyel ile ilgili konvansiyonel çalışmalara dayalı klinik dışı veriler, insanlar için özel bir tehlikeolmadığını ortaya koymaktadır. Tekrarlı doz toksisite çalışmalarında fare, sıçan ve köpeklerde karaciğer ağırlık artışı gözlenmiştir; genellikle hepatik sentrilobular hipertropi ile birlikte görülür. Tedavininkesilmesinden sonra geri dönüş gözlenmiştir. Sıçanlarda ve farelerde yürütülen karsinojeniteçalışmalarında karaciğer tümörü insidansında artış gözlenmiştir. Bu hepatik bulgular hepatikmikrozomal enzimlerin indüksiyonu ile bağlantılıdır, bu etki Pirfenidon alan hastalarda 15 gözlenmemiştir. Bu bulguların insanlar ile ilişki olduğu düşünülmemektedir. İnsan 2043 mg/gün dozunun 37 katı olan 1500 mg/kg/gün doz uygulanan dişi sıçanlarda uterus tümörlerinde istatistiksel olarak belirgin artış gözlenmiştir. Hastalık gelişme mekanizması ileilgili çalışmaların sonuçlarına göre, uterus tümörlerinin görülmesi muhtemelen sıçanlardakitüre spesifik endokrin metabolizmasını içeren kronik dopamin-aracılı seks hormondengesizliğinden dolayıdır, bu da insanlarda bulunmamaktadır. Reprodüktif toksikoloji çalışmalarında, dişi veya erkek sıçan fertilitesinde veya yavruların postnatal gelişimi üzerinde advers etki görülmemiştir ve sıçanlarda (1000 mg/kg/gün) veyatavşanlarda (300 mg/kg/gün) teratojenite görülmemiştir. Hayvanlarda pirfenidonun ve/veyametabolitlerinin plasental transferi, pirfenidonun ve/veya metabolitlerinin amniyotik sıvıdabirikmesi potansiyeli ile birlikte ortaya çıkar. Yüksek dozlarda (>450 mg/kg/gün) sıçanlardaöstrus döngüsünde uzama ve düzensiz döngülerin insidansında yükselme gözlenmiştir. Yüksekdozlarda (>1000 mg/kg/gün) sıçanlarda hamilelik süresinde uzama ve fötal yaşam şansındaazalma gözlenmiştir. Emziren sıçanlardaki çalışmalar, pirfenidonun ve/veya metabolitlerininsüte atılımının, pirfenidonun ve/veya metabolitlerinin sütte birikmesi potansiyelleri olduğunugöstermiştir. Pirfenidon standart testler grubunda mutajenik veya genotoksik aktivite göstermemiştir ve UV maruziyet altında test edildiğinde mutajenik değildir. UV maruziyet altında test edildiğindePirfenidon, Çin hamsterı akciğer hücresinde fotoklastojenik miktar tayininde pozitif çıkmıştır. Oral yoldan pirfenidon verilen kobaylarda, UVA / UVB ışığına maruz bırakıldıklarında fototoksisite ve iritasyon geliştiği kaydedilmiştir. Güneş koruyucu uygulamasıyla fototoksisitelezyonlarının şiddeti minimize edilmiştir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiDibazik kalsiyum fosfat Kroskarmelloz sodyumMagnezyum stearat Size No:2 Mor opak gövde, mor opak kapak sert jelatin kapsül (Sığır jelatini, Sunset Yellow (E110), Brilliant Blue (E133), Eritrosin (E127), Titanyum dioksit (E171)) 6.2. GeçimsizliklerBilinen herhangi bir geçimsizliği bulunmamaktadır. 6.3. Raf ömrü24 ay 6.4. Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklığında saklayınız. 16 6.5. Ambalajın niteliği ve içeriğiŞeffaf tristar ultra PVC-PE-PVDC- Al Blister 360 sert kapsüllük blister ambalajlarda takdim edilmektedir. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelikleri ne uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİNOBEL İLAÇ SANAYİİ ve TİCARET AŞ. Saray Mah. Dr. Adnan Büyükdeniz Cad. No: 14 Ümraniye 34768 İSTANBULTel: (216) 633 60 00Fax: (216) 633 60 01-02 8. RUHSAT NUMARASI2019/267 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 02.04.2019 Ruhsat yenileme tarihi: - 10. KÜB'ÜN YENİLENME TARİHİ17 |
İlaç BilgileriPirfect 200 Mg Sert KapsülEtken Maddesi: Pirfenidon Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.