Haemonine 500 Iu/5 Ml Iv Enjeksiyonluk Çözelti Hazırlamak İçin Toz ve Çözücü Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADIHAEMONINE 500 IU/5 mL IV enjeksiyonluk çözelti hazırlamak için toz ve çözücü Steril, apirojen 2. KALİTATİF VE KANTİTATİF BİLEŞİMİEtkin Madde:İnsan koagülasyon faktörü IX HAEMONINE toz flakonu, 5 mL enjeksiyonluk su ile sulandırıldıında yaklaşık 100 IU/mL insan koagülasyon faktörü IX ihtiva eder. Potens (IU) Avrupa Farmakopesi bir aşamalı pıhtılaşma testi kullanılarak belirlenir. HAEMONINE'nın spesifik aktivitesi > 70 IU/mg proteindir. Yardımcı maddeler:1 ml sulandırılmış çözelti: Sodyum sitrat: 5.16 mg Sodyum klorür: 9.12 mg Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk toz ve çözücü Sulandırıldıktan sonra gözle görülür partikül içermeyen berrak ya da hafif opak çözelti elde edilir. 4. KLİNİK ÖZELLİKLER4.1. Terapötik ¦EendikasyonlarHemofili B (konjenital Faktör IX eksiklii) hastalarında kanamanın profilaksisi ve tedavisinde endikedir. HAEMONINE yetişkinlerde, ergenlerde ve 6 yaş ve üzeri çocuklarda endikedir. 4.2. Pozoloji ve uygulama şekliTedaviye mutlaka hemofili tedavisi konusunda uzman bir doktorun gözetiminde başlanmalıdır. Pozoloji / uygulama sıklıı ve süresi:Uygulanacak doza ve tekrarlanan infüzyonların sıklıına rehberlik etmesi için tedavinin seyri boyunca, faktör IX seviyelerinin uygun şekilde belirlenmesi önerilmektedir. Her hastanın faktör
1 IX'a yanıtı farklı yarılanma ömürleri ve iyileşme süreleri ile deişiklik gösterebilir. Vücut aırlıına balı doz normalin altındaki veya üzerindeki kiloya sahip hastalarda ayarlamagerektirebilir. Özellikle büyük cerrahi girişimlerde, koagülasyon analizi (plazma faktör IXaktivitesi) vasıtasıyla ikame tedavisinin hassas şekilde izlenmesi zorunludur. Doz ve ikame tedavisinin süresi, faktör IX eksikliinin şiddetine, kanamanın yeri ve derecesi ile hastanın klinik durumuna balıdır. Uygulanan faktör IX birimi sayısı, faktör IX ürünleri için Dünya Salık Örgütü tarafından onaylanmış mevcut Uluslararası Birim (IU) cinsinden ifade edilir. Faktör IX'in plazmadakiaktivitesi, yüzde (normal insan plazmasına göre) olarak veya Uluslararası Birim (plazmadakifaktör IX için uluslararası bir standarda göre) cinsinden ifade edilir. Faktör IX aktivitesinin bir Uluslararası Birimi (IU) bir mL normal insan plazmasında bulunan faktör IX miktarıdır. İhtiyaç halinde ilaç alımıGerekli faktör IX dozunun hesaplanması, şu ampirik bulguya dayanmaktadır: Vücut aırlıının kg'ı başına 1 IU faktör IX, plazma faktör IX aktivitesini normal aktiviteye göre %1-2 arttırır.Gerekli doz aşaıdaki formül kullanılarak bulunur: Gerekli IU= vücut aırlıı (kg) x istenen faktör IX artışı (%) (IU/dL) x 0.8Uygulanacak dozun miktarı ve uygulama sıklıı her bir vakadaki klinik etkinlie göre uyarlanmalıdır. Aşaıda belirtilen hemorajik durumların ortaya çıkması halinde, faktör IX aktivitesi, bu süre içinde, belirtilen plazma aktivitesi düzeyinin (normalin %'si veya IU/dL) altına düşmemelidir.Aşaıdaki tablo, kanama vakaları ve ameliyatlarda kullanılacak dozlar için kılavuz olarakkullanılabilir:
