Onaxan® 2,5 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIONAXAN® 2,5 mg film kaplı tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Rivaroksaban 2,5 mg Yardımcı maddeler:Laktoz monohidrat (inek sütü kaynaklı) 35,70 mg Yardımcı maddelerin tam listesi için 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tablet. Açık sarı renkli, bir yüzü üçgen ve 2.5 baskılı, yuvarlak bikonveks film kaplı tablet. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarONAXAN®, tek başına asetilsalisilik asit (ASA) ya da ASA ile birlikte tienopiridinler (klopidogrel veya tiklopidin) ile kombinasyon şeklinde, akut koroner sendrom (AKS) (STelevasyonsuz ya da ST elevasyonlu miyokard infarktüsü ya da unstabil angina) sonrasıhastalarda kardiyovasküler (KV) ölüm, miyokard infarktüsü (MI) ve stent trombozununönlenmesinde endikedir (bkz. Bölüm 4.2 Pozoloji ve uygulama şekli). ONAXAN®, asetilsalisilik asit (ASA) ile birlikte uygulandığında, yüksek iskemik olay riski taşıyan koroner arter hastalığı (KAH) veya semptomatik periferik arter hastalığı (PAH) olanyetişkin hastalarda aterotrombotik olayların önlenmesinde endikedir. 4.2 Pozoloji ve uygulama şekliPozoloji ve uygulama sıklığıÖnerilen doz günde iki kez 2,5 mg'dir. AKSGünde iki kez ONAXAN® 2,5 mg alan hastalar ayrıca 75-100 mg/gün ASA dozu ya da 75 mg/gün klopidogrel veya standart günlük tiklopidin dozuna ek olarak 75-100 mg/gün ASA dozualmalıdır. KAH veya PAHGünde iki kez ONAXAN® 2,5 mg alan hastalar aynı zamanda 75-100 mg ASA/gün dozu da almalıdır. 1 / 27Uygulama süresiAKSTedavi, düzenli olarak hasta bazında değerlendirilmeli; iskemik olay riski kanama riski karşısında tartılmalıdır. 24 aya kadar olan deneyimler sınırlı olduğundan, tedavinin 12 aydanuzun süreyle devam ettirilmesi hasta bazında değerlendirilmelidir (bkz. Bölüm 5.1Farmakodinamik özellikler). KAH veya PAHTedavi süresi, düzenli değerlendirmelere göre her bir hasta için belirlenmeli ve kanama risklerine karşı trombotik olay riski göz önüne alınmalıdır. Uygulama şekliONAXAN® oral kullanım içindir. ONAXAN® yemeklerle birlikte ya da ayrı alınabilir (bkz. Bölüm 4.5 Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri ve Bölüm 5.2 Farmakokinetik özellikler). Tabletleribütün olarak yutamayan hastalar için ONAXAN® tablet kullanımdan hemen önce ezilip su veyaelma püresi gibi yumuşak gıdalarla karıştırılarak oral yoldan kullanılabilmektedir. Ezilen ONAXAN® tablet g astrik tüp yoluyla da (sondanın mideye doğru şekilde yerleştirildiği onaylandıktan sonra) verilebilir. Ezilen tablet, gastrik tüp aracılığıyla az miktarda su içindeuygulanmalı ve ardından tüp su ile yıkanmalıdır. (bkz. Bölüm 5.2 Farmakokinetik özellikler). AKSONAXAN® ile tedavi, AKS olayının (revaskülarizasyon prosedürleri dahil) stabilizasyonundan sonra mümkün olan en kısa sürede; hastanın hastaneye kabulünden en erken 24 saat sonra ve parenteral antikoagülasyon tedavisinin normalde kesileceği zamanbaşlatılmalıdır. KAH veya PAH:Akut trombotik olay veya vasküler prosedürü olan ve ikili antiplatelet tedavisine ihtiyaç duyan hastalarda günde iki kez ONAXAN® 2,5 mg uygulamasının sürdürülmesi, olayın veyaprosedürün türüne ve antiplatelet rejimine bağlı olarak değerlendirilmelidir. ASA artıklopidrogel/tiklopidinle kombine olarak günde iki kez rivaroksaban 2,5 mg'ın güvenliliği veetkililiğine dair sadece yakın zamanda AKS'si olan hastalarla çalışma yapılmıştır (bkz. Bölüm4.1 Terapötik endikasyonlar). KAH veya PAH hastalarında günde iki kez rivaroksaban 2,5 mgile kombine olarak ikili antiplatelet tedavisine ilişkin çalışma yapılmamıştır (bkz. Bölüm 4.4Özel kullanım uyarıları ve önlemleri ve Bölüm 5.1 Farmakodinamik özellikler). Bir doz atlanması halinde, hasta bir sonraki programlanmış zamanda tavsiye edilen şekilde normal dozu almaya devam etmelidir. Eksik dozu telafi etmek için aynı anda iki dozalınmamalıdır. Tedavinin Vitamin K Antagonistlerinden (VKA) ONAXAN®'a değiştirilmesi:VKA tedavisi gören hastalarda tedavinin ONAXAN®'a değiştirilmesinde, ONAXAN® alınmasının ardından Uluslararası Normalizasyon Oranı (INR) değerleri yalancı yükselmegösterecektir. INR, ONAXAN®'ın antikoagülan aktivitesinin ölçümü için geçerli bir ölçümdeğildir ve bu nedenle kullanılmamalıdır (bkz. Bölüm 4.5 Diğer tıbbi ürünlerle etkileşimler vediğer etkileşim şekilleri). Tedavinin ONAXAN® 'dan Vitamin K antagonistlerine (VKA) değiştirilmesi:ONAXAN® tedavisinden VKA tedavisine geçiş sırasında yetersiz antikoagülasyon olasılığı mevcuttur. Alternatif herhangi bir antikoagülana geçişte sürekli ve yeterli antikoagülasyon 2 /Tlsağlanmalıdır. ONAXAN®'ın INR yükselmesine katkıda bulunabileceğine de dikkat edilmelidir. ONAXAN® tedavisinden VKA tedavisine geçiş yapılan hastalarda INR>2.0olana dek VKA eşzamanlı olarak verilmelidir. Değişim periyodunun ilk iki gününde VKA'nınstandart başlangıç dozu kullanılmalı ve onu INR testi rehberliğinde VKA dozu takip etmelidir.Hastalar hem ONAXAN® ve hem de VKA kullanırken ONAXAN®'ın bir sonraki dozundanönce INR ölçülmeli, ancak bu ölçüm dozdan 24 saat önce ölçülmemelidir. ONAXAN®'ınkesilmesinin ardından son dozdan 24 saat sonra INR testi güvenilir biçimde yapılabilir (bkz.Bölüm 4.5 Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri ve Bölüm 5.2Farmakokinetik özellikler). Tedavinin parenteral antikoagülanlardan ONAXAN'a değiştirilmesi:Halihazırda parenteral antikoagülan ile tedavi edilen hastalarda parenteral antikoagülan kesilir ve ONAXAN® tedavisine parenteral ilacın (örn. düşük molekül ağırlıklı heparin) bir sonrakiplanlanan dozundan 0 ila 2 saat önce ya da sürekli uygulanan parenteral ilacın (örn. intravenözfraksiyone olmayan heparin) kesilme zamanında başlanır. Tedavinin ONAXAN® 'dan parenteral antikoagülanlara değiştirilmesi:İlk parenteral antikoagülan dozu bir sonraki ONAXAN® dozunun alınacağı zamanda uygulanmalıdır. Özel popülasyonlara ilişkin ek bilgiler Böbrek yetmezliği:Ciddi böbrek yetmezliği olan hastalardan (Kreatinin klerensi 15-29 ml/dak) elde edilen kısıtlı klinik veriler, bu hasta popülasyonunda rivaroksaban plazma düzeylerinin anlamlı derecedearttığına işaret etmektedir. Bu nedenle, ONAXAN® bu hasta popülasyonunda dikkatlikullanılmalıdır (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Kreatinin klerensi <15 ml/dak olan hastalarda ONAXAN® kullanımı önerilmemektedir (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri ve Bölüm 5.2 Farmakokinetik özellikler). Hafif (kreatinin klerensi: 50-80 ml/dak) veya orta derecede (kreatinin klerensi: 30-49 ml/dk.) derecede böbrek yetmezliği olan hastalarda doz ayarlaması gerekmemektedir (bkz. Bölüm 5.2Farmakokinetik özellikler). Karaciğer yetmezliği:ONAXAN®, Child Pugh B ve C derecesinde sirozu olan hastalar dahil olmak üzere koagülopati ve klinik açıdan anlamlı kanama riski ile ilişkili karaciğer hastalığı olan hastalardakontrendikedir (bkz. Bölüm 4.3 Kontrendikasyonlar ve 5.2 Farmakokinetik özellikler). Pediyatrik popülasyon:Rivaroksabanın güvenlilik ve etkililiği 0 ile 18 yaş arası çocuklarda belirlenmemiştir. Bu konuda veri bulunmamaktadır. Bu nedenle, 18 yaşından küçüklerde ONAXAN® kullanımıönerilmemektedir. Geriyatrik popülasyon:Doz ayarlaması gerekmemektedir (bkz. Bölüm 4.4. Özel kullanım uyarılan ve önlemleri ve Bölüm 5.2 Farmakokinetik özellikler). Kanama riski, yaş ilerledikçe artar (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). 3 / 27Diğer:Cinsiyet:Doz ayarlaması gerekmemektedir (bkz. Bölüm 5.2 Farmakokinetik özellikler). Vücut ağırlığı:Doz ayarlaması gerekmemektedir (bkz. Bölüm 4.4. Özel kullanım uyarılan ve önlemleri ve bkz. Bölüm 5.2 Farmakokinetik özellikler). 4.3 KontrendikasyonlarONAXAN® aşağıdaki hastalarda kontrendikedir: Rivaroksabana ya da tablet içeriğindeki diğer maddelere karşı aşırı duyarlılığı olanhastalarda (bkz. Bölüm 6. Farmasötik özellikler), Klinik olarak anlamlı aktif kanaması olan hastalarda, Mevcut veya yeni geçirilmiş gastrointestinal ülserasyon, kanama riski yüksek malignneoplazm varlığı, yeni geçirilmiş beyin veya omurilik yaralanmaları, yeni geçirilmişbeyin, omurilik veya oftalmoloji ameliyatı, yakın zamanda gelişmiş olan intrakraniyalkanama, bilinen veya şüphelenilen özefagus varisleri, arteriyovenöz, malformasyonlar,vasküler anevrizmalar veya majör intraspinal ya da intraserebral vasküler anomalilergibi majör kanama bakımından anlamlı risk teşkil eden lezyon veya durumlar, Açık bir santral venöz veya arteriyel kateterin açık kalması için gereken dozlardafraksiyone olmayan heparin (UFH) kullanılan durumlar veya antikoagülan tedavidegeçiş yapılan özel koşullar (bkz. Bölüm 4.2 Pozoloji ve uygulama şekli) hariç olmaküzere, herhangi bir diğer antikoagülan ilaçla [örn. UFH, düşük molekül ağırlıklıheparinler (enoksaparin, dalteparin vb.), heparin türevleri (fondaparinuks vb.) oralantikoagülanlar (varfarin, apiksaban, dabigatran eteksilat, edoksaban vb.)] eşzamanlıtedavi (bkz. Bölüm 4.5 Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri), Önceden inme veya geçici iskemik atak (GİA) geçirmiş olan hastalarda AKS tedavisiiçin antiplatelet tedavisiyle eşzamanlı tedavi, (bkz. Bölüm 4.4 Özel kullanım uyarılarıve önlemleri), Önceden hemorajik veya lakünar inme veya bir ay içinde inme geçirmiş hastalardaKAH/PAH tedavisi ile eş zamanlı ASA tedavisi (bkz. Bölüm 4.4 Özel kullanım uyarılarıve önlemleri), Child Pugh B ve C'li siroz hastaları dahil olmak üzere koagülopati ve klinik açıdananlamlı kanama riskiyle ilişkili hepatik hastalık (bkz. Bölüm 5.2 Farmakokinetiközellikler), Gebelik ve emzirme döneminde (bkz. Bölüm 4.6 Gebelik ve laktasyon). 4.4 Özel kullanım uyarıları ve önlemleriAKS hastalarında, Rivaroksaban 2,5 mg'ın etkililiği ve güvenliliği tek başına ASA antiplatelet ajanlarıyla kombine olarak veya ASA + klopidrogel/tiklopidinle araştırılmıştır. Prasugrel veyatikagrelor vb. diğer antiplatelet ajanlarıyla kombine tedavi hakkında çalışma yapılmamıştır,dolayısıyla önerilmemektedir. Yüksek iskemik olay riski olan KAH/PAH hastalarında, Rivaroksaban 2,5 mg'ın etkililiği ve güvenliliği ASA ile kombine olarak incelenmiştir. Antikoagülasyon uygulamasına paralel klinik gözetim tedavi dönemi boyunca önerilmektedir. 4 / 27Kanama riski: Diğer antikoagülanlarla olduğu gibi, ONAXAN® alan hastaların kanama belirtileri açısından dikkatle gözlemlenmesi gerekir. Kanama riskinin arttığı durumlarda dikkatle kullanılmasıtavsiye edilir. Ciddi kanama meydana gelirse ONAXAN® uygulaması kesilmelidir (bkz. Bölüm4.9 Doz aşımı ve tedavisi). Klinik çalışmalarda, mukozal kanamalar (yani burun, dişeti, gastrointestinal, anormal vajinal veya artan adet kanaması dahil genito üriner kanamaa) ve anemi, tekli ve çift anti-platelettedavisinin yanı sıra uzun süreli rivaroksaban tedavisi sırasında daha sık görülmüştür. Bu yüzden,yeterli klinik gözetime ek olarak, hemoglobin/hematokrite yönelik laboratuvar testleri, okültkanamayı tespit etmek ve uygun olduğu kararı verildiği üzere açık kanamanın klinik açıdananlamının miktarını tayin etmek için çok değerli olabilir. Aşağıda ayrıntılı açıklandığı üzere, pek çok hasta alt grubu yüksek kanama riski altındadır. Buna ilaveten, bu hastalar tedavi başlatıldıktan sonra kanama komplikasyonları ve anemi belirti vesemptomları açısından dikkatlice izlenmelidir (bkz. Bölüm 4.8 İstenmeyen etkiler). Hemoglobin ya da kan basıncında herhangi bir açıklanamayan düşüş varsa, kişi kanama odağının tespiti açısından kapsamlı olarak aranmalıdır. Rivaroksaban ile tedavi rutin maruziyet takibi gerektirmemesine rağmen, kalibre edilmiş kantitatif bir anti-faktör Xa testi ile ölçülmüş rivaroksaban seviyeleri, rivaroksaban maruziyetibilgisinin doz aşımı ve acil cerrahi gibi klinik kararları almaya yardım edebileceği istisnaidurumlarda faydalı olabilir (bkz. Bölüm 5.1 Farmakodinamik özellikler ve Bölüm 5.2Farmakokinetik özellikler). Böbrek yetmezliği:Ciddi böbrek yetmezliği olan hastalarda (kreatinin klerensi < 30 ml/dak.) rivaroksaban plazma düzeyleri, kanama riskinde artışa yol açabilecek şekilde anlamlı derecede (ortalama 1,6 kat)yükselebilir. ONAXAN®, kreatinin klerensi 15-29 ml/dak olan hastalarda dikkatlikullanılmalıdır Kreatinin klerensi <15 ml/dak. olan hastalarda kullanılması önerilmemektedir(bkz. Bölüm 4.2 Pozoloji ve uygulama şekli ve Bölüm 5.2 Farmakokinetik özellikler). Rivaroksaban'ın plazma konsantrasyonlarını artıran diğer ilaçlan eş zamanlı olarak alan böbrek yetmezliği olan hastalarda ONAXAN® dikkatli kullanılmalıdır (bkz. Bölüm 4.5 Diğer tıbbiürünlerle etkileşimler ve diğer etkileşim şekilleri). Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri:Azol-antimikotikler (örn. Ketokonazol, itrakonazol, vorikonazol ve pasokonazol) veya HIV proteaz inhibitörleri (örn. ritonavir) ile eş-zamanlı sistemik tedavi gören hastalarda ONAXAN®kullanımı tavsiye edilmez. Bu etkin maddeler hem CYP3A4'ün, hem de P-gp'nin güçlüinhibitörleridir ve bu nedenle, bu ilaçlar rivaroksaban plazma konsantrasyonunu klinik olarakönemli derecede (ortalama 2,6 kat) arttırabilir; bu da kanama riskinde yükselmeye yol açabilir(bkz. Bölüm 4.5 Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri). Hastalar, steroidal olmayan anti-inflamatuvar tıbbi ürünler (NSAID'ler), asetilsalisilik asit, trombosit (ASA) agregasyon inhibitörleri veya seçici serotonin geri alım inhibitörleri (SSRI)veya serotonin norepinefrin geri alım inhibitörleri (SNRI) gibi hemostazı etkileyen tıbbiürünlerle eş zamanlı olarak tedavi edilirken dikkatli olunmalıdır. Ülseratif gastrointestinalhastalık riski olan hastalarda uygun bir proflaktik tedavi düşünülebilir (bkz. Bölüm 4.5 Diğertıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri ve Bölüm 5.1 Farmakodinamik özellikler). 5 / 27ONAXAN® ve ASA veya ONAXAN® ve ASA + klopidogrel/tiklopidin tedavisi uygulanan hastalara, sadece faydanın kanama riskine ağır basması halinde NSAID'lerle kronik eşzamanlıtedavi uygulanmalıdır. Diğer kanama riski faktörleri:ONAXAN®, diğer antitrombotikler gibi, kanama riskinin arttığı aşağıdaki durumlarda önerilmemektedir: Konjenital ya da edinilmiş kanama bozuklukları Kontrolsüz ciddi arteriyel hipertansiyon Aktif ülser olmaksızın kanama komplikasyonuna yol açabilecek diğer gastrointestinalhastalıklar (ör. inflamatuar bağırsak hastalığı, özofajit, gastrit ve gaströzofageal reflühastalığı) Vasküler retinopati Bronşiektazi ya da pulmoner kanama öyküsüAşağıdaki AKS ve KAH/PAH hastalarında dikkatli kullanılmalıdır: - >75 yaş, tek başına ASA veya ASA + klopidogrel veya tiklopidin ile eşzamanlıuygulandığında, Tedavinin fayda risk dengesi düzenli olarak ayrı ayrı değerlendirilmelidir. - vücut ağırlığı düşük olan (60 kg'den az), tek başına ASA veya ASA + klopidogrel veyatiklopidin ile eşzamanlı uygulandığında. - Şiddetli semptomatik kalp yetmezliği olan KAH hastaları. Çalışma verileri bu tür hastalarınrivaroksaban tedavisinden daha az fayda sağlayabileceğini göstermektedir (bkz. Bölüm 5.1Farmakodinamik özellikler). Prostetik kalp kapakçığı olan hastalar:Rivaroksaban, yakın zamanda transkateter aort kapak replasmanı (TAVR) geçirmiş hastalarda tromboprofilaksi için kullanılmamalıdır. Prostetik kalp kapakçıkları olan hastalardarivaroksabanın güvenliliği ve etkililiğine ilişkin çalışma yapılmamıştır; dolayısıylarivaroksabanