Fanhdi 1000 Iu/10 Ml Iv Enjeksiyonluk Çözelti Hazırlamak İçin Liyofilize Toz ve Çözücü Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonlarıTÜFAM'a bildirmeleri beklenmektedir. Bakınız Bölüm 4.8. Advers reaksiyonlar nasılraporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIFANHDI 1000 I.U./10 mL IV Enjeksiyonluk Çözelti Hazırlamak İçin Liyofilize Toz ve Çözücü Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her flakonda; 1000 I.U. insan koagülasyon faktörü (Faktör VIII) içeren enjeksiyon için liyofilize toz bulunur. Ürün, 10 mL enjeksiyonluk suda çözüldükten sonra, yaklaşık olarak 100 I.U./mL insan koagülasyon faktörü VIII içerir. Potens I.U. olarak, Avrupa Farmakopesi'ndeki kromojenik tayin metodu ile saptanır. FANHDFnin spesifik aktivitesi, dozuna bağlı olarak en az 10 I.U./mg protein'dir. Yardımcı maddeler:Yardımcı maddeler için 6,1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyon için liyofilize toz Beyaz ya da açık sarı renkli toz içeren flakon ve enjeksiyonluk su içeren enjektör. Çözelti berrak veya hafif opelesan olmalıdır. 4. KLİNİK ÖZELLİKLER4.1. Terapötik EndikasyonlarFANHDI, hemofili A hastalığına (konjenital faktör VIII eksikliği) bağlı olarak ortaya çıkan kanamanın tedavisi veya profilaksisinde endikedir. FANHDI, yetersiz olan pıhtılaşmafaktörünün yerini geçici olarak alarak, kanamalı olayların engellemesine ya da kontrol altınaalınmasına; acil ya da planlı cerrahi müdahalelere olanak verir. 4.2. Pozoloji ve uygulama şekliPozoloji/Uygulama sıklığı ve süresi:Tedavi hemofili konusunda uzman bir doktor tarafından başlatılmalı ve devam ettirilmelidir. FANHDFnin tedavi dozu ve süresi her hastanın gereksinimine göre ayarlanmalıdır. Faktör VIII'ün ünite sayısı, Uluslararası Ünite (I.U.) olarak belirtilir. Plazmadaki faktör VIII aktivitesi yüzde olarak (normal insan plazmasına göreceli olarak) veya I.U. şeklinde(plazmadaki faktör VIII için uluslararası standarda göreceli olarak) tanımlanır. 1 / 10 Faktör VIII aktivitesinin 1 I.U.'si, normal insan plazmasının 1 mL' si içindeki faktör VIII miktarına eşdeğerdir. Faktör VIII'in gerekli dozunun hesaplanması; vücut ağırlığının kg'ıbaşına 1 I.U. faktör VIII'in plazma faktör VIII aktivitesini, normal aktivitenin yaklaşık olarak %2 kadar yükselttiği şeklindeki ampirik bulguya dayanmaktadır.Gerekli dozaj aşağıdaki formül kullanılarak hesaplanabilir: Gerekli faktör VIII ünitesi (I.U.) = Vücut ağırlığı (kg) x istenen faktör VIII artışı (%) (I.U./mL) x 0.5 FANHDI ile tedavi sırasında hastaların plazma FVIII düzeyleri saptanmalı ve monitörize edilmelidir. Bu özellikle cerrahi durumlarda önemlidir.