Daha youn hemartroz, kas 30 -60İnfüzyon, 3-4 gün veya daha uzun kanaması veya hematom süreyle 24 saatte bir tekrarlanır, arı ve akut iş görememe hali geçinceye kadar _ devam edilir._Hayati tehlike yaratan 60-100Tehlike geçinceye kadar 8-24 saatte bir kanamalar infüzyon tekrarlanır. Cerrahi Operasyon_Minör (diş çekimi dahil) 30- 60İyileşinceye kadar en az 1 gün, 24 saatte bir uygulanır. --Bu bolgo. güvenli elektronik imza ile imzalanmıştır.-.--Belge DoMâjöTodu: lZW56MOFyaklUZW56ZlAxSl80d00W56_BelgeY&rad& yg&friiyilt8şme Şal^fekASaya ys2 (Operasyon öncesi kadar infüzyon 8-24 saatte bir ve sonrası)tekrarlanır, daha sonra %30-%60 (IU/dL) faktör IX aktivitesi salanıncaya kadar en az 7gün daha devam edilir. Profilaksi:Aır hemofili B vakalarında, kanamanın uzun süreli profilaksisi için, normal dozlar 3-4 günlük aralıklarla, vücut aırlıının her bir kilogramı için 20-40 IU faktör IX'dur. Bazı durumlarda,özellikle daha genç hastalarda, daha kısa doz aralıkları veya daha yüksek doz uygulamasıgerekebilir. Uygulama şekli:Ürün intravenöz yolla uygulanır. Uygulamadan önce tıbbi ürünün sulandırılmasına ilişkin talimatlar için Bölüm 6.6'ya bakınız. Uygulama hızının 5 mL/dakika olan maksimum infüzyonhızını aşmaması önerilir. Özel popülasyona ilişkin ek bilgiler:Böbrek / Karacier yetmezlii:HAEMONINE'nın böbrek ve karacier yetmezlii olan hastalardaki güvenlilik ve etkililii incelenmemiştir. Pediyatrik popülasyon:6 yaşından küçük çocuklarda HAEMONINE kullanımına dair yeterli veri bulunmamaktadır. Geriyatrik popülasyon: Dozaj ve infüzyon hızı hastanın durumuna göre ayarlanır.4.3. KontrendikasyonlarÜrünün etken madde ya da Bölüm 6.1'de listelenen herhangi bir bileşene veya heparine karşı aşırı duyarlılıı olan hastalarda kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleriVirüs güvenlii:HAEMONINE, insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibi,çeşitli hastalıklarayol açabilenenfeksiyon yapıcı ajanlar içerebilirler.HAEMONINE 'da Varyant Creutzfeldt-Jacob hastalıının bulaşma riski teorik olarak minimumken, klasik Creutzfeldt-Jacob hastalıının bulaşma riski hiçbirkanıtla desteklenmez. Alınan önlemlere ramen, bu tür ürünler halen potansiyelolarak hastalık bulaştırabilir.Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıpkalmadıının izlenmesi,belirli virüsenfeksiyonlarının halihazırda varlıının test edilmesi ve belirli virüslerin yokedilmesi ve/veya inaktivasyonu ile azaltılmıştır. Bütün bu önlemlere ramen,bu ürünler hala potansiyel olarak hastalık bulaştırabilirler. Ayrıca, henüzbilinmeyen enfeksiyon yapıcı ajanların bu ürünlerin içerisinde bulunma ihtimali mevcuttur.HIV, HBV, HCV gibi zarflı virüsler1 ve HAV gibi zarflı olmayan virüslerBelge Dorulama Kodu:lZW56M0ryaklUZW5üZlAxSIIY3ZlAxZW56-Delge Takıp Adı-esnıttps://www.turkıye.gov.tr/saglık-tıtek-ebys-3 için etkili önlemlerin alınmasına dikkat edilmelidir. Parvovirüs B19 gibi zarflı olmaya virüslere karşı alınan tedbirler sınırlı sayıda olabilir. Parvovirüs B19enfeksiyonu, gebelikte (fetal infeksiyon) ve immünyetmezlikya dakırmızıkan hücre üretiminde artış olan hastalarda tehlikeli olabilir (hemolitik an emi gibi).Doktor, bu ilacı hastaya reçete etmeden veya uygulamadan önce hastası ile risk veyararlarını tartışmalıdır._Ayrıca; HAEMOMINE kullanılması gerekiyorsa hekim tarafından, hastalık yapıcı etkenlerin hastaya bulaşmasını önlemek için uygun aşıların (Hepatit A, Hepatit B vb.) yaptırılması önerilebilir. Aşırı duyarlılıkHAEMONINE ile alerjik tipte aşırı duyarlılık reaksiyonları görülebilir. Ürün faktör IX dışında eser miktarlarda insan proteini içerir. Hastalara, bu tip semptomlar görüldüünde, ürünkullanımının derhal durdurmaları ve doktora başvurmaları tavsiye edilmelidir. Hastalar,kurdeşen, bütün vücudu saran ürtiker, göüs sıkışması, hırıltılı soluma, hipotansiyon veanafilaksiyi de içine alan aşırı duyarlılık reaksiyonlarının erken belirtileri konusundabilgilendirilmelidir. Şok durumunda şok için standart tedavi uygulanmalıdır. İnhibitörlerİnsan koagülasyon faktör IX ürünleri ile tekrarlı tedaviden sonra hastalar, nötralize edici ajanların (inhibitör) gelişip gelişmediinin saptanması açısından izlemelidir. İnhibitörler, uygun biyolojiktestler kullanılarak Bethesda Birimi (BU) cinsinden kantifiye edilmelidir. Literatürde, faktör IX inhibitörleri ile alerjik reaksiyonların görülmesi arasında bir korelasyon olduunu gösteren raporlar bulunmaktadır. Bu nedenle, alerjik reaksiyon gösteren hastalar,inhibitör mevcutiyeti açısından deerlendirilmelidir. Faktör IX inhibitörü taşıyan hastalarda,tekrarlanan faktör IX uygulamasında anafilaksi riskinin daha yüksek olduu akılda tutulmalıdır. Faktör IX ürünleri ile olası alerjik reaksiyon riski nedeniyle, faktör IX başlangıç uygulaması, tedavi eden hekimin kararına göre, alerjik reaksiyonlara gerekli müdahelenin yapılabilecei birmerkezde, tıbbi gözetim altında yapılmalıdır. TromboembolizmPotansiyel tromboembolik komplikasyon riskinden dolayı, karacier hastalıı olanlarda, ameliyat sonrasında, yeni doanlarda veya trombotik olay veya dissemine (yaygın) intravaskülerkoagülasyon (DIC) riski taşıyan hastalarda bu ürün uygulanırken, uygun biyolojik testlerletrombotik semptomlar ve konsumptif koagülopatinin erken belirtilerini saptayabilmek için klinikgözetim uygulanmalıdır. Bu durumların her birinde, HAEMONINE uygulamasının potansiyelyararı, yukarıda belirtilen komplikasyonlara göre hesaplanmalıdır. Aditif veya sinerjistik farmakodinamik etkilerden dolayı, antifibrinolitik ajanların anti-inhibitör koagülan kompleksi veya faktör IX kompleksi ile birlikte kullanımı tromboz riskini artırabilir. Kardiyovasküler olaylarMevcut kardiyovasküler risk faktörlerine sahip hastalarda, faktör IX ile tedavi kardiyovasküler riski artırabilir. Belge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys4 Kateter ilişkili komplikasyonlarEer bir santral venöz erişim aracı gerekliyse, lokal enfeksiyonlar, bakteriyemi ve kateter bölgesi trombozu dahil santral venöz erişim aracı ile ilişkili komplikasyonlara dikkat edilmelidir. Bulaşıcı ajanlarİnsan kanı ya da plazmasından üretilen tıbbi ürünlerin kullanımının neden olduu enfeksiyonları önlemek için alınan standart önlemler, baışçıların seçimini, bireysel baışların ve plazmahavuzlarının spesifik enfeksiyon markerleri için taramadan geçirilmesini ve virüslerin etkisizhale getirilmesi/elimine edilmesi için etkin üretim basamaklarının dahil edilmesini içerir. Bunaramen, insan kanından ya da plazmasından hazırlanan ilaçlar hastalara uygulandıında,enfeksiyona neden olan ajanların bulaşma olasılıı tamamen ortadan kaldırılamaz. Bu durumayrıca bilinmeyen ya da sonradan ortaya çıkan virüsler ve dier patojenler için de geçerlidir. Alınan önlemlerin, insan immün yetmezlii virüsü (HIV), hepatit B (HBV) virüsü, hepatit C (HCV) virüsü gibi zarflı virüsler ve zarfsız hepatit A virüsü (HAV) için etkili olarak kabul edilir. Alınan önlemler parvovirüs B19 gibi zarfsız virüslere karşı sınırlı deerde olabilir. Parvovirüs B19 enfeksiyonu, hamile kadınlar (fetal enfeksiyon) ve immün yetmezlii veya artmış eritropoeziolan (örn.; hemolitik anemi) kişiler için ciddi olabilir. Hasta ve ürün serisi arasındaki ilişkiyi belirlemek için her bir HAEMONINE dozunun alımında ürünün adının ve seri numarasının kaydedilmesi önemle önerilmektedir. Pediyatrik popülasyon:Listelenen uyarılar ve önlemler hem 6 yaş ve üzeri çocuklar hem yetişkinler için geçerlidir. (Bkz. Bölüm 4.2) Bu ilaç 2000 IU'lik her dozunda maksimum 4.9 mmol (113 mg) sodyum ihtiva eder. Bu durum kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır. 4.5. Dier tıbbi ürünler ile etkileşimler ve dier etkileşim şekilleriHerhangi bir etkileşim çalışması yürütülmemiştir. İnsan koagülasyon faktörü IX'un, dier ilaçlarla etkileştiine dair herhangi bir bildirim bulunmamaktadır. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karacier yetmezlii:Etkileşim çalışması yapılmamıştır. Geriyatrik popülasyon:Etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:Etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategori: C Bu belge Belge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys 5 Çocuk dourma potansiyeli bulunan kadınlar/ doum kontrolü (Kontrasepsiyon)HAEMONINE'nın, çocuk dourma potansiyeli bulunan kadınlarda kullanımına ilişkin özel bir öneri veya, tedavi sırasında veya sonrasında doum kontrolünün gerekli olduuna dairherhangi bir bilgi söz konusu deildir. Gebelik dönemi:HAEMONINE'nın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut deildir. Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doum /ve-veya/ doum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (Bkz. Bölüm 5.3).İnsanlara yönelik potansiyel risk bilinmemektedir. Bu nedenle, HAEMONINE gebelik ve emzirme dönemi boyunca yalnız açık şekilde endike olduu durumlarda kullanılmalıdır. Laktasyon dönemi:Faktör IX'un insan sütüyle atılıp atılmadıı bilinmemektedir. Faktör IX'un süt ile atılımı hayvanlar üzerinde araştırılmamıştır. Emzirmenin durdurulup durdurulmayacagına ya daHAEMONINE tedavisinin durdurulup durdurulmayacaına/tedaviden kaçınılıpkaçınılmayacaına ilişkin karar verilirken, emzirmenin çocuk açısından faydası veHAEMONINE tedavisinin çocuk açısından faydası dikkate alınmalıdır. Üreme yetenei/Fertilite:Faktör IX'un üreme yetenegi ve fertilite üzerindeki etkisi ile ilgili hayvan deneyleri mevcut degildir. 4.7. Araç ve makine kullanımı üzerindeki etkilerHAEMONINE'nın araç ve makine kullanımı üzerinde etkisi yoktur veya ihmal edilebilir düzeydedir. 4.8. İstenmeyen etkilerGüvenlilik profilinin özetiAşırı duyarlılık reaksiyonları veya anafilaktik reaksiyonlar (anjiyoödem, enfeksiyon bölgesinde yanma ve batma hissi, souk terleme, sıcak basması, genel ürtiker, baş arısı, kurdeşen,hipotansiyon, letarji, bulantı, huzursuzluk, taşikardi, göüste rahatsızlık hissi, karıncalanma,kusma, hırıltılı nefes) nadiren gözlenmiştir. Bazı vakalarda, bu reaksiyonlar şiddetli anafilaksiye ilerlemiş ve faktör IX inhibitörlerinin gelişmesi ile zamansal ilişki içinde olmuşlardır (Bkz. Bölüm 4.4). Faktör IX inhibitörleri olan veya alerjik reaksiyon hikayesi olan hemofili B hastalarında baışıklık toleransı indüksiyonu girişiminin ardından nefrotik sendrom rapor edilmiştir. HAEMONINE, alerjik reaksiyona ve kan pıhtılaşma sistemini etkileyebilecek şekilde düşük kan hücresi sayısına neden olabilen miktar tayin limiti (0.1 IU/mL) altında eser miktarda hepariniçerebilir. Heparin kaynaklı alerjik reaksiyonlar içeren geçmişi olan hastalar heparin içerenilaçları kullanmaktan kaçınmalıdırlar. Hemofili B hastaları, faktör IX'a karşı nötralize edici antikorlar (inhibitörler) geliştirebilirler. . Bu belge, güvemi elektronik iıuza.ıle imzalanmıştır*..Bdge Dogg£akuıtü6\inhibJtörJefv0löşur§gv3duÂum5kendini3lyeterıâiz\ckliniks:ya»ıttıfllarik.eaitertir^k-Böyle6 vakalarda, uzman bir hemofili merkezi ile temasa geçilmesi önerilir. En yüksek risk düşük saflıktaki ürünlerde olmak üzere faktör IX ürünlerinin uygulamasının ardından bir tromboembolik atak riski mevcuttur. Düşük saflıktaki faktör IX ürünlerininkullanımı miyokard enfarktüsü, dissemine intravasküler koagülasyon, venöz tromboz vepulmoner embolizm vakaları ile ilişkilendirilmiştir. Yüksek saflıkta faktör IX kullanımı nadirenbu tür yan etkiler ile ilişkili olmuştur. Bulaşıcı ajanlara ilişkin güvenlilik bilgisi için Bölüm 4.4'e bakınız. İstenmeyen etkilerin sıklıkları aşaıdaki şekilde deerlendirilmiştir. Çok yaygın (>1/10); yaygın (> 1/100 ila < 1/10); yaygın olmayan (>1/1.000 ila < 1/100); seyrek (>1/10.000 ila < 1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketletahmin edilemiyor). Klinik çalışmalarda HAEMONINE ile advers ilaç reaksiyonlarının sıklıı (sıklıklar tedavi edilen her bir hasta için hesaplanmıştır (n=36)):
Seçilmiş advers reaksiyonların tanımıFaktör IX inhibisyonuİnhibe edici antikorların gelişmesi hemofili B hastalarının tedavisinde bilinen bir komplikasyondur. Şimdiye kadar önceden tedavi görmüş hastalarla deneyim mevcut deildir.Klinik geliştirme süresinde, 1493 maruziyet günü boyunca önceden tedavi görmüş hastalarda(n=36) hiçbir faktöre IX inhibitör indüksiyonu gözlenmemiştir. Pediyatrik popülasyon:Advers reaksiyonların sıklıı, tip ve şiddeti hem 6 yaş ve üzeri çocuklar hem yetişkinler için geçerlidir. (Bkz. Bölüm 4.2) Belge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys7 Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak salar. Salık meslei mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr[email protected]. Doz aşımı ve tedavisiHerhangi bir doz aşımı vakası bildirilmemiştir. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antihemorajikler: kan koagülasyon faktörü IX. ATC Kodu: B02BD04 Faktör IX, moleküler aırlıı 68.000 Dalton olan tek zincirli bir glikoproteindir. K-vitaminine baımlı bir pıhtılaşma faktörüdür ve karacierde sentezlenir. Faktör IX, intrensek koagülasyonyolaında faktör XIa ile ve ekstrensek yolakta faktör VII/doku faktörü kompleksi ile aktiveedilir. Aktive edilmiş faktör IX, aktive edilmiş faktör VIII ile kombinasyon halinde, faktör X'uaktive eder. Aktive edilmiş faktör X protrombini trombine dönüştürür. Daha sonra trombin,fibrinojenin fibrine dönüştürür ve pıhtı oluşur. Hemofili B, faktör IX düzeylerindeki azalmadankaynaklanan, cinsiyete balı kalıtsal bir kan pıhtılaşması bozukluudur ve spontan olarak veyakaza veya cerrahi travma sonucunda eklemlerde, kaslarda veya iç organlarda önemli miktardakanamaya neden olur. İkame tedavisi ile faktör IX'un plazmadaki düzeyi arttırılır. Böyleliklefaktör IX eksiklii geçici olarak düzeltilir ve kanama eilimi düzeltilir. Pediyatrik popülasyon:6 yaşından küçük çocuklarda HAEMONINE kullanımını tavsiye etmek için yeterli bilgi bulunmamaktadır. 5.2. Farmakokinetik özelliklerGenel özellikler13 hasta ile gerçekleştirilen bir farmakokinetik çalışması aşaıdaki sonuçları vermiştir: Emilim:İntravenöz uygulama sonrası, uygulanan faktör IX miktarının tamamı dolaşımda tespit edilebilir. İntravenöz uygulama sonrası absorbsiyon tam ve hızlıdır. Daılım ve biyotransformasyon:Bifazik bir model kullanılarak, ortalama başlangıç yarılanma ömrü ilk vizitte 2,2 ± 1,9 saat ve 3. ayda 3,1 ± 2,9 saat olmuştur. Ortalama terminal yarılanma ömrü ilk vizitte 28,5 ± 12,1 saat v 3.ayda 30,1 ± 14,7 saat olarak hesaplanmıştır. HAEMONINE'nin artımlı geri kazanımı ilk vizitteve 3. ayda sırasıyla %69,8 ± 21,6 ve %72,2 ± 22,2 olmuştur. Bu da ilk vizitte 0,015 ± 0,005 IU/mlmL /IU/kg vücut aırlıı ve 3. ayda 0,016 ± 0,005 IU/mL /IU/kg vücut aırlıı artımlı gerikazanıma karşılık gelmektedir. Belge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys8 Eliminasyon:HAEMONINE'mn dier farmakokinetik parametreleri aşaıda olduu gibidir: Eri altında kalan alan (EAA): yaklaşık 25 IU saat/mL; Ortalama kalış süresi (MRH): yaklaşık 33 saat;Klirens: yaklaşık 200 mL /saat. Dorusallık / Dorusal olmayan durumlar:Eliminasyonu dozla orantılı olarak lineerdir. 5.3. Klinik öncesi güvenlilik verileriÜrün yalnız insan plazması türevi proteinler başka bir deyişle endojen faktör IX ile özdeş yüksek saflıkta koagülasyon faktör IX içerir. Bir Ames testindeki klinik öncesi çalışmalar ürününmutajenik potansiyeline ilişkin bir bulgu göstermemiştir. HAEMONINE deişik tavşanmodellerinde anormal toksisite ve trombojenik potansiyel için test edilmiştir. Sonuçlartoksikolojik veya trombojenik potansiyele ilişkin hiçbir bulgu ortaya koymamıştır. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiToz: Arginin Lizin, Sodyum sitrat, Sodyum klorür Çözücü: Enjeksiyonluk su 6.2. GeçimsizliklerBu tıbbi ürün, dier tıbbi ürünlerle karıştırılmamalıdır. İnsan koagülasyon faktörü IX'in bazı infüzyon ekipmanlarının iç yüzeylerine adsorpsiyonu sonucu tedavi hataları oluşabileceinden, yalnızca verilen infüzyon setleri kullanılmalıdır. 6.3. Raf ömrü24 ay. Sulandırıldıktan sonra hemen kullanılmalı, dondurulmamalıdır. 6.4. Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklıında saklayınız. Ürünü, dış karton kutusu içinde saklayınız ve ışıktan koruyunuz. 6.5. Ambalajın nitelii ve içerii1 paket HAEMONINE 500 IU/5 mL IV enjeksiyonluk çözelti hazırlamak için toz ve çözücü aşaıdakileri içerir:Tip I (Av. Far.) klorobütil kauçuk tıpalı Tip I cam (Av. Far.) flakon içerisinde toz Tip I (Av. Far.) bromobütil kauçuk tıpalı Tip I cam (Av. Far.) flakon içerisinde çözücü (5 mL)Aynı paket 1 adet tek kullanımlık enjektör (5 mL), 1 adet çift filtreli transfer sistemi ve bir adetkelebek kanül içerir. Belge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys9 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve dier özel önlemlerTüm üretimin basamaklarında mutlak sterilitenin salandıından emin olunmalıdır. <
