Signifor 0.6 Mg/1 Ml Enjeksiyonluk Çözelti Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır?1. BEŞERİ TIBBİ ÜRÜNÜN ADISIGNIFOR 0.6 mg / 1 ml enjeksiyonluk çözelti Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Pasireotid diaspartat 0,752 mg (0,6 mg Pasireotid serbest baza karşılık gelmektedir) Yardımcı maddeler:Sodyum hidroksit yeteri miktar (pH 4,2) Yardımcı maddelerin tam listesi için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti. Bir ucundan kesilen 1 ml renksiz cam ampul içinde berrak, renksiz bir çözeltidir. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarCerrahi müdahalenin bir tedavi seçeneği olmadığı ya da başarısız olduğu, Cushing hastalığı olan erişkin hastaların tedavisinde endikedir. 4.2 Pozoloji ve uygulama şekliUygulama sıklığı ve süresi:SIGNIFOR'un önerilen başlangıç dozu, günde iki kere subkutan (s.c.) enjeksiyon yoluyla 0,6 mg'dır. SIGNIFOR tedavisine başlandıktan iki ay sonra hastalar, klinik fayda açısından değerlendirilmelidir. Üriner serbest kortizol [UFC] düzeylerinde klinik açıdan anlamlıazalmanın olduğu hastalar, fayda elde ettikleri müddetçe SIGNIFOR ile tedavi almaya devametmelidir. 0,6 mg dozaj hasta tarafından iyi tolere edildiği sürece, tedaviye verilen yanıta göredozu 0,9 mg'a yükseltmek düşünülebilir. günde iki kez 0,9 mg'a doz artışı düşünülebilir. İkiay tedavi sonrası SIGNIFOR'a yanıt vermeyen hastalar için tedavinin bırakılmasıdüşünülmelidir. Şüpheli advers reaksiyonların tedavisi, SIGNIFOR dozunun geçici olarak azaltılmasını gerektirebilir. Günde iki kere 0,3 mg'lık adımlarla doz azaltımı önerilmektedir. 1Bir SIGNIFOR dozu atlanırsa, bir sonraki enjeksiyon planlandığı saatte uygulanmalıdır. Atlanan bir dozu telafi etmek için çift doz uygulanmamalıdır. Uygulama şekli:Subkutan (s.c.) enjeksiyon şeklinde uygulanır. Hastalar, SIGNIFOR'un subkutan yolla nasıl enjekte edileceğine dair hekimden ya da bir sağlık profesyonelinden talimat almalıdır. Arka arkaya iki enjeksiyon için aynı enjeksiyon bölgesinin kullanılması önerilmemektedir. Enflamasyon ya da iritasyon belirtileri gösteren bölgelerde uygulamadan kaçınılmalıdır.Subkutan enjeksiyon için tercih edilen enjeksiyon bölgeleri, uyluk kemiğinin üst kısmı vekarındır (göbek ve bel çizgisi hariç). Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek fonksiyonu bozulmuş olan hastalarda doz ayarlaması gerekli değildir (bkz. bölüm 5.2). Karaciğer yetmezliği:Hafif şiddette bozulmuş karaciğer fonksiyonuna (Child-Pugh A) sahip hastalarda doz ayarlaması gerekli değildir. Orta şiddette bozulmuş karaciğer fonksiyonu (Child-Pugh B) olanhastalar için önerilen başlangıç dozu günde iki kere 0,3 mg'dır (bkz. bölüm 5.2). Orta şiddettekaraciğer yetmezliği olan hastalar için maksimum önerilen doz günde iki kere 0,6 mg'dır.SIGNIFOR şiddetli karaciğer yetmezliği (Child Pugh C) olan hastalarda kullanılmamalıdır(bkz. bölüm 4.3 ve 4.4). Pediyatrik popülasyon:SIGNIFOR'un18 yaşın altındaki çocuk ve ergen hastalarda güvenliği ve etkililiği ile ilgili veri mevcut değildir. Geriyatrik popülasyon:65 yaşından büyük hastalarda SIGNIFOR kullanımı ile ilgili veriler sınırlıdır; fakat yaşlı hastalarda bir doz ayarlamasının gerekli olduğuna işaret eden kanıtlar mevcut değildir (bkz.bölüm 5.2) 4.3 Kontrendikasyonlar- Etkin maddeye ya da bölüm 6.1'de listesi yer alan yardımcı maddelerin herhangi birine aşırıduyarlılığı ve, - Şiddetli karaciğer yetmezliği (Child Pugh C) olan hastalarda kontrendikedir. 4.4 Özel kullanım uyarıları ve önlemleriGlukoz metabolizmasıPasireotid ile tedavi edilen sağlıklı gönüllüler ve hastalarda sıklıkla kan glukoz düzeylerinde değişiklikler gözlenmiştir. Pasireotid ile yapılan klinik çalışmalara katılanlarda hiperglisemive daha seyrek olarak hipoglisemi gözlenmiştir (bkz. Bölüm 4.8). Hiperglisemi derecesinin, prediyabetik durumlar ya da tanı almış diyabeti olan hastalarda daha yüksek olduğu görülmektedir. Pivot çalışma sırasında HbAlc düzeyleri anlamlı düzeydeyükselmiş ve stabilize olmuş, fakat başlangıç değerlerine geri dönmemiştir (bkz. bölüm 4.8). 2Günde iki kez 0,9 mg dozu uygulanan hastalarda, hiperglisemi nedeniyle daha fazla çalışmadan ayrılma ve daha yüksek şiddetli advers olaylar bildirme oranı bildirilmiştir. Hiperglisemi gelişimi, insülin (özellikle doz sonrası dönemde) ve ayrıca inkretin hormonların (yani, glukagon benzeri peptid-1 [GLP-1] ve glukoza bağlı insülinotropik polipeptid [GIP])salgısında azalma ile bağlantılı görünmektedir. Glisemik durum (açlık plazma glukozu/hemoglobin Â1c [AKŞ/HbA1c]), pasireotid ile tedaviye başlanmadan önce değerlendirilmelidir. Tedavi sırasında AKŞ /HbA1c takibindemevcut kılavuzlar takip edilmelidir. Kan glukozu ve/veya AKŞ değerlendirmelerinin hastatarafından takibi, ilk iki ila üç ayda haftada bir ve ardından klinik olarak endike olduğu şekildeperiyodik olarak ve yine dozda herhangi bir artırmadan sonraki ilk iki ila dört haftadayapılmalıdır. Tedavinin bırakılmasından 4 hafta sonra FGP ve 3 ay sonra HbA1cmonitörizasyonu yapılmalıdır. Eğer SIGNIFOR ile tedavi edilen bir hastada hiperglisemi gelişirse, hiperglisemi tedavisine yönelik bilinen tedavi kılavuzları takip edilerek anti-diyabetik tedavinin başlatılması ya daayarlanması önerilmektedir. Eğer uygun tıbbi tedaviye rağmen kontrol edilemeyenhiperglisemi devam ederse, SIGNIFOR dozu azaltılmalı ya da tedavi bırakılmalıdır (bkz.Bölüm 4.5). SIGNIFOR kullanan, diyabet öyküsü olan ve olmayan hastalarda pazarlama sonrası ketoasidoz vakaları görülmüştür. Şiddetli metabolik asidoz ile uyumlu belirti ve bulgulargörülen hastalar, diyabet geçmişine bakılmaksızın ketoasidoz yönünden değerlendirilmelidir. Glisemik kontrolü zayıf olan hastalarda (anti-diyabetik tedavi görürken HbA1c değerleri >%8 olarak tanımlanmaktadır), pasireotid tedavisi başlatılmadan önce ve tedavi sırasında diyabetkontrolü ve izlemi yoğunlaştırılmalıdır. Karaciğer testleriPasireotid ile tedavi edilmiş hastalarda, aminotransferazlarda hafif ve geçici yükselmeler yaygın olarak gözlenmiştir. Birkaç vakada ALT'de (alanin aminotransferaz) 3 x normalin üstsınırından (ULN) daha yüksek ve bilirubinde 2 x ULN'den daha yüksek eşzamanlıyükselmeler de gözlenmiştir (bkz. bölüm 4.8). SIGNIFOR ile tedaviden önce ve tedavisırasında bir, iki, dört, sekiz ve on ikinci haftadan sonra karaciğer fonksiyonunun takibiönerilmektedir. Bundan sonra karaciğer fonksiyonu klinik açıdan gerekli olduğu durumlardatakip edilmelidir. Transaminaz düzeyleri artan hastalar bulgunun doğrulanması için ikinci bir karaciğer fonksiyonu değerlendirmesi ile takip edilmelidir. Eğer bulgu doğrulanırsa, hastaya, değerlertedavi öncesi düzeylere gerileyene kadar sık sık karaciğer fonksiyonu takibi yapılmalıdır.Hasta sarılık geliştirirse veya klinik açıdan anlamlı karaciğer fonksiyon bozukluğuna işareteden semptomlar ortaya çıkarsa (AST [aspartat aminotransferaz] ya da ALT'de 5 x ULN yada daha yüksek sürekli bir artış veya 3 x ULN'den daha yüksek ALT veya AST yükselmeleri2 x ULN'den daha yüksek bilirubin yükselmeleri ile eşzamanlı meydana gelirse) pasireotid iletedavi bırakılmalıdır. Pasireotid ile tedavinin bırakılmasını takiben, hastalar durumlarıdüzelene kadar takip edilmelidir. Tedavi yeniden başlatılmamalıdır. Kardiyovasküler bağlantılı olaylar3Pasireotid kullanımı ile bağlantılı bradikardi bildirilmiştir (bkz. Bölüm 4.8). Kalp hastalığı ve/veya bradikardi için risk faktörleri olan hastalar (örn., klinik açıdan anlamlı bradikardi yada akut miyokard infarktüsü öyküsü, yüksek dereceli kalp bloku, konjestif kalp yetmezliği(NYHA Sınıf III ya da IV), stabil olmayan angina, devamlı (sustained) ventriküler taşikardi,ventriküler fibrilasyon) dikkatli bir şekilde takip edilmelidir. Beta-blokörler, kalsiyum kanalblokörleri ya da elektrolit dengesini kontrol eden ajanlar gibi ilaçlarda doz ayarlaması gerekliolabilir. Sağlıklı gönüllüler üzerinde yapılan iki çalışmada pasireotidin EKG üzerinde QT aralığını uzattığı gösterilmiştir. Bu uzamanın klinik önemi bilinmemektedir. Cushing hastalığı olan hastalar üzerinde gerçekleştirilen klinik çalışmalarda, 201 hastanın ikisinde >500 msn QTcF gözlenmiştir. Bu epizotlar sporadiktir ve klinik sonuç gözlenmedenbir kez meydana gelmiştir. Bu çalışmalarda ya da diğer hasta popülasyonları üzerinde yapılanklinik çalışmalarda torsade de pointes epizotları gözlenmemiştir. Pasireotid, aşağıdakiler gibi QT uzaması geliştirme için anlamlı risk altında olan hastalarda dikkatli kullanılmalıdır: konjenital uzun QT sendromu kısa zaman önce meydana gelmiş miyokard infarktüsü, konjestif kalp yetmezliği, stabilolmayan angina ya da klinik açıdan anlamlı bradikardi dahil olmak üzere kontroledilemeyen ya da önemli kalp hastalığı QT uzamasına yol açtığı bilinen anti-aritmik tıbbi ürünlerin ya da diğer maddelerinalınması hipokalemi ve/veya hipomagnezemi QTc aralığındaki olası bir etkinin izlenmesi önerilir ve SIGNIFOR ile tedavi başlatılmadan önce, tedavi başladıktan bir hafta sonra ve sonrasında klinik açıdan endike olduğu durumlardaEKG uygulanmalıdır. Hipokalemi ve/veya hipomagnezemi SIGNIFOR tedavisine başlanmadan önce düzeltilmelidir ve tedavi sırasında periyodik olarak izlenmelidir. HipokortizolizmSIGNIFOR ile tedavi Cushing hastalığında ACTH (adrenokortikotropik hormon) salgısının hızlı bir şekilde baskılanmasına yol açar. Hızlı ve tam ya da tama yakın ACTH baskılanmasıdolaşımdakikortizoldüzeylerindebir azalmaya neden olabilirve geçici hipokortizolizm/hipoadrenalizme yol açabilir. Dolayısıyla hastaları hipokortizolizm ile bağlantılı işaretler ve semptomlar (örn., güçsüzlük, yorgunluk, anoreksi, mide bulantısı, kusma, hipotansiyon, hiperkalemi, hiponatremi ya dahipoglisemi) açısından takip etmek ve bilgilendirmek gerekmektedir. Belgelenmişhipokortizolizm durumunda, geçici ekzojen steroid (glukokortikoid) replasman tedavisive/veya doz azaltımı ya da SIGNIFOR ile tedavinin kesilmesi gerekli olabilir. Safra kesesi ve bağlantılı olaylar4Kolelitiyazis (safra kesesi taşı), somatostatin analoglarının uzun süreli kullanımı ile bağlantılı iyi bilinen bir advers ilaç reaksiyonudur ve pasireotid ile yapılan klinik çalışmalarda sıklıklabildirilmiştir (bkz. Bölüm 4.8). SIGNIFOR kullanan hastalarda pazarlama sonrası kolanjit vakaları görülmüştür, bu vakaların çoğu, safra taşı komplikasyonu olarak bildirilmiştir. SIGNIFOR tedavisinden önce veSIGNIFOR tedavisi sırasında 6 ila 12 ay aralıklarla safra kesesinin ultrasonla incelenmesiönerilmektedir. SIGNIFOR ile tedavi edilen hastalarda safra taşlarının varlığı büyük orandaasemptomatiktir; semptomatik taşlar klinik uygulamaya göre tedavi edilmelidir. Hipofiz hormonlarıPasireotidin farmakolojik aktivitesi somatostatini taklit ettiğinden, ACTH dışındaki hipofiz hormonlarının inhibisyonu da olasılıdır. Dolayısıyla, SIGNIFOR ile tedavi başlatılmadan önceve tedavi sırasında periyodik olarak hipofiz fonksiyonunun (örn., TSH/serbest T4, GH/IGF-1)takibi yapılmalıdır. Kadın fertilitesi üzerindeki etkiCushing hastalığı olan kadın hastalarda serum kortizol düzeylerinin azaltılması veya normalleştirilmesinin terapötik faydaları fertiliteyi olasılıkla eski haline getirebilir. Gebekalma potansiyeline sahip kadın hastalara, Signifor ile tedavi süresince yeterli doğum kontrolüuygulamaları tavsiye edilmelidir (bkz. bölüm 4.6). Böbrek yetmezliğiBağlanmamış ilaca maruziyetteki artış sebebiyle SIGNIFOR, şiddetli böbrek yetmezliği ya da son evre böbrek hastalığı olan kişilerde dikkatle kullanılmalıdır (bkz. bölüm 5.2). Yardımcı maddelerBu tıbbi ürün her dozunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında sodyum içermez. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriPasireotid üzerinde etkilere yol açacak, beklenen farmakokinetik etkileşimlerP-gp inhibitörü verapamilin subkutan pasireotidin farmakokinetiği üzerindeki etkisi sağlıklıgönüllülerde yürütülen bir ilaç-ilaç etkileşimi çalışmasında test edilmiştir. Pasireotidinfarmakokinetiğinde (maruziyet hızı veya boyutu) bir değişiklik gözlenmemiştir.Diğer tıbbi ürünler üzerinde etkilere yol açacak, beklenen farmakokinetik etkileşimlerPasireotid siklosporinin bağıl biyoyararlanımını düşürebilir. SIGNIFOR ve siklosporinineşzamanlı uygulanması, ilacın terapötik düzeylerinin korunması amacıyla siklosporin dozundabir ayarlamanın yapılmasını gerektirebilir.Beklenen farmakodinamik etkileşimlerQT aralığını uzatan tıbbi ürünler:Sınıf Ia antiaritmikler (örn., kinidin, prokainamid, disopiramid), sınıf III antiaritmikler (örn., amiodaron, dronedaron, sotalol, dofetilid, ibutilid), belirli antibakteriyeller (intravenözeritromisin, pentamidin enjeksiyonu, klaritromisin, moksifloksasin), belirli antipsikotikler(örn., klorpromazin, tioridazin, flufenazin, pimozid, haloperidol, tiaprid, amisülpirid,sertindol, metadon), belirli antihistaminikler (örn., terfenadin, astemizol, mizolastin),antimalaryaller (örn., klorokin, halofantrin, lumefantrin), belirli antifungaller (ketokonazol, 5şampuan içinde olduğu durumlarda hariç) gibi, QT aralığında uzamaya yol açan ürünleri eşzamanlı olarak alan hastalarda pasireotid dikkatle kullanılmalıdır (ayrıca bkz. bölüm 4.4). Bradikardik tıbbi ürünler: Pasireotid ile beta blokörler (örn., metoprolol, karteolol, propranolol, sotalol), asetilkolinesteraz inhibitörleri (örn., rivastigmin, fizostigmin), belirli kalsiyum kanalblokörleri (örn., verapamil, diltiazem, bepridil), belirli antiaritmatikler gibi bradikardik tıbbiürünleri bir arada alan hastalarda, özellikle tedavinin başında olmak üzere, kalp hızının kliniktakibi önerilmektedir (ayrıca bkz. bölüm 4.4). İnsülin ve antidiyabetik tıbbi ürünler: Pasireotid ile eşzamanlı uygulandığında, insülin ve antidiyabetik tıbbi ürünlerin (örn., metformin, liraglutid, vildagliptin, nateglinid) dozunda ayarlamalar (azaltma veya artırma)gerekli olabilir (ayrıca bkz. bölüm 4.4). Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin veri bulunmamaktadır. Pediyatrik popülasyon:Pediyatrik popülasyonlara ilişkin veri bulunmamaktadır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi C'dir. Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)SIGNIFOR kullanan kadınların tedavi süresince etkin bir doğum kontrol yöntemi kullanmaları tavsiye edilir. Gebelik dönemiPasireotidin gebe kadınlarda kullanımı hakkında sınırlı veri mevcuttur. Hayvanlar üzerinde yapılan çalışmalar üreme toksisitesine işaret etmiştir (bkz. bölüm 5.3). Pasireotidin gebeliksüresince ve çocuk doğurma potansiyeli bulunan ve doğum kontrolü uygulamayan kadınlardakullanılması önerilmemektedir (bkz. bölüm 4.4). Laktasyon dönemiPasireotidin insanda anne sütüne geçip geçmediği bilinmemektedir. Sıçanlardan elde edilen veriler pasireotidin süte geçtiğini göstermiştir (bkz. bölüm 5.3). SIGNIFOR bebek emzirenkadınlarda kullanılmamalıdır. Üreme yeteneği/FertiliteSıçanlar üzerinde gerçekleştirilen çalışmalarda, dişi üreme parametreleri üzerinde etkiler gösterilmiştir (bkz. bölüm 5.3). Bu etkilerin insanlar açısından klinik ilgisi bilinmemektedir. 4.7 Araç ve makine kullanımı üzerindeki etkiler6SIGNIFOR'un araç ve makine kullanma yeteneği üzerindeki etkileri yok sayılabilecek derecede azdır. Hastalar, yorgunluk veya başağrısı hissetmeleri durumunda, araç ve makinekullanırken dikkatli olmaları konusunda uyarılmalıdır. 4.8 İstenmeyen etkilerGüvenlilik profilinin özetiFaz II ve Faz III çalışmalarda toplam 201 Cushing hastalığı olan hasta SIGNIFOR almıştır. SIGNIFOR'un güvenlilik profili, hipokortizolizm oluşumu ve hiperglisemi derecesi dışındasomatostatin analog sınıfı ile tutarlıdır. Aşağıda tarif edilmiş olan veriler, bir Faz III çalışmada, 162 Cushing hastalığı olan hastanın SIGNIFOR'a maruziyetini yansıtmaktadır. Çalışmaya girişte hastalar günde iki kere (b.i.d.)0,6 mg ya da 0,9 mg SIGNIFOR dozları alacak şekilde randomize edilmiştir. Hastalarınortalama yaşı 40'tır ve hastaların büyük kısmı kadınlardan oluşmaktadır (%77,8). Hastalarınçoğunda (%83,3) persistan ya da nükseden Cushing hastalığı vardır ve her iki tedavi grubundabirkaç hastaya (<%5) daha önce hipofiz radyoterapisi uygulanmıştır. Birincil etkililik vegüvenlilik analizinin veri kesme tarihine kadar tedaviye medyan maruziyet 10,37 aydır (0,03ila 37,8) ve hastaların % 66'sında en az altı aylık maruziyet vardır. Hastaların %57,4'ünde 1. ve 2. derece ADR (Advers İlaç Reaksiyonu)'ler bildirilmiştir. Hastaların %35,8'inde 3. derece yan etkiler ve hastaların %2,5'inde 4. derece yan etkilergözlenmiştir. 3. ve 4. derece yan etkiler çoğunlukla hiperglisemi ile ilişkilidir. En yaygın yanetkiler (insidans >%10) şunlardır: ishal, mide bulantısı, abdominal ağrı, kolelitiyazis,enjeksiyon bölgesi reaksiyonları, hiperglisemi, diyabetes mellitus, yorgunluk, glikozilehemoglobin miktarında artış. Yan etkiler MedDRA primer Sistem Organ Sınıfı'na göre listelenmektedir. Her bir Sistem Organ Sınıfı içinde yan etkiler sıklığa göre sıralanmış ve en sık reaksiyonlar ilk olarakgösterilmiştir. Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila<1/100); seyrek (>1/10.000 ila <1/1000); çok seyrek (<1/10.000), bilinmiyor (eldekiverilerden hareketle tahmin edilemiyor). Cushing hastalığı olan hastalar üzerinde gerçekleştirilen bir Faz III çalışmadaki ve pazarlama sonrası deneyimlerden elde edilen advers ilaç reaksiyonları aşağıda belirtilmektedir. Kan ve lenf sistemi hastalıklarıYaygın olmayan: Anemi Endokrin hastalıklarıYaygın: Adrenal yetmezlik Metabolizma ve beslenme hastalıklarıÇok yaygın: Hiperglisemi, diyabetes mellitus Yaygın: İştah azalması, tip 2 diyabetes mellitus, bozulmuş glukoz toleransı Bilinmiyor: Diyabetik ketoasidoz Sinir sistemi hastalıklarıYaygın: Baş ağrısı, baş dönmesi 7Kardiyak hastalıklarYaygın: Sinüs bradikardisi, QT uzaması Vasküler hastalıklarYaygın: Hipotansiyon Gastrointestinal hastalıklarÇok yaygın: İshal, abdominal ağrı, mide bulantısı Yaygın: Kusma, üst abdominal ağrı Hepato-bilier hastalıklarÇok yaygın: Kolelitiyazis Yaygın: Kolesistit*, kolestaz Deri ve deri altı doku hastalıklarıYaygın: Alopesi, prurit Kas-iskelet bozukluklar, bağ doku ve kemik hastalıklarıYaygın: Miyalji, artralji Genel bozukluklar ve uygulama bölgesine ilişkin rahatsızlıklarÇok yaygın: Enjeksiyon yeri reaksiyonu, yorgunluk AraştırmalarÇok yaygın: Glikozile hemoglobinde artış Yaygın: Gama-glutamiltransferazda artış, alanin aminotransferazda artış, artmış aspartat aminotransferaz, lipazda artış, kan glukozunda artış, kan amilazda artış, protrombin zamanında uzama * Kolesistit akut kolesistiti içerir. istenmeyen etkilerden bazılarına ilişkin açıklamaGlukoz metabolizması bozuklukları: Yüksek glukoz seviyeleri, Cushing hastalığı olan hastalar üzerinde gerçekleştirilen Faz III çalışmada en sık bildirilen 3. derece laboratuvar anormalliğidir (hastaların %23,2'si). Prediyabetik ya da diyabetik hastalarla karşılaştırıldığında, çalışmaya girişte normal glisemisiolan hastalarda ortalama HbA1c artışları daha az belirgindir (Tablo 1).
Ortalama açlık plazma glukoz (FPG) düzeylerinin, yaygın olarak tedavinin ilk ayında artış gösterdiği ve takip eden aylarda azaldığı ve kararlı hale geldiği gözlenmiştir. Açlık plazmaglukozu ve HbA1c değerleri genel olarak pasireotidin bırakılmasını takiben 28 günlük 8dönemde azalmış, fakat başlangıç değerlerinin üzerinde kalmıştır. Uzun vadeli veriler mevcut değildir. Başlangıçta HbA1c değeri >%7 olan ya da randomizasyon öncesinde antidiyabetiktıbbi ürünler alan hastalarda, diğer hastalara göre, açlık plazma glukozu ve HbAlcdeğerlerinde daha yüksek ortalama değişiklikler yönünde bir eğilim olmuştur. Hiperglisemi vediyabetes mellitus advers reaksiyonları, sırasıyla 5 (%3,1) ve 4 (%2,5) hastada çalışmanınbırakılmasına neden olmuştur. SIGNIFOR'un insani amaçlı ilaca erken erişim programıdahilinde bir ketoz olgusu ve bir ketoasidoz olgusu bildirilmiştir. SIGNIFOR ile tedavi edilmiş hastalarda kan glukoz düzeylerinin takip edilmesi önerilmektedir (bkz. bölüm 4.4). Gastrointestinal bozukluklar: Diğer somatostatin analoglarıyla olduğu gibi, SIGNIFOR kullanımıyla bağlantılı olarak sık sık gastrointestinal bozukluklar bildirilmiştir. Bu olaylar genellikle düşük derecelidir, girişimgerektirmez ve tedavinin sürdürülmesi neticesinde iyileşmiştir. Enjeksiyon yeri reaksiyonları: Cushing hastalığı olan kişilerde yapılan Faz III çalışmaya kaydolmuş hastaların %13,6'sında enjeksiyon yeri reaksiyonları bildirilmiştir. Enjeksiyon yeri reaksiyonları diğerpopülasyonlarda yapılan klinik çalışmalarda da bildirilmiştir. En sık bildirilen olaylar lokalağrı, eritem, hematom, hemoraji ve prurittir. Bu olaylar kendi kendine ortadan kalkmıştır vegirişim gerekmemiştir. Karaciğer enzimleri: Somatostatin analoglarının kullanımı ile birlikte karaciğer enzimlerinde geçici yükselmeler bildirilmiştir; bu durum aynı zamanda pasireotid ile yapılan klinik çalışmalardaki hastalardada gözlenmiştir. Yükselmeler genellikle asemptomatiktir, düşük derecelidir ve tedavinindevam ettirilmesini takiben ortadan kalkmaktadır. Birkaç vakada ALT'de 3 x ULN'den dahayüksek ve bilirubinde 2 x ULN'den daha yüksek eşzamanlı yükselmeler gözlenmiştir. Tümeşzamanlı yükselme vakaları SIGNIFOR ile tedavi başlatıldıktan sonraki on gün içindebelirlenmiştir. Hastalar klinik sekel olmadan iyileşmiş ve karaciğer fonksiyon testi sonuçlarıtedavi bırakıldıktan sonra başlangıç düzeylerine dönmüştür. SIGNIFOR ile tedavi başlatılmadan önce ve tedavi sırasında karaciğer enzimlerinin izlenmesi önerilmektedir (bkz. bölüm 4.4). Pankreatik enzimler: Klinik çalışmalarda pasireotid alan hastalarda lipaz ve amilazda asemptomatik yükselmeler gözlenmiştir. Yükselmeler büyük oranda düşük derecelidir ve tedavinin sürdürülmesineticesinde ortadan kalkmıştır. Pankreatit; kolelitiyazis ve akut pankreatit arasındaki ilişkinedeniyle somatostatin analoglarının kullanımı ile bağlantılı potansiyel bir adversreaksiyondur. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye 9Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. ([email protected];4.9 Doz aşımı ve tedavisiSağlıklı gönüllülerde günde iki kere 2,1 mg'a kadar olan dozlar sağlıklı gönüllüler üzerinde kullanılmış ve en sık gözlenen advers reaksiyon ishal olmuştur. Doz aşımı durumunda semptomlar ortadan kalkana kadar hastanın klinik durumuna uygun destekleyici tedavinin başlatılması önerilmektedir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamiik özelliklerFarmakoterapötik grup: Hipofiz ve hipotalamus hormonları ve analogları, somatostatin ve analogları ATC kodu: H01CB05 Etki mekanizmasıPasireotid yeni bir sikloheksapeptid, enjektabl somatostatin analogudur. Doğal peptid hormonlar somatostatin-14 ve somatostatin-28 (Somatotropin Salım İnhibitör Faktör [SRIF]olarak da bilinmektedir) ve diğer somatostatin analoglar gibi, pasireotid de farmakolojikaktivitesini somatostatin reseptörlerine bağlanarak göstermektedir. Beş insan somatostatinreseptör alt tipi bilinmektedir: hsst 1, 2, 3, 4 ve 5. Bu reseptör alt tiplerinin ekspresyonunormal fizyolojik koşullarda farklı dokularda gerçekleştirilmektedir. Somatostatin analoglarıhsst reseptörlerine farklı potenslerle bağlanmaktadır (Tablo 2). Pasireotid beş hsst'nin dördüneyüksek afinite ile bağlanmaktadır.
Farmakodinamik etkilerSomatostatin reseptörleri özellikle, Cushing hastalığındaki adrenokortikotropik hormonların (ACTH) aşırı salgılandığı nöroendokrin tümörler dahil olmak üzere birçok dokuda ekspreseedilmektedir. İn vitroKlinik etkililik ve güvenlilikPersistan ve nükseden Cushing hastalığında ya da cerrahinin endike olmadığı veya cerrahiyi reddeden de novo hastalıkta on iki ay süren ve SIGNIFOR'un farklı doz düzeylerinin 10güvenlilik ve etkililiğinin değerlendirildiği bir faz III, çok merkezli, randomize çalışma gerçekleştirilmiştir. Çalışmaya başlangıç UFC (Üriner Serbest Kortizol) düzeyi >1,5 x ULN (normalin üst sınırı) olan 162 hasta kaydedilmiştir; hastalar, 1:1 oranında SIGNIFOR 0,6 mg s.c. b.i.d. ya da 0,9mg s.c. b.i.d. alacak şekilde randomize edilmiştir. Üç aylık tedaviden sonra, 24 saatlikortalama UFC değeri < 2 x ULN ve daha düşük ya da başlangıç değerlerine eşit olan hastalar 6. aya kadar randomize dozda körlenmiş tedaviye devam etmiştir. Bu kriterleri sağlamamışolan hastaların körlüğü kaldırılmış ve doz 0,3 mg b.i.d. arttırılmıştır. Çalışmadaki ilk 6 aydansonra hastalar ilave bir 6 aylık açık etiketli tedavi dönemine girmiştir. Eğer 6. ayda yanıtaulaşılamazsa ya da yanıt açık etiketli tedavi döneminde sürdürülmezse, dozajın 0,3 mg s.c.b.i.d. arttırılmasına izin verilmiştir. Hastalara uygulanan maksimum doz 1,2 mg s.c. b.i.d.olmuştur. Doz, tolerabilite nedeniyle çalışma sırasında herhangi bir zamanda 0,3 mg b.i.d.adımlarla azaltılabilir. Birincil etkililik sonlanım noktası her bir tedavi kolunda 6 aylık tedaviden sonra ortalama 24 saatlik UFC düzeyleri normale dönmüş (UFC < ULN) ve bu dönemde (randomize doza göre)bir doz artışı yapılmamış hastaların oranı olarak belirlenmiştir. İkincil sonlanım noktaları,diğerlerinin yanında aşağıdakilerde başlangıca göre değişiklikleri kapsamaktadır: 24 saatlikUFC, plazma ACTH, serum kortizol düzeyleri, Cushing hastalığının klinik işaretleri vesemptomları. Tüm analizler randomize doz gruplarına dayalı olarak gerçekleştirilmiştir. Başlangıç demografikleri iki randomize doz grubu arasında iyi dengelenmiştir ve hastalık epidemiyolojisi ile tutarlıdır. Hastaların ortalama yaşı yaklaşık olarak 40'tır ve büyük orandakadınlardan oluşmaktadır (%77,8). Hastaların çoğunda persistan ya da nükseden Cushinghastalığı (%83,3) vardır ve her iki tedavi grubunda birkaç hastaya (<%5) daha önce hipofizradyoterapisi uygulanmıştır. Başlangıç 24 saatlik UFC'nin ortalama değerindeki belirgin farklılıklar dışında iki randomize doz grubu arasında başlangıç özellikleri iyi dengelenmiştir (0,6 mg b.i.d. için 1156 nmol/24saat ve 0,9 mg b.i.d. grubu için 782 nmol/24 saat); normal aralık 30 ila 145 nmol/24 saat). Sonuçlar: 6. ayda, pasireotid 0,6 mg b.i.d. ve 0,9 mg b.i.d.'ye randomize edilmiş hastaların %14,6 (%95 GA 7,0 ila 22,3) ve %26,3'ünde (%95 GA 16,6 ila 35,9) ortalama UFC düzeylerinin normaledöndüğü gözlenmiştir. Çalışma 0,9 mg b.i.d. grubu için birincil etkililik hedefini sağlamıştır;çünkü %95 GA'nın alt sınırı önceden belirlenmiş %15 sınırından daha yüksektir. 0,9 mg dozkolundaki yanıt, başlangıçta daha düşük ortalama UFC'si olan hastalar için daha yüksekgörünmektedir. 6. ayda yanıt verenlerin büyük kısmı (%55,6) 12. ayda da yanıt vermiştir. 12.ayda yanıt verme oranı 6. aydaki orana benzerdir; 0,6 mg b.i.d. ve 0,9 mg b.i.d.'de sırasıyla%13,4 ve %25,0. Destekleyici bir etkililik analizi gerçekleştirilmiş ve hastalar, 3. aydaki yukarı titrasyondan bağımsız olarak 3 yanıt kategorisi altında sınıflandırılmıştır: tam kontrol altında (UFC < 1,0 xULN), kısmen kontrol altında (UFC >1,0 x ULN; fakat başlangıca kıyasla UFC'de > %50azalma) ya da kontrol altında değil (tüm diğer hastalar). 6. ayda tam kontrol altında ve kısmenkontrol altında olarak sınıflandırılan yanıt veren hastaların oranı, tüm randomize hastalarınsırasıyla %34 ve %41'ini (sırasıyla 0,6 mg b.i.d. ve 0,9 mg b.i.d.) oluşturmuştur. 1. ve 2. ayda 11kontrol altında olmayan hastalar 6. ve 12. aylarda da büyük olasılıkla (%90) kontrol altında olmamıştır. Her iki doz grubunda SIGNIFOR, 1 aylık tedaviden sonra ortalama UFC'de hızlı ve kalıcı bir azalmaya yol açmıştır ve bu durum tedavi süresince sürdürülmüştür (Şekil 1). 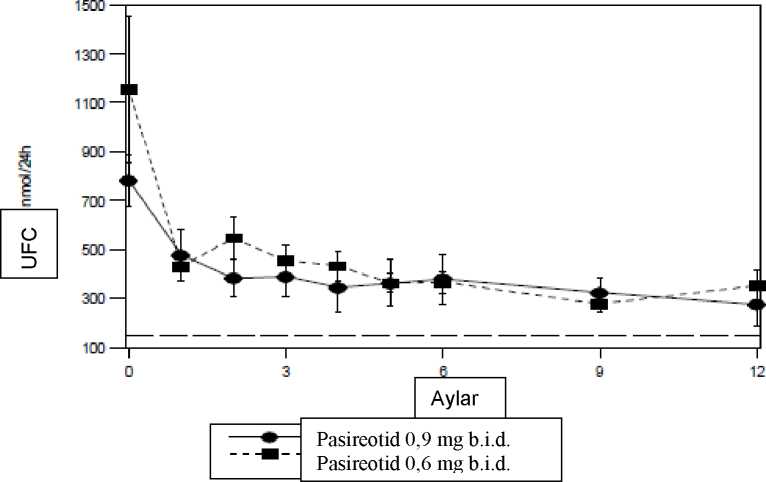 Başlangıç değerleri ile karşılaştırıldığında 6. ve 12. ayda ortalama ve medyan UFC düzeylerinde genel değişim yüzdesine göre kalıcı azalmaların olduğu gösterilmiştir (Tablo 3).Ayrıca her bir doz grubu için her bir zaman noktasında ortalama serum kortizol ve plazmaACTH düzeylerindeki azalmalar gözlenmiştir.
6. ayda her iki doz grubunda, oturur durumda sistolik ve diyastolik kan basıncı, BMI (beden-kütle indeksi) ve toplam kolesterolde klinik açıdan anlamlı azalmalar gözlenmiştir. Bu parametrelerdeki genel azalmaların, UFC'si normale dönmüş hastalarda daha yüksek olmaeğilimi vardır. 12. ayda da benzer eğilimler gözlenmiştir. 125.2 Farmakokinetik özelliklerEmilim:Sağlıklı gönüllülerde, pasireotid hızlı bir şekilde emilir ve 0,25-0,5 saat içinde pik plazma konsantrasyonlarına ulaşılır. Cmaks ve EAA, tek ve çoklu dozları takiben yaklaşıkolarak dozla orantılıdır.İnsanlarda pasireotidin biyoyararlanımını değerlendirmek üzere çalışma yapılmamıştır. Dağılım:Sağlıklı gönüllülerde pasireotid, yüksek bir görünür dağılım hacmiyle (Vz/F >100 L) yaygın bir şekilde dağılmaktadır. Kan ve plazma arasındaki dağılım konsantrasyondanbağımsızdır ve pasireotidin büyük oranda (%91) plazmada konumlandığına işaret etmektedir .Plazma proteini bağlama oranı orta düzeydedir (%88) ve konsantrasyondan bağımsızdır.İn vitro verilere göre pasireotidin dışa akım taşıyıcısı P-gp'nin bir substratı olduğu görülmektedir; Pasireotid BCRP (meme kanseri direnç proteini), OCT1 (organik katyontaşıyıcısı 1) ya da OATP (organik anyon-taşıyıcı polipeptidler) 1B1, 1B3 ya da 2B1'insubstratı değildir. Terapötik doz düzeylerinde pasireotid UGT1A1, OATP, 1B1 ya da 1B3, P-gp, BCRP, MRP2 ve BSEP inhibitörü değildir. Biyotransformasyon:Pasireotid metabolik açıdan oldukça stabildir ve in vitro veriler pasireotidin CYP450'nin majör enzimlerinden hiçbirisinin substratı, inhibitörü veyaindükleyicisi olmadığını göstermiştir. Sağlıklı gönüllülerde pasireotid plazma, idrar ve dışkıdatemel olarak değişmemiş formda bulunur.Eliminasyon:Pasireotid büyük oranda hepatik klerens yoluyla (biliyer atılım) elimine edilmektedir; renal yolun küçük bir katkısı vardır. Bir insan farmakokinetik (ADME)çalışmasında, radyoaktivite dozunun %55,9 ± 6,63'ü doz uygulamasından sonra ilk 10 güniçinde geri kazanılır; bu değer, radyoaktivitenin dışkıdaki %48,3 ± 8,16'lık kısmından veidrardaki %7,63 ± 2,03'lük kısmından oluşmaktadır.Sağlıklı gönüllülerde ve Cushing hastalığı olan hastalarda pasireotidin klerensi (CL/F) sırasıyla ~7,6 litre/saat ve ~3,8 litre/saat'tir. EAA birikim oranları doğrultusunda, sağlıklıgönüllülerde hesaplanan etkili yarı ömür (t1/2,eff) yaklaşık 12 saat bulunmuştur. Doğrusallık/doğrusal olmayan durum:Cushing hastalığı olan hastalarda pasireotid, günde iki kere 0,3 mg ila 1,2 mg aralığında da, doğrusal ve zamandan bağımsız farmakokinetik göstermektedir. Popülasyon farmakokinetiğianalizi, Cushing hastalığı olan hastalarda kararlı durumun %90'ına C maks ve EAA bazındasırasıyla yaklaşık 1,5 ve 15 gün sonra ulaştığını düşündürmektedir. Hastalardaki karakteristik özelliklerBöbrek yetmezliği olan hastalar:Renal klerensin insanlarda pasireotidin eliminasyonuna küçük bir katkısı bulunmaktadır. Böbrek fonksiyonunda bozukluk olan hastalarda 900 |ig pasireotidin tek doz subkutanuygulaması ile yürütülen bir klinik çalışmada hafif, orta ve şiddetli böbrek yetmezliği ya dason evre böbrek hastalığının (SEBH) toplam pasireotid plazma maruziyetine anlamlı bir etkisiolmamıştır. Böbrek yetmezliği olan hastalarda bağlanmamış plazma pasireotid maruziyeti(EAAinf,u), gönüllü kontrol olgularıyla karşılaştırıldığında yükselmiştir (hafif: %33; orta:%25, şiddetli: %99, SEBH: %143). 13Karaciğer yetmezliği olan hastalar:Karaciğer fonksiyon bozulmuş olan (Child-Pugh A, B ve C) gönüllüler üzerinde gerçekleştirilen bir klinik çalışmada, orta ve şiddetli karaciğer yetmezliği olan gönüllüler(Child-Pugh B ve C) normal karaciğer fonksiyonuna sahip gönüllülerden anlamlı oranda dahayüksek maruziyet sergilemiştir. Eşdeğişken etkisi (yaş, beden-kütle indeksi ve albümin) içindüzeltme yapıldıktan sonra kontrol grubu ile karşılaştırıldığında orta ve şiddetli karaciğeryetmezliği olan gruplarda sırasıyla EAAinf %60 ve %79 artmış, Cmaks %67 ve %69 artmış veCL/F %37 ve %44 azalmıştır. Yaşlı hastalar (65 yaş ve üzeri):Cushing hastaları üzerinde yapılan bir popülasyon farmakokinetik analizinde yaşın bir eşdeğişken olduğu bulunmuştur. Artan yaşla birlikte toplam vücut klerensinin azaldığı vefarmakokinetk (FK) maruziyetlerinin arttığı görülmüştür. İncelenmiş yaş aralığı olan 18 ila 73arasında, 12 saatlik bir doz aralığı için kararlı durumda eğri altındaki alanın (EAASS) 41yaşındaki tipik bir hastanın %86'sı ila %111'i arasında olduğu öngörülmektedir. Budeğişkenlik orta düzeydedir ve etkinin gözlendiği geniş yaş aralığı göz önüne alındığında çokönemli olmadığı düşünülmüştür. 65 yaşın üzerinde Cushing hastalığı olan hastalar ile ilgili veriler sınırlıdır; fakat daha genç hastalarla karşılaştırıldığında güvenlilik ve etkililikte klinik açıdan anlamlı farklılıklara işaretetmemektedir. Pediyatrik hastalar:Pediyatrik hastalarda çalışma gerçekleştirilmemiştir. Demografikler:SIGNIFOR üzerinde yapılan popülasyon farmakokinetik analizleri, ırk ya da cinsiyetin farmakokinetik parametrelerini etkilemediğini göstermektedir. Beden ağırlığının, Cushing hastalığı olan hastalar üzerinde yapılan popülasyon farmakokinetik analizinde bir eşdeğişken olduğu bulunmuştur. 60-100 kg aralığında EAAss değerinde artankilo ile birlikte oluşan düşüşün yaklaşık %27 olacağı öngörülmekte olup bu fark ortadüzeylidir ve minör klinik öneme sahiptir. 5.3 Klinik öncesi güvenlilik verileriKlinik olmayan güvenlilik çalışmaları, güvenlilik farmakolojisi, tekrarlı doz toksisitesi, genotoksisite, karsinojenik potansiyel, üreme ve gelişim üzerindeki toksisiteyi kapsamaktadır.Tekrarlı toksisite çalışmalarında gözlenen bulguların büyük kısmı geri dönüşümlüdür vepasireotidin farmakolojisine bağlanabilir. Klinik olmayan çalışmalardaki etkiler, yalnızcamaksimum insan maruziyetinin yeteri kadar üzerinde olduğu düşünülen maruziyetlerdegözlenmiştir; bu durum elde edilen bulguların klinik kullanım açısından fazla bir öneme sahipolmadığını göstermektedir. İn vitro ve in vivo deneylerde pasireotid genotoksik etki göstermemiştir. Sıçanlarda ve transgenik farelerde gerçekleştirilen karsinojenisite çalışmalarında karsinojenik potansiyel belirlenmemiştir. 14Pasireotid, erkek sıçanlarda fertiliteyi etkilememiştir fakat pasireotidin farmakolojisinden de bekleneceği gibi, dişi sıçanlarda anormal veya asiklik sikluslar ve korpus luteum veimplantasyon yerlerinin sayısında azalma görülmüştür. Sıçanlarda ve tavşanlarda, maternaltoksisiteye yol açan dozlarda embriyo toksisitesi görülmüş ancak herhangi bir teratojenpotansiyel tespit edilmemiştir. Sıçanlardaki prenatal ve postnatal çalışmada, pasireotid, doğumsancısı ve doğumu etkilememiş ancak pinna ayrılması oluşumunda hafif bir gecikme veyavrularda beden ağırlığında düşüşe neden olmuştur. Hayvanlardaki mevcut toksikoloji verileri, pasireotidin süte geçtiğini göstermiştir. 6. FARMASÖTİK BİLGİLER6.1 Yardımcı maddelerin listesiMannitol Tartarik asitSodyum hidroksitEnjeksiyonluk suAzot* *(işlem esnasında uzaklaştırılmadadır)6.2GeçimsizliklerDiğer ürünlerle geçimlilik verileri mevcut değildir. Enjeksiyonluk pasireotid çözeltisi diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3 Raf ömrü36 ay 6.4 Saklamaya yönelik özel tedbirler30°C'nin altındaki oda sıcaklığında saklayınız. Işıktan korumak için orijinal ambalajında saklayınız. 6.5 Ambalajın niteliği ve içeriği1 ml'lik, kırarak açılan, renksiz, hidrolitik sınıf I (Ph. Eur., USP) cam ampul 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİRecofarma İlaç ve Hammaddeleri San. ve Tic. Ltd. Şti. Sarıyer / İstanbul Tel: 0 212 401 91 00 8. RUHSAT NUMARASI:2021/230 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİ:İlk ruhsat tarihi:Ruhsat yenileme tarihi:1510. KÜB'ÜN YENİLENME TARİHİ:13.08.2021 16 |
İlaç BilgileriSignifor 0.6 Mg/1 Ml Enjeksiyonluk ÇözeltiEtken Maddesi: Pasireotid Diaspartat Kullanma talimatı ve kısa ürün bilgileri |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.