Convolyn 200 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİV Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8. Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADICONVOLYN® 200 mg Film Kaplı Tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir film tablet 200 mg Favipiravir içermektedir. Yardımcı madde(ler):Yardımcı maddeler için 6.1.'e bakınız. 3. FARMASÖTİK FORMFilm Tablet Sarı renkli, yuvarlak, bikonveks film kaplı tabletlerdir. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarYeni veya yeniden ortaya çıkan influenza virüs enfeksiyonları (diğer anti-influenza virüs ajanlarının etkili olmadığı veya yetersiz olduğu olgularla sınırlıdır) tedavisinde endikedir. 4.2 Pozoloji ve uygulama şekli Pozoloji/uygulama sıklığı ve süresi:Erişkinlerde doz aşağıdaki şekilde verilir: 1. gün: sabah 1600 mg (8 tablet) ve akşam 1600 mg (8 tablet) olmak üzere günde iki kez 2-5. günler: sabah 600 mg (3 tablet) ve akşam 600 mg (3 tablet) olmak üzere günde iki kezverilir. Kullanım süresi 5 gündür. Uygulama şekli:Oral yoldan kullanım içindir. Aç veya tok karnına alınır. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek yetmezliğinde kullanımı konusunda veri mevcut değildir. Ancak gut hastaları ya da gut öyküsü olan hastalarda ve hiperürisemili hastalarda (kan ürik asit seviyesi artabilir vesemptomlar ağırlaşabilir) dikkatli kullanılmalıdır (Bkz. Bölüm 4.4). Karaciğer yetmezliği:Karaciğer yetmezliğinde favipiravir plazma düzeylerinde artış gözlendiğinden dikkatli kullanılmalıdır (Bkz Bölüm^.2)' güvenli eiektronik imza üe imzalanmıştır.
1 / 15 Pediyatrik popülasyon:Favipiravir çocuklarda denenmemiştir. Pediyatrik popülasyonda kullanımı mevcut değildir. Geriyatrik popülasyon:Yaşlıların fizyolojik fonksiyonları yavaşladığından CONVOLYN®, bu hastaların genel durumlarını izleyerek dikkatli verilmelidir. 4.3 Kontrendikasyonlar Favipiravir veya ilacın içerdiği maddelerin (bölüm 6.1'de listelenmiştir) herhangi birinekarşı aşırı duyarlılığı olduğu bilinen kişilerde, Gebe iseniz veya gebelik şüphesi durumunda (hayvan çalışmalarında erken embriyonikölümler ve teratojenisite gözlenmiştir) kontrendikedir (Bkz. Bölüm 4.6). 4.4 Özel kullanım uyarıları ve önlemleri Hayvan çalışmalarında erken embriyonik ölümler ve teratojenisite gözlenmesisebebiyle gebe ya da gebelik şüphesi olan kadınlarda favipiravir kullanılmamalıdır(Bkz. Bölüm 4.3 ve 4.6). Çocuk doğurma potansiyeli bulunan kadınlara favipiravir uygulamadan önce negatifgebelik testi onaylanmalıdır. Tedavi sırasında ve tedavi sonlanımından sonraki 7 günsüresince eşi ile birlikte en etkili kontrasepsiyon metodunun kullanılması ve tüm risklerkonusunda hasta bilgilendirilmelidir (Bkz. Bölüm 4.6). Tedavi sırasında gebelikşüphesi olur ise tedavi derhal kesilmeli ve doktora başvurulmalıdır. Favipiravir sperm içerisinde dağılır. Bu tıbbi ürün erkek hastalara uygulanırken tedavisırasında ve tedavi sonlanımından sonraki 7 gün süresince en etkili kontrasepsiyon metodunun (erkekler kondom kullanmalıdır) kullanılması ve tüm riskler konusunda hasta bilgilendirilmelidir. Ayrıca gebe kadınlar ile cinsel ilişkiye girilmemesikonusunda hasta bilgilendirilmelidir (Bkz Bölüm 4.6) CONVOLYN® ile tedaviye başlamadan önce etkililiği ve riskleri (fetusa maruziyetsonucu riskler de dahil olmak üzere) hastalara ve aile bireylerine anlatılmalıdır. Kullanımdan önce favipiravir kullanımının gerekliliği dikkatli bir şekilde incelenmelidir. Favipiravir yalnızca yeni ya da yeniden ortaya çıkan bir influenza virüs salgınında diğer anti-influenza virüs ajanlarının yetersiz ya da etkisiz kaldığı durumlarda ve devletin influenza virüslerine karşı önlem olarak bu tıbbi ürünü kullanım kararı alması halindekullanılır. Bu tıbbi ürün uygulanırken, influenza virüsleri gibi virüslere karşı önlem ileilişkili devlet talimatını içeren güncel bilgi sağlanmalıdır ve yalnızca uygun hastalarareçete edilmelidir. Favipiravir bakteriyel enfeksiyonlara karşı etkili değildir. Favipiravirin çocuklarda kullanımı mevcut değildir. Tedavi, influenza benzeri semptomların görülmesinden hemen sonra başlatılmalıdır. Favipiravir yeni veya yeniden ortaya çıkan virüs enfeksiyonları için kullanılmamıştır. Advers olaylar ve klinik çalışma sonuçlarına ilişkin bilgiler onaylanan dozdan daha düşük dozlarlayürütülen Japon klinik çalışmalarına dayanmaktadır. Favipiravirin onaylanmış dozaj ile etkililiğini ve güvenliliğini incelemek için herhangi bir klinik çalışma yapılmamıştır. Onaylanan dozaj, influenza virüsü enfeksiyonu olan hastalarda Belge DoHY3 t Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebysplasebo kontrollü bir faz I / II klinik çalışmasının sonuçlarına ve Japon ve denizaşırı çalışmalardan elde edilen farmakokinetik verilere dayanılarak tahmin edilmiştir. Japonya 2 / 15 dışında yapılan farmakokinetik çalışmada karaciğer fonksiyon bozukluğu olan hastalarda favipiravir plazma seviyesinin arttığı bildirilmiştir (Bknz. Bölüm 5.2). Gut hastaları ya da gut öyküsü olan hastalarda ve hiperürisemili hastalarda kan ürik asit seviyesi artabilir ve semptomlar ağırlaşabilir. Bu hastalarda dikkatli kullanılmalıdır. Nedensel ilişkibilinmemekle birlikte, favipiravir dahil anti-influenza virüs ajanlarının uygulanmasından sonraanormal davranış gibi psikonörotik semptomlar bildirilmiştir. Çocukların ve reşit olmayanların tedavisi için, düşme gibi anormal davranışlardan kaynaklanan bir kaza durumunda önleyici bir yaklaşım olarak, hastalara / ailelerine, antiinfluenza virüsajanları ile tedaviye başlandıktan sonra (i) anormal davranış gelişebildiği ve (ii) dolayısıylaebeveynlerin çocukların / reşit olmayanların evde tedavi edildiklerinde en az 2 gün boyuncayalnız bırakılmamaları gerektiği konusunda bilgilendirilmelidir. İnfluenza ensefalopatisi ileilişkili benzer semptomlar bildirildiğinden, yukarıdaki ile aynı talimat verilmelidir. İnfluenza virüs enfeksiyonları bakteriyel enfeksiyonlarla komplike olabilir veya influenza benzeri semptomlarla karışabilir. Bakteriyel enfeksiyon mevcudiyetinde veya şüphesidurumunda, anti-bakteriyel ajanların verilmesi gibi uygun önlemler alınmalıdır. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriFavipiravir, sitokrom P-450 ile metabolize olmayıp, aldehid oksidaz ile metabolize olmakta ve kısmen de ksantin oksidaz ile metabolize edilmektedir. Favipiravir, aldehid oksidazı veCYP2C8'i inhibe etmekte, fakat sitokrom P-450'yi uyarmamaktadır (Bkz. Bölüm 5.2).Favipiravir, aşağıdaki ilaçlarla birlikte kullanımında dikkatli olunmalıdır:
2C9Bu2Cf9güv2B6ekt2E11imyeie3A4l
' hynUySftö ~--Bste.T; sitfenmiş'meaSolit;l'CYPiA2;k;2C8; 2C9, ?2Cf9ek2MlS:İJ2fefVe5®^Z^ifl^nfi!6,İİÖr aktivite göstermiştir, Favipiravirin CYP üzerinde indükleyici etkisi gözlenmemiştir. 3 / 15 İlaç etkileşim klinik çalışmaları: Birlikte kullanılan ilaçların favipiravir farmakokinetiği üzerine etkileri
4 / 15
Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin herhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:Pediyatrik popülasyona ilişkin herhangi bir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: X Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli bulunan kadınlarda tedaviye başlamadan önce gebelik testi yapılarak sonucun negatif olduğu tespit edilmelidir. Kişiye riskler çok iyi bir şekilde anlatılmalıve tüm riskler ile ilgili hasta bilgilendirilmelidir. Tedavi sırasında ve tedavinin bitimindensonraki 7 gün boyunca en etkin kontraseptif yöntem kullanılmalıdır. Tedavi sırasında gebelikşüphesi olur ise tedavi derhal kesilmeli ve doktora başvurulmalıdır.
5 / 15 Gebelik dönemiFavipiravir gebelik veya gebelik şüphesi durumunda kullanılmamalıdır. Hayvan çalışmalarında klinik maruziyete benzer ve ondan daha düşük düzeylerde maruziyet durumunda erkenembriyonik ölüm (ratlar) ve teratojenisite (maymun, fare, rat ve tavşan) gözlenmiştir. Favipiravir'in gebelik döneminde uygulandığı takdirde ciddi doğum kusurlarına yol açtığından şüphelenilmektedir. Favipiravir gebelik döneminde kontrendikedir. Laktasyon dönemiLaktasyon döneminde CONVOLYN® verildiğinde, emzirmeye ara verilmelidir. CONVOLYN®'in majör metaboliti hidroksile formu anne sütünde bulunmuştur. Üreme yeteneği/FertiliteFavipiravir sperm içerisinde dağılır. Bu tıbbi ürün erkek hastalara uygulanırken tedavi sırasında ve tedavi sonlanımından sonraki 7 gün süresince en etkili kontrasepsiyon metodunun (erkeklerkondom kullanmalıdır) kullanması gerekliliği ve tüm riskler ile ilgili hasta bilgilendirilmelidir. Hayvan çalışmalarında ratlarda (12 haftalık) ve küçük köpeklerde (7-8 aylık) testislerde histopatolojik değişiklikler ve farelerde (11 haftalık) spermlerinde anormal bulgularbildirilmiştir. Tedavi sonlandırılmasını takiben iyileşme veya iyileşme eğilimi gözlenmiştir. 4.7 Araç ve makine kullanımı üzerindeki etkilerCONVOLYN® görme bulanıklığına yol açabileceğinden ilaç alımı esnasında araç ve makine kullanımı konusunda uyarılmalıdır. Favipiravir dahil anti-influenza virüs ajanlarınınuygulanmasından sonra anormal davranış gibi psikonörotik semptomlar bildirildiğinden hastalararaç ve makine kullanma konusunda uyarılmalıdır. 4.8 İstenmeyen etkilerTüm ilaçlarda olduğu gibi, CONVOLYN®'in içeriğinde bulunan maddelere duyarlı olan kişilerde yan etkiler olabilir. CONVOLYN® klinik çalışmalarda onaylanmış doz ile verilmemiştir. Japonya'da klinik çalışmalarda ve global faz III çalışmada (onaylanmış dozdan daha düşük doz düzeylerindeyürütülen çalışmalar) güvenlilik değerlendirmesinde 501 hastadan 100'ünde (%19,96) adversreaksiyonlar gözlendi. Major advers reaksiyonlar, 24 hastada (%4,79) gözlenen kan ürik asitdüzeylerinde artış, 24 hastada (%4,79) diare, 9 hastada (%1,80) gözlenen nötrofil düzeylerindeazalma, 9 hastada (%1,80) gözlenen AST (GOT) düzeylerinde artış, 8 hastada (%1,60)gözlenen ALT (GPT) düzeylerinde artıştır. Diğer anti-influenza virüsü ajanları (benzer ilaçlar) ile aşağıdaki klinik olarak anlamlı advers reaksiyonlar bildirilmiştir. Hastalar dikkatle izlenmeli ve herhangi bir anormallik gözlenirse,tedavi kesilmeli ve uygun önlemler alınmalıdır:
6 / 15 Advers ilaç reaksiyonları aşağıda tanımlanan sıklığa göre listelenmiştir: Çok yaygın (>1/10); yaygın (>1/100 ile <1/10), yaygın olmayan (>1/1.000 ile <1/100); seyrek (>1/10.000 ile <1/1.000); çok seyrek (<1/10.000); bilinmiyor (eldeki verilerle tahminedilemiyor). Advers etkiler klinik çalışmalardan havuzlanmış analizlere göre tablolarda uygun kategorilere eklenmiştir. Her bir sıklık grubunda advers etkiler azalan ciddiyet sırasıyla listelenmiştir. Kan ve lenf sistemi hastalıkları:Bilinmiyor: Beyaz kan hücresi sayısında azalma, nötrofil sayısında azalma, trombosit sayısında azalma Bağışıklık sistemi hastalıkları:Bilinmiyor: Şok, anafilaksi Psikiyatrik hastalıklar:Bilinmiyor: Nörolojik ve psikiyatrik belirtiler (bilinç bozukluğu, anormal davranış, delirium, halüsinasyon, sanrı, konvülsiyon vb.) Solunum göğüs bozuklukları ve mediastinal hastalıklar:Bilinmiyor: Zatürre Gastrointestinal hastalıklar:Bilinmiyor: Hemorajik kolit Hepato-biliyer hastalıklar:Bilinmiyor: Hepatit fulminan, karaciğer fonksiyon bozukluğu, sarılık Deri ve deri altı doku hastalıkları:Bilinmiyor: Toksik epidermal nekroliz (TEN), okülomukokutanöz sendrom (Stevens-Johnson sendromu) Böbrek ve idrar yolu hastalıkları:Bilinmiyor: Akut böbrek hasarı Aşağıdaki advers reaksiyonlar, Japon klinik çalışmalarında ve küresel faz III klinik çalışmada gözlenen advers reaksiyonlardır (onay dozundan daha düşük doz seviyeleriyle yapılançalışmalar). Bu advers reaksiyonlar meydana gelirse, semptomlara göre uygun önlemleralınmalıdır. Kan ve lenf sistemi hastalıkları:Yaygın: Nötrofil sayısında azalma, beyaz kan hücresi sayısında azalma Yaygın olmayan: Beyaz kan hücresi sayısında artış, retikülosit sayısında azalma, monosit artışı Göz hastalıkları:Yaygın olmayan: Bulanık görme, göz ağrısı Kulak ve iç kulak hastalıkları:Yaygın olmayan: Vertigo
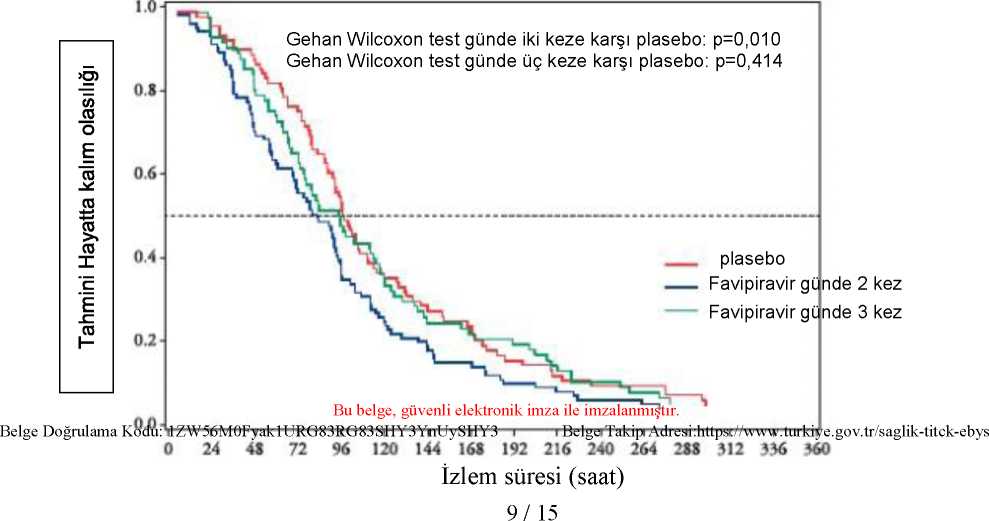
7 / 15 Kardiyak hastalıklar:Yaygın olmayan: Supraventriküler ekstrasistoller Metabolizma ve beslenme hastalıkları:Yaygın: Kanda ürik asit artışı, kan trigliserit artışı Yaygın olmayan: İdrarda glikoz varlığı, kan potasyumunda azalma Solunum göğüs bozuklukları ve mediastinal hastalıklar:Yaygın olmayan: Astım, orofarenjeal ağrı, rinit, nazofarenjit, tonsilde polip Gastrointestinal hastalıklar:Yaygın: İshal (%4,79) Yaygın olmayan: Bulantı, kusma karın ağrısı, karın rahatsızlığı, duodenum ülseri, hematokezya, gastrit, disguzi Hepato-biliyer hastalıklar:Yaygın: AST (GOT) artışı, ALT (GPT) artışı, y-GTP artışı Yaygın olmayan: Kan ALP artışı, kan bilirubin artışı Deri ve deri altı doku hastalıkları:Yaygın olmayan: Pigmentasyon, morarma, döküntü, egzama, kaşıntı Araştırmalar:Yaygın olmayan: İdrarda kan, kan CPK düzeylerinde artış Bu advers reaksiyonların hepsi Japon klinik çalışmalarında ve küresel faz III klinik çalışmada gözlenen advers reaksiyonlar olup onay dozundan daha düşük doz seviyeleriyle yapılançalışmalardır. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta:[email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9 Doz aşımı ve tedavisiCONVOLYN® aşırı dozda alınması durumunda toksik yan etkilere yol açabilir. Özel bir antidotu olmadığından semptomatik tedavi yapılmalıdır. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grubu: Sistemik kullanım için antiviraller-Direk etkili antiviraller ATC kodu: J05AX27 Etki mekanizmasıFavipiravirin hücrelerde bir ribosil trifosfat formuna (favipiravir RTP) metabolize olduğu ve favipiravir RTP'nin influenza virüs replikasyonunda yer alan RNA polimerazı seçici olarakinhibe ettiği düşünülmektedir. İnsan DNA polimerazları a, P ve y'ya karşı aktivite ile ilgili BelgeDooıaiıâk,Tavipfr5avirıRTPI^06e^moi/LyâHüz3erinde ]inffi^!1tıörveıtiihgöstermeuz1f;p!fzerrindfe,%19:,F-8 / 15 13.5 inhibitör etki ve y üzerinde %11.7-41.2 inhibitör etki gösterir. İnsan RNA polimeraz II üzerindeki favipiravir RTP'nin inhibitör konsantrasyonu (IC50) 905 pmol/L'dir. İn vitroantiviral aktiviteFavipiravir tip A ve tip B influenza virüs laboratuvar suşlarına 0,014-0,55 pg/mL EC50 değerlerinde antiviral aktivite göstermiştir. Adamantanlara (amantadin, rimantadin), oseltamivire veya zanamivire dirençli suşlar da dahil olmak üzere mevsimsel tip A ve tip B influenza virüslerine karşı EC50değerleri sırasıyla0,030,94 ve 0,09-0,83 pg/mL idi. Yüksek patojenik suşları da (H5N1 ve H7N9) kapsayacak şekilde domuz orijinli ve kuş orijinli tip A influenza virüslerine (adamantan, oseltamivir veya zanamivire dirençli suşlar dahil olmaküzere) karşı EC50değerleri 0,06-3,53 pg/mL idi. Adamantanlara, oseltamivire ve zanamivire dirençli tip A ve tip B influenza virüslerine karşı EC50değerleri 0,09-0,47 pg/mL idi ve herhangi bir çapraz direnç gözlenmemiştir. DirençFavipiravir varlığında 30 pasajdan sonra A tipi influenza virüslerinin favipiravire duyarlılığında herhangi bir değişiklik gözlenmemiş ve dirençli virüs seçilmemiştir. Global faz III çalışması dadahil olmak üzere klinik çalışmalarda favipiravir dirençli influenza virüsleri oluşması ile ilgilibilgi elde edilmemiştir. Klinik çalışmalarJapon olmayan kişilerde yapılan çalışmalarTip A veya tip B influenza hastalarında plasebo kontrollü faz I/II çalışma gerçekleştirildi (1800 mg/800 mg günde 2 kez, oral yolla 1. gün günde 2 kez 1800 mg, sonraki 4 gün günde 2 kez 800mg; 2400 mg/600 mg günde 3 kez, 1. gün günde 3 kez olmak üzere 2400 mg+ 600 mg+ 600mg ve sonraki 4 gün günde 3 kez 600 mg). * Primer değerlendirme kriterine** bakıldığında;Favipiravir 1800 mg/800 mg günde 2 kez (101 hasta) influenza semptomlarının azalmasınakadar geçen sürede plaseboya göre (88 hasta) belirgin azalma gösterdi (p=0,01). Favipiravir 2400 mg/600 mg günde 3 kez (82 hasta) ile bu azalma gözlenmedi (p=0,414). Şekil 1: İnfluenza semptomlarının hafiflemesine kadar geçen süre *Favipiravir onaylı dozu 1 gün boyunca günde iki kez 1600 mg, ardından 4 gün boyunca günde iki kez 600 mg'dır. **6 primer influenza semptomunu (öksürük, boğaz ağrısı, baş ağrısı, nazal konjesyon, kas ağrısı, yorgunluk) ve vücut ısısını azaltmak için geçen süre A tipi veya B tipi influenza hastalarında primer sonlanım noktasının primer influenza semptomlarının hafiflediği süre olan plasebo kontrollü iki faz III çalışma yürütülmüştür. (1 günboyunca günde iki kez 1800 mg, ardından 4 gün boyunca günde iki kez 800 mg favipiravir oraluygulama [1800 mg /800 mg BID]; Çalışma 1 ve Çalışma 2) Favipiravir'in onaylanmış dozu1 gün boyunca günde iki kez 1600 mg oral, ardından 4 gün boyunca günde iki kez 600 mgoraldır. Çalışmanın birincil sonlanım noktası 6 primer influenza semptomunu (öksürük, boğazağrısı, baş ağrısı, burun tıkanıklığı, vücut ağrıları ve yorgunluk) ve vücut sıcaklığını hafifletmekiçin gereken süredir. Hafifleme ise 6 grip semptomunun tamamının ya hiç bulunmadığı ya dahafif olduğu ve ateşin düzeldiği, her ikisinin de en az 21,5 saat devam ettiği koşul olaraktanımlanmıştır. Çalışma sonuçları aşağıda sunulmuştur.
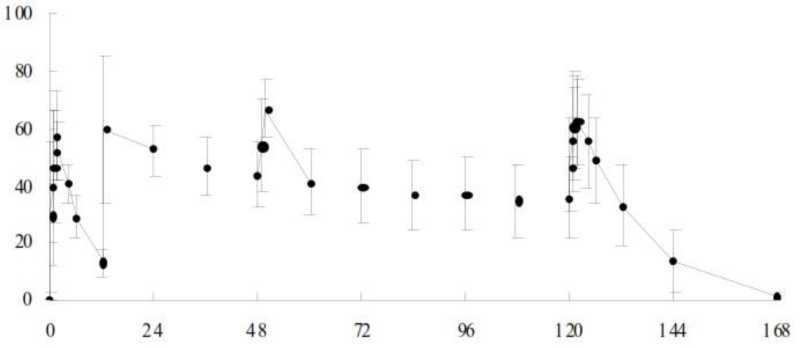
Şekil 2: Primer değerlendirme kriterinin Kaplan-Meier eğrisi ile gösterilmesi (ITT popülasyon, Çalışma 1) %
ı o -
T\\plaseboFavipiravirVA6ÛO İzlem süresi (saat)
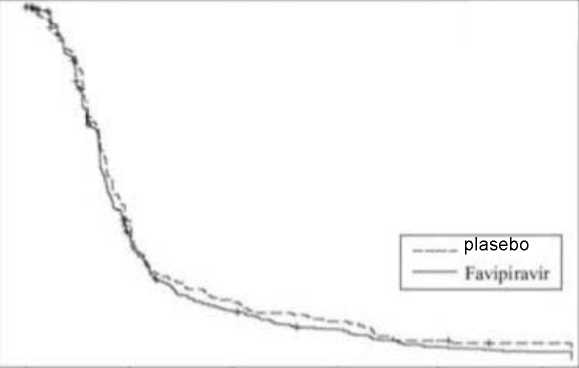
10 / 15 Şekil 3: Primer değerlendirme kriterinin Kaplan-Meier eğrisi ile gösterilmesi (ITT popülasyon, Çalışma 2)
 *Favipiravir onaylı dozu 1 gün boyunca günde iki kez 1600 mg, ardından 4 gün boyunca günde iki kez 600 mg'dır. **6 primer influenza semptomunu (öksürük, boğaz ağrısı, baş ağrısı,nazal konjesyon, kas ağrısı, yorgunluk) ve vücut ısısını azaltmak için geçen süre. Hafifleme, 21,5 saat boyunca 6influenza semptomunun hepsinin kaybolması veya hafif gözlenmesi ve ateşin geçmesi olaraktanımlanmıştır. Global faz III klinik çalışmasıTip A veya tip B influenza hastalarında yürütülen global faz III klinik çalışmasında (640 hasta: Japonyada 467 hasta, Korede 55 hasta, Tayvanda 118 hasta) favipiravir (erişkinlerde onaylanandozdan farklı dozaj*) ile oseltamivir fosfat (5 gün boyunca günde 2 kez 75 mg) karşılaştırıldı.Primer influenza semptomlarının azalmasına kadar geçen ortalama süre** (%95 GA),favipiravir kolunda (377 hasta) 63,1 saat (55,5-70,4) ve oseltamivir fosfat kolunda (380 hasta)51,2 saatti (45,9-57,6). Primer influenza semptomlarının azalmasına kadar geçen süredefavipiravirin oseltamivir fosfata hazard oranı (%95 GA) 0.818 idi (0,7070,948) ve favipiravirinetkililiği gösterilemedi (p=0,007, log-rank testi). *1. Gün 1200+400 mg ve takiben diğer 4 gün günde 2 kez 400 mg aldılar. Favipiravir onaylı dozu 1 gün boyunca günde iki kez 1600 mg, ardından 4 gün boyunca günde iki kez 600 mg'dır. **Çalışma ilacını almayı takiben 7 primer influenza semptomunu (öksürük, boğaz ağrısı, baş ağrısı, nazal konjesyon, kendini ateşli hissetme, kas ağrısı, yorgunluk) hafifletmek için geçensüre (tüm semptomların 1 veya daha aşağısında olduğu zaman). Hafifleme, hekim tarafındanişaretlenen hastanın günlüğündeki tüm skorların 1 veya altında olmasını takiben 21,5 saatboyunca değişmeden kalması olarak tanımlanmıştır. Japon olmayanlarda yürütülen faz II klinik çalışmasıTip A veya tip B influenza hastalarında yürütülen plasebo kontrollü faz II çalışma gerçekleştirildi (1000 mg/400 mg günde 2 kez, oral olarak 1. gün favipiravir günde 2 kez 1000 Bdge Do^Mw ^ı^-olarak!- Ganfavipiravir günde 2 kez 1200 mg ve sonraki 4 gün günde 2 kez 800 mg)*. Primer influenza 11 / 15 semptomlarının azalmasına kadar geçen süre** (%95 GA), 1000 mg/400 mg günde 2 kez grubunda (88 hasta) 100,4 saat (82,4-119,8), 1200 mg/800 mg günde 2 kez kolunda(121 hasta) 86,5 saat (79,2-102,3) ve plasebo kolunda (124 hasta) 91,9 saatti (70,3-105,3).Favipiravir ile plasebo grupları arasında belirgin bir farklılık gözlenmedi (p>0,05). *Favipiravir onaylı dozu 1 gün boyunca günde iki kez 1600 mg, ardından 4 gün boyunca günde iki kez 600 mg'dır. **6 primer influenza semptomunu (öksürük, boğaz ağrısı, baş ağrısı, nazal konjesyon, kas ağrısı, yorgunluk) ve vücut ısısını azaltmak için geçen süre. Hafifleme, tüm skorların 1 veyaaltında ve vücut ısısı 20-65 yaş kişilerde <380C ve 65 yaş ve üzeri kişilerde <37,80C olmasınıtakiben 21,5 saat boyunca değişmeden kalması olarak tanımlanmıştır. 5.2 Farmakokinetiközellikler Genel özelliklerEmilim:Aşağıdaki tablo 8 sağlıklı yetişkinde 1 gün boyunca günde iki kez 1600 mg, daha sonra 4 gün boyunca günde iki kez 600 mg, ardından 1 gün boyunca günde bir kez 600 mg (1600 mg /600mg BID) oral uygulamadan sonra favipiravir'in farmakokinetik parametrelerini göstermektedir.
12 / 15 Aldehid oksidaz aktivitesi az olan sağlıklı bir erişkine çoklu favipiravir oral uygulamasını takiben değişmeyen ilacın EAA değeri 1.gün 1452,73 gg.saat/mL ve 7.günde 1324,09gg.saat/mL idi*. *1. Gün 1200 mg+400 mg,2-6. Günler günde 2 kez 400 mg ve 7. Gün günde tek doz 400 mg. Favipiravir onaylı dozu 1 gün boyunca günde iki kez 1600 mg, ardından 4 gün boyunca günde iki kez 600 mg'dır. Dağılım:Japon olmayanlarda sonuçlarFavipiravir 20 sağlıklı erişkine 1. gün günde 2 kez 1200 mg ve sonraki 4 gün günde 2 kez 800 mg (1200/800 mg günde 2 kez)* oral olarak verildiğinde semende ilacın geometrik ortalamakonsantrasyonu 3. günde 18,341 gg/mL, ve tedavinin bitmesini takiben ikinci günde 0,053gg/mL idi. Tedaviden 7 gün sonra tüm kişilerde semen düzeyleri sınır değerlerin altına indi(0,02 gg/mL). Semendeki ilaç konsantrasyonunun plazmadaki konsantrasyona oranı 3.gün 0,53 ve tedaviden sonraki 2. gün 0,45 idi. * Favipiravir onaylı dozu 1 gün boyunca günde iki kez 1600 mg, ardından 4 gün boyunca günde iki kez 600 mg'dır. 0,3-30 gg/mL dozunda serum protein bağlanma oranı %53,4 -54,4 idi (in-vitro veriler). Hayvan verileriMaymunlara tek doz C-favipiravir oral olarak verildiğinde, geniş bir şekilde dokulara dağıldı. Her dokunun radyoaktivitesi uygulamadan sonra 0,5 saatte zirve yaptı ve plazmadakiradyoaktiviteye paralel değişim gösterdi. Akciğerlerdeki radyoaktivitenin plazmadakiradyoaktiviteye oranı ilacın alımını takiben 0,5 saatte 0,51 idi ve ilaç enfeksiyon yeri olarakdüşünülen respiratuvar dokulara hızlı bir şekilde dağıldı. Böbreklerdeki radyoaktiviteplazmadaki radyoaktiviteden yaklaşık 2,66 kat daha yüksekti. Her dokudaki radyoaktivite(kemik hariç) ilacın alınmasından 24 saat sonra zirve değerin %2,8 ve daha altına kadarinmiştir. Biyotransformasyon:Favipiravir sitokrom P-450 ile metabolize edilmez, çoğunlukla aldehid oksidaz (AO) ile metabolize edilir ve kısmen ksantin oksidaz (XO) ile hidroksillenmiş bir formada metabolizeolur. İnsan karaciğer mikrozomlarını kullanan çalışmalarda hidroksilat oluşumu AOaktivitesinde bireyler arası maksimum 12 kat değişim ile 3,98 ila 47,6 pmol/mg protein/dakikaarasında değişmektedir. Hidroksilatlı formdan farklı bir metabolit olarak insan plazması veidrarında bir glukuronat konjugatı gözlenmiştir. Eliminasyon:Favipiravir esas olarak hidroksillenmiş bir form olarak idrar ile atılır ve az miktarda değişmemiş ilaç gözlenir. 6 sağlıklı yetişkinle yapılan bir 7 günlük oral çoklu doz çalışmasında1, sonuygulamadan sonraki 48 saat boyunca değişmemiş ilacın ve hidroksillenmiş formun kümülatifidrar atılım oranı sırasıyla %0,8 ve %53,1 olmuştur. Doğrusallık/doğrusal olmayan durum:Veri yoktur. Hastalardaki karakteristik özelliklerBöbrek yetmezliği:Veri yoktur. Karaciğer yetmezliği:İlk gün günde 2 kez 1200 mg ve sonraki 4 gün günde 2 kez 800 mg alan hafif ve orta karaciğer yetmezliği olan hastalar (Child Pugh sınıflandırması A ve B; her grupta 6 hasta) sağlıklıgönüllüler ile karşılaştırıldığında; hafif karaciğer yetmezliği olan hastalarda 5. günde C maksmaksve EAA sırasıyla2.1 ve 6,3 kat daha yüksek gözlenmiştir. 5.3 Klinik öncesi güvenlilik verileriİnfluenza virüs A (H7N9), A (H1N1) pdm09 veya A (H3N2) ile aşılı fare enfeksiyon modellerinde favipiravir <60 mg/kg/gün dozunda oral olarak 5 gün verilmesini takiben akciğerdokularındaki virüs titrelerinde azalma gözlenmiştir. İnfluenza virüs A (H3N2) veya A (H5N1) ile aşılı fare enfeksiyon modellerinde favipiravir 30 mg/kg/gün dozunda oral olarak 5 gün verilmesini takiben terapötik etki gözlenmiştir. İnfluenza virüs A (H3N2) aşılı fare enfeksiyon modellerinde favipiravir 30 mg/kg/gün dozunda 14 gün verilmesini takiben terapötik etki gözlenmiştir. Hayvan çalışmalarında klinik maruziyete benzer ve ondan daha düşük düzeylerde maruziyet durumunda erken embriyonik ölüm (ratlar) ve teratojenisite (maymun, fare, rat ve tavşan)gözlenmiştir. Jüvenil köpeklerde (8 haftalık) yapılan 1 aylık çalışmada letal dozdan daha düşük dozda (60 mg/kg/gün) verilmesinden 20 gün sonra ölüm vakaları gözlenmiştir. Jüvenil hayvanlarda ( 6günlük ratlar ve 8 haftalık köpeklerde) anormal yürüyüş, iskelet sistemi kas liflerinde atrofi vepapiller kaslarda dejenerasyon/nekroz/mineralizasyon bildirilmiştir. Hayvan çalışmalarında ratlarda (12 haftalık) ve küçük köpeklerde (7-8 aylık) testislerde histopatolojik değişiklikler ve farelerde (11 haftalık) spermlerinde anormal bulgularbildirilmiştir. Uygulama sonlandırıldıktan sonra iyileşme veya iyileşmeye eğilim gözlenmiştir. 6. FARMASÖTİK ÖZELLİKLER6.1 Yardımcı maddelerin listesiMikrokristalin Selüloz pH 101 Krospovidon XL-10Povidon K30Prejelatinize NişastaKolloidal Silikon DioksitSodyum Stearil FumaratOpadry Yellow 03B620070 Opadry Yellow 03B620070 bileşirai: hipr°mel!°z 2910 6oP, titanyum dtotat, talk, polietilen Belge DolRG83SHY3YnUySHY3 Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys14 / 15 6.2 GeçimsizliklerGeçerli değil. 6.3 Raf ömrü24 ay. 6.4 Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriği40 film kaplı tablet, Şeffaf PVC/ PVDC Alüminyum Folyo blister ambalajda, karton kutu kullanma talimatı ile birlikte ambalajlanır. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller 'Tıbbi Atıkların Kontrolü Yönetmeliği' ve 'Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmelikleri'ne uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİSanta Farma İlaç San. A.Ş. Okmeydanı, Boruçiçeği Sok. No:16 34382 Şişli - İSTANBUL0212 220 64 000212 222 57 59 8. RUHSAT NUMARASI:2020/205 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİRuhsat tarihi: 11.10.2020 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ
15 / 15 1ünde 1200 mg+400 mg, daha sonra 2.ve 6.günlerde günde iki kez 400 mg, ardından Belge Don ¦Belge Takin Adresı:https://.www.turkiye.gov.tr/saglik-titqk-.ebvs7.günde günde bir kez 400 mg. Onaylanmış favipiravir dozu, 1 gün boyunca günde iki kez 1600 mg oral, ardından 4 gün boyunca günde iki kez 600 mg oraldir. 13 / 15 |
İlaç BilgileriConvolyn 200 Mg Film Kaplı TabletEtken Maddesi: Favipiravir Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.