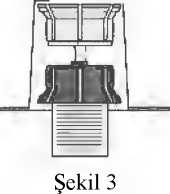
Konsantrenin çözündürülmesi: Açılmamış flakonlardaki çözücü (enjeksiy onluksu) ve tozu oda sıcaklıına (15°C-25°C) getirin.Eer ısıtma için bir su banyosu kullanılıyorsa,suyun flakonların kapakları veya stoperleri iletemas etmediinden kesin olarak emin olun. Aksitakdirde ilaçta bulaşma oluşabilir. Kauçuk tıpaların merkezi bölümlerini açıktabırakmak için her iki flakonun da kapaklarıçıkarınız. Ürün ve çözücü flakonlarının kauçuktıpalarının dezenfekte edildiinden emin olunuz. Transfer sisteminin ambalajını çıkartınız (2).Transfer sisteminin mavi kısmını, dikpozisyondaki çözücü içeren flakonayerleştiriniz (3). Transfer sistemi ambalajının kalan kısmınıçıkarınız. Şimdi transfer sisteminin saydamkısmı görünür olacaktır. Ürün flakonunu düz bir yüzeye koyunuz. Transfer sistemini batırdıınız çözücü flakonubaş aşaı çeviriniz. Adaptörün saydamkısmının sivri ucunu ürün flakonunun tıpasınadoru itiniz (4) Ürün flakonundaki basınççözücünün ürün flakonuna akmasınısalayacaktır (5) Transfer sisteminin mavikısmını çözücü flakon ile birlikte derhalçıkarınız. Çözücü flakonunu üzerindeki transfersisteminin mavi kısmı ile birlikte atınız. (6)Ürün flakonunun hafifçe kendi etrafındaçevrilmesi tozun çözünmesine yardımcıolacaktır. Sert şekilde çalkalamayınız, her türlüköpük oluşumundan kaçınınız! Çözücü berrakveya hafif opak görünümde olacaktır. Kullanıma hazır çözelti çözündürüldüktenhemen sonra kullanılmalıdır. Bulanık ve gözlegörünür partiküller içeren çözeltilerikullanmayınız. Belge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:lıttps://www.turkiye.gov.tr/saglik-titck-ebys
10
Şekil 7  Enjeksiyon: Tozu yukarıda tanımlandıı şekildeçözüldükten sonra transfer sisteminin saydamkısmı ile Luer-Lock balantısı bulunan kapalıenjektörü substrat flakonuna doru itiniz (7). Bu çözünmüş ilacı enjektöre kolayca çekmenizi salayacaktır. Transfer sistemi kendi dahilifiltresine sahip olduundan dolayı ayrı bir filtregerekli deildir. Transfer sisteminin saydam kısmı ile flakonuenjektörden dikkatli şekilde ayırınız. Kapalıkelebek ineyi kullanarak ilacı yavaşintravenöz enjeksiyon ile derhal uygulayınız.Enjeksiyon hızı 2-3 mL /dakikayıgeçmemelidir. Kelebek ine kullanıldıktan sonra, koruyucukapak ile emniyete alınmalıdır. Kullanılmamış olan ürünler ya da atık materyaller 'Tıbbi ürünlerin kontrolü yönetmelii' ve 'Ambalaj ve Ambalaj Atıklarının Kontrolü yönetmelii'ne uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİMaxicells İlaç San. A.Ş. Oruç Reis Mahallesi Tekstilkent Cad. No: 12 A/233 Esenler /İSTANBULTel: 0 212 438 30 30Faks: 0 212 438 29 29 8. RUHSAT NUMARASI2021/166 9. İLK RUHSAT TARİHİ/ RUHSAT YENİLEME TARİHİİlk ruhsat tarihi:18.06.2021 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİBelge Dorulama Kodu: !ZW56M0FyaklUZW56ZlAxSHY3ZlAxZW56 Belge Takip Adresi:lıttps://www.turkiye.gov.tr/saglik-titck-ebys11 |
İlaç BilgileriHaemonine 500 Iu/5 Ml Iv Enjeksiyonluk Çözelti Hazırlamak İçin Toz ve ÇözücüEtken Maddesi: İnsan Koagülasyon Faktörü Ix Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.