Tedavi dönemi süresince, faktör VIII düzeyinin saptanması, infüzyonların tekrarlanma sıklığı ile uygulanan doza rehber olacaktır. Özellikle majör cerrahi müdahale durumunda,koagülasyon analizi (faktör VIII aktivitesi) aracılığı ile yapılan terapinin tam olarakgözlenmesi zorunludur. ProfilaksiŞiddetli hemofili A hastalarında, kanamaya karşı uzun süreli profilaksi için FANHDI 2-3 gün aralıklar ile 20-40 I.U./kg dozlarında uygulanmalıdır. Bazı vakalarda, özellikle gençhastalarda daha kısa süreli aralıklar ya da daha yüksek dozlar gerekebilir. Uygulama şekli:Tarif edildiği şekilde hazırlanan ürün uygulamadan önce ürün oda ya da vücut sıcaklığına kadar ısıtılmalıdır. 2 / 10 FANHDI yalnız intravenöz kullanım içindir. Uygulama hızı 10 mL/dk'yı geçmemelidir. Çözelti berrak veya hafif opelesan olmalıdır. Bulanıklık veya çökelti içeren çözeltiler kullanılmayıp imha edilmelidir. Çözülerek kullanılan ürünler uygulamadan önce, partikül yönünden gözle kontrol edilmelidir. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karaciğer yetmezliği:Böbrek/karaciğer yetmezliği olan insanlar için hiçbir veri mevcut değildir. FANHDI, sadece tıbbi kararda açıkça belirtildiyse hastalara uygulanmalıdır. Pediyatrik popülasyon:Klinik çalışmaların yetersizliği nedeniyle, 6 yaşından küçük çocuklarda FANHDI kullanımı önerilmemektedir. Geriyatrik popülasyon:Geriyatrik popülasyon için hiçbir veri mevcut değildir. FANHDI, sadece tıbbi kararda açıkça belirtildiyse yaşlı hastalara uygulanmalıdır. 4.3. KontrendikasyonlarÜrünün bileşimindeki etkin maddeye ya da yardımcı maddelerden herhangi birisine aşırı duyarlılığı olan hastalarda kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleri_FANHDI, insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibi, çeşitli hastalıklara yolaçabilen enfeksiyon yapıcı ajanlar içerebilirler. FANHDI'de Varyant Creutzfeldt-Jacobhastalığının bulaşma riski teorik olarak minimumken, klasik Creutzfeldt-Jacobhastalığının bulaşma riski hiçbir kanıtla desteklenmez. Alınan önlemlere rağmen, bu türürünler halen potansiyel olarak hastalık bulaştırabilir.Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıp kalmadığının izlenmesi, belirli virüs enfeksiyonlarınınhalihazırda varlığının test edilmesi ve belirli virüslerin yok edilmesi ve/veyainaktivasyonu ile azaltılmıştır. Bütün bu önlemlere rağmen, bu ürünler hala potansiyelolarak hastalık bulaştırabilirler. Ayrıca, henüz bilinmeyen enfeksiyon yapıcı ajanlarınbu ürünlerin içersinde bulunma ihtimali mevcuttur.HIV, HBV, HCV gibi zarflı virüsler ve HAV gibi zarflı olmayan virüsler için etkili önlemlerin alınmasına dikkat edilmelidir. Parvovirüs B19 gibi zarflı olmayan virüslerekarşı alınan tedbirler sınırlı sayıda olabilir. Parvovirüs B19 enfeksiyonu, gebelikte (fetalinfeksiyon) ve immün yetmezlik ya da kırmızı kan hücre üretiminde artış olanhastalarda tehlikeli olabilir (hemolitik anemi gibi).Doktor, bu ilacı hastaya reçete etmeden veya uygulamadan önce hastası ile risk ve yararlarını tartışmalıdır.3 / 10 Protein içeren ürünlerin intravenöz uygulanmaları ile alerjik tipte aşırı duyarlık reaksiyonları ortaya çıkabilmektedir. FANHDI, faktör VIII'ün dışında çok az miktarda insan proteini içerir.Hastalar; yaygın ürtiker, göğüsde sıkışma, hipotansiyon, anafilaksi vb. içeren aşırı duyarlığınerken işaretleri konusunda bilgilendirilmelidir. Bu semptomların oluşması halinde uygulamahemen durdurulmalı ve doktorla temasa geçilmelidir. Şok oluşması durumunda, şok tedavisiiçin geçerli tıbbi standartlar yerine getirilmelidir. İnhibitörlerFaktör VIII'e karşı nötralize edici antikor (inhibitörler) oluşumu, hemofili A hastalarının tedavisinde bilinen bir komplikasyondur. Bu inhibitörler genellikle faktör VIII prokoagülanaktiviteye yönelik olan IgG immünoglobulinleridir ve modifiye tetkik kullanılarak her mlplazmada Bethesda Ünitesi (BU) olarak ölçülür. İnhibitör gelişme riski, faktör VIII'emaruziyetin yanı sıra hastalığın şiddeti ile ilişkilidir ve bu risk ilk 50 maruziyet gününde enyüksek seviyededir; ancak risk yaygın görülmemesine rağmen yaşam boyu devam eder. İnhibitör gelişiminin klinik önemi inhibitör titresine bağlı olacaktır; düşük titrenin teşkil ettiği yetersiz klinik yanıt riski, yüksek titreli inhibitörlere kıyasla daha az olacaktır. Genel olarak, koagülasyon faktörü VIII ürünleri ile tedavi edilen tüm hastalar, uygun klinik gözlem ve laboratuvar testleri ile inhibitörlerin gelişimi açısından dikkatle izlenmelidir. Eğerbeklenen faktör VIII aktivitesinin plazma düzeylerine ulaşılamazsa veya yeterli doz ilekanama kontrol altına alınamazsa faktör VIII inhibitörü varlığı açısından test yapılmalıdır.İnhibitör düzeyleri yüksek olan hastalarda faktör VIII tedavisi etkili olmayabilir ve diğertedavi seçenekleri dikkate alınmalıdır. Böyle hastaların tedavisi hemofili ve faktör VIIIinhibitörleri tedavisi hasta tedavisinde deneyimli hekimler tarafından yönlendirilmelidir. Düzenli olarak insan plazma-türevi faktör VIII ürünleri uygulanan hastalar için uygun aşılama (hepatit A ve B'ye karşı) önerilmektedir. Biyolojik tıbbi ürünlerin takip edilebilirliğinin sağlanması için uygulana ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. Sodyum içeriğiBu tıbbi ürün her dozunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında sodyum içermez. Ancak pozolojiye ve hastanın vücut ağırlığına bağlı olarak, hasta birdenfazla şişe alabilir. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önündebulundurulmalıdır. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriFANHDI'nin ilaç veya tıbbi ürünlerle etkileşmeleri konusunda herhangi bir bulgu olmamasına rağmen diğer ilaçlarla karıştırılması önerilmemektedir. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (kontrasepsiyon)Kadınlarda hemofili A'nın nadir görülmesine dayalı olarak, çocuk doğurma potansiyeli/kontrasepsiyon bulunan kadınlarda faktör VIII kullanıma ilişkin deneyim 4 / 10 bulunmamaktadır. Kontraseptif yöntemlerle etkileşimi bilinmemektedir. Bu nedenle, faktör VIII sadece açıkça endike olduğu taktirde kullanılmalıdır. Gebelik dönemiFaktör Vlll'in gebelikte kullanım güvenilirliğine dair kontrollü klinik çalışmalar yapılmamıştır. Kadınlarda hemofili A oluşumunun azlığı nedeniyle, gebelik dönemi süresince faktör VIII kullanım deneyimi bulunmamaktadır. Bundan dolayı, faktör VIII konsantreleri gebelik döneminde, mutlak zorunlu görülmesi durumunda kullanılmalıdır. Laktasyon dönemiKadınlarda hemofili A oluşumunun azlığı nedeniyle, emzirme dönemi süresince faktör VIII kullanım deneyimi bulunmamaktadır. Bundan dolayı, faktör VIII konsantreleri laktasyon döneminde mutlak zorunlu görülmesi durumunda kullanılmalıdır. Üreme yeteneği/fertiliteFANHDI ile hayvan üreme çalışmaları yürütülmemiştir. İnsanlarda üreme yeteneği/fertiliteyi etkileyip etkilemediği bilinmemektedir. 4.7. Araç ve makine kullanımı üzerindeki etkilerFaktör VIII ürünlerinin araba sürme ya da makine kullanma yeteneği üzerinde olumsuz etkisi olduğuna dair hiçbir veri yoktur. 4.8. İstenmeyen etkilerGüvenlilik profili özetiAşırı duyarlılık veya alerjik reaksiyonlar (bunların arsında anjiyoödem, infüzyon bölgesinde yanma ve batma, titreme, sıcak basması, yaygın ürtiker, baş ağrısı, kurdeşen, kan basıncındadüşme, letarji, bulantı, huzursuzluk, taşikardi, göğüste sıkışma hissi, karıncalanma, kusma,hırıltılı solunum yer alabilir) nadiren gözlenmiştir ve bazı olgularda şiddetli anafilaksiye kadarilerleyebilir (şok dahil). Çok ender olarak, vücut sıcaklığında yükselme gözlenmiştir. FANHDI de dahil olmak üzere faktör VIII ile tedavi edilmiş hemofili A hastalarında nötralize edici antikorlar (inhibitörler) gelişebilir (Bkz. Bölüm 5.1). Bu tür inhibitörler oluşursa, durum,yetersiz klinik yanıt şeklinde kendini gösterebilir. Bu gibi durumlarda uzman hemofilimerkezleriyle bağlantı kurulması önerilmektedir. 7000 infüzyondan fazla Fanhdi alan 164 hastanın dahil edildiği çeşitli klinik çalışmalar yapılmıştır. Bu çalışmalardan elde edilen veriler advers olayların insidansı düşük olduğu içinürünün iyi bir tolerabilitiye sahip olduğunu gösterir. Araştırmacıların belgelendirmesine göreiki advers olay, potansiyel olarak çalışılan medikal ürün ile ilişkilendirilmiştir. İnfüzyonsonrası iki ateş nöbetinden oluşmaktadır. 5 / 10 Advers reaksiyonların tablolaştırılmış listesiAşağıda verilen tablo, MedDRA sistem organ sınıflandırmasına (SOC ve Tercihli Terim Düzeyi) uygundur. Görülme sıklıkları şu yaklaşıma göre değerlendirilmiştir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek(<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Advers reaksiyonlar her bir sıklık grubunda azalan ciddiyet sırasına göre verilmiştir.
* Sıklık, şiddetli hemofili A hastalarının yer aldığı, tüm FVIII ürünleri ile yapılmış çalışmalara dayanmaktadır.TGH'ler = Daha önce tedavi görmüş hastalar, HTGH'ler = Daha önce tedavi görmemiş hastalarEnfeksiyon ajanlarının bulaşma riski tümüyle ortadan kaldırılamaz. Viral güvenliği için 4.4'e bakınız. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr;[email protected]. Doz aşımı ve tedavisiDoz aşımı vakaları rapor edilmediği için doz aşımı sonuçları bilinmemektedir. 5. FARMAKOLOJİK ÖZELLİKLER5.1.Farmakodinamik özelliklerFarmakoterapik grup: Antihemorajikler: Kan koagülasyon faktörü VIII ATC kodu: B02BD02 Etki mekanizması: FANHDI'deki Faktör VIII, von Willebrand faktörü ile kompleks olarak sunulmaktadır. Faktör VIII/von Willebrand faktörü kompleksi farklı fizyolojik işlevlere sahip iki molekülden (Faktör VIII ve von Willebrand faktörü) oluşur. Bir hemofili hastasına uygulandığında, Faktör VIII hastanın kan dolaşımında von Willebrand faktöre bağlanır. 6 / 10 Aktif Faktör VIII, Faktör X'un aktif Faktör X'a dönüşmesini hızlandırarak, aktif Faktör IX için bir kofaktörü olarak hareket eder. Aktif Faktör X protrombini trombine dönüştürür.Ardından trombin, fibrinojeni fibrine dönüştürür ve pıhtı oluşur. Hemofili A, azalan Faktör VIII kompleksi seviyesi sebebiyle, cinsiyete bağlı kalıtsal bir kan pıhtılaşma hastalığıdır ve eklemlerde, kaslarda, ya da iç organlarda ya kendiliğinden ya dakaza ya da cerrahi travmanın sonucu olarak şiddetli kanama ile sonuçlanır. Yerine koymatedavisi ile Faktör VIII plazma seviyesi artar, böylece geçici olarak Faktör VIII eksikliğiningiderilmesi ve kanama eğiliminin giderilmesi sağlanır. Faktör VlII'in koruyucu protein rolüne ek olarak, von Willebrand vasküler yaralanma bölgelerine trombosit tutunmasına aracılık eder ve trombosit agregasyonunda rol oynar. Onaylı endikasyonlar için 6 yaşından küçük çocuklarda yürütülen klinik araştırmalardan yeterli veri elde edilememiştir. İmmün tolerans indüksiyon (ITI) verileri, FVIII'e inhibitör geliştiren pediyatrik ve yetişkin hemofili A hastalarından elde edilmiştir. İmmün tolerans elde etmek için değişken prognozasahip primer ve kurtarma (rescue) ITI hastalarının geniş bir spektrumunu içeren retrospektifbir çalışmadan 57 hasta ve prospektif çalışmalardan 14 hasta değerlendirmeye alınmıştır.Veriler, Fahndi'nin immün toleransı indüklemek için kullanıldığını göstermektedir.Toleransın elde edildiği hastalarda, FVIII konsantresi ile profilaktik veya ihtiyaç tedavisindekanama önlenebilir veya kontrol altına alınabilir. 5.2. Farmakokinetik özelliklerGenel Özellikler:Emilim:FANHDI ile yürütülen klinik çalışmalardan elde edilen in vivogeri kazanım, uygulanan IU/kg başına yaklaşık 2,1 ± 0,4 IU/dL'ye eşdeğer (kromojenik metdotla gerçekleştirilen tayin) olan105,5 ± %18,5'tir.Dağılım:Ortalama tutulma zamanı (MRT) 19,9 ± 4,1 saattir. Plazma konsantrasyon-zaman eğrisinin altında kalan alan (EAA) 19,3 ± 3,8 IU saat/mL'dir. Biyotransformasyon:Faktör VIII, bir protein olup, dolayısıyla endojen proteinlere benzer şekilde metabolize edilir. Eliminasyon:FANHDI ile yürütülen klinik çalışmalardan elde edilen insan FVIII'in yarılanma ömrü 14,18 ± 2,55 saattir. Klirens 2,6 ± 0,6 mL/saat/kg'dır. Doğrusallık/Doğrusal olmayan durum:Sağlıklı bir veri mevcut değildir. 5.3. Klinik öncesi güvenlilik verileriİnsan plazma koagülasyon faktörü VIII, insan plazmasının normal bir bileşeni olup, endojen faktör VIII gibi davranır. Yüksek dozlar aşırı yükleme ile sonuçlandığı için tek doz toksisitetesti bağımsızdır. 7 / 10 Hayvanlar üzerinde yapılan tekrarlanan doz toksisite testleri, heterolog proteine antikor gelişimiyle etkileşmesine bağlı olarak uygulanamaz. Vücut ağırlığının kilogramı başına önerilen insan dozu çok defalar uygulansa bile, laboratuvar hayvanlarında toksik etki göstermemektedir. Klinik deneyler, insan plazma koagulasyon faktörü VIII'in tümörijenik ve mutajenik etkileri hakkında ipucu sağlamazken, deneysel çalışmalar, özellikle heterolog türlerde zorunlusayılmamaktadır. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiHistidin İnsan albüminiArjinin Enjeksiyonluk su 6.2. GeçimsizliklerÜrün diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3. Raf ömrü36 aydır. 6.4.Saklamaya yönelik özel uyarılar30°C altındaki sıcaklıklarda saklayınız. Ürün dondurulmamalıdır. Donmuş ürün çözülüp kullanılmamalıdır. Fiziksel ve kimyasal in-use stabilitesi 25°C'de 12 saattir. Mikrobiyolojik açıdan ürün hemen kullanılmalıdır. Eğer hemen kullanılmayacaksa, saklama süreleri ve koşulları kullanıcınınsorumluluğunda olup, seyreltme kontrolü ve valide edilmiş aseptik koşullarda yapılmadıkçasaklama süresi 2°C - 8°C'de 24 saatten fazla olmamalıdır. 6.5. Ambalajın niteliği ve içeriğiHer kutuda; 1000 I.U. liyofilize faktör VIII içeren klorobütil kauçuk tıpalı, alüminyum kapüşonlu, polipropilen kapaklı 1 adet 20 ml Tip II cam flakon ve bromobütil kauçuk pistontıpalı, sentetik poliizopren kapak uçlu, 10 ml enjeksiyonluk su içeren 1 adet 10 ml Tip I camenjektör bulunmaktadır. Ürünün rekonstitüsyonu ve uygulanması için; 1 flakon adaptörü, 1 mikrofiltre, 2 adet alkollü bez ve 1 infüzyon seti, FANHDI ambalajı ile birlikte sunulur. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Ürünlerin Kontrol Yönetmeliği ve 'Ambalaj ve Ambalaj Atıklarının Kontrol Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİDem İlaç San. ve Tic. A.Ş. Dem Plaza İnönü Mah. Kayışdağı Cad. No:172 34755 Ataşehir / İSTANBUL 8 / 10 Tel: 0 216 4284029 Fax: 0 216 4284086 8. RUHSAT NUMARASI104 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 04.06.2013 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİAŞAĞIDAKİ BİLGİLER BU İLACI UYGULAYACAK SAĞLIK PERSONELİ İÇİNDİR. Çözeltinin hazırlanması:1- Flakon ve enjektörü 30°C üzerine çıkmayacak şekilde ısıtınız. 2- Çözücüyü içeren enjektöre piston yerleştiriniz. 3- Filtreyi ambalajdan çıkarınız. Enjektör başlığını çıkarınız ve enjektörü filtreye tutturunuz. 4- Flakon adaptörünü ambalajından çıkarınız, enjektör ve filtreye tutturunuz. 5- Flakonun plastik başlığını çıkarınız ve kauçuk tıpanın yüzeyini bir antiseptik ile siliniz. 6- Adaptör iğnesi ile tıpayı deliniz. 7- Tüm çözücüyü enjektörden flakona aktarınız. 8- Toz tümüyle çözününceye kadar flakonu hafifçe çalkalayınız. Bütün parenteralçözeltilerde olduğu gibi, çözünme tümüyle olmazsa ya da parçacıklar kalırsakullanmayınız. 9- Enjektör/filtreyi, flakon/adaptörden, vakum serbest bırakarak ayırınız. 10- Flakonu ters çeviriniz ve çözeltinin enjektöre geçmesini sağlayınız. 11- Hastanın enjeksiyon yapılacak bölgesini hazırlayınız, enjektörü flakondan ayırınız vesteril bir iğne ya da kelebek seti kullanarak ürünü enjekte ediniz. 12- Enjeksiyonu damara 3 mL/dk hızda yapınız. Vazomotor reaksiyonları önlemek amacıylauygulama hızı 10 mL/dk'yı geçmemelidir. Uygulama setini tekrar kullanmayınız. Ürünün kullanılmayan kısmı veya atık malzeme, ilgili yönetmeliğe uygun olarak imha edilir. 9 / 10 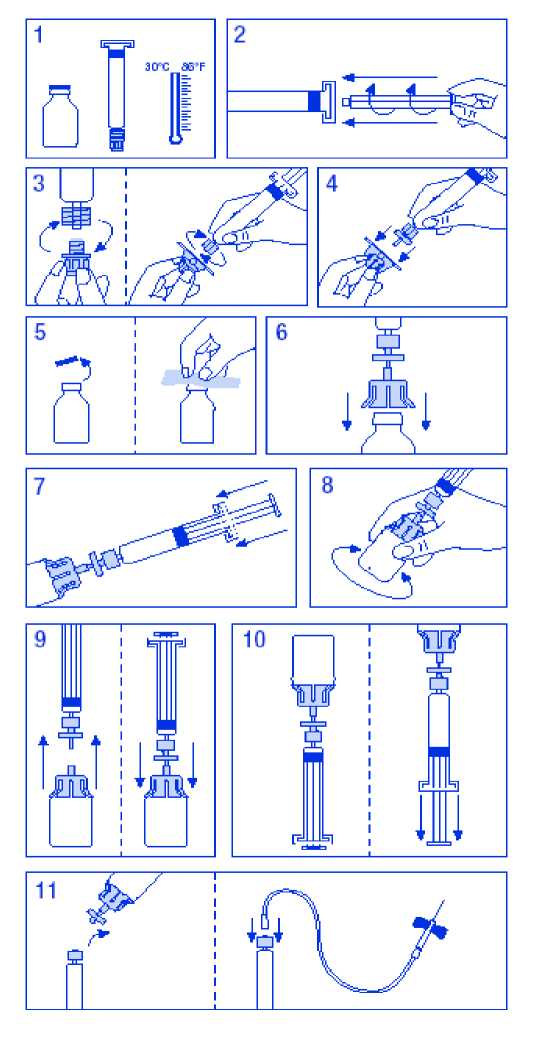
10 / 10 |
İlaç BilgileriFanhdi 1000 Iu/10 Ml Iv Enjeksiyonluk Çözelti Hazırlamak İçin Liyofilize Toz ve ÇözücüEtken Maddesi: İnsan Koagülasyon Faktörü (faktör Viii) Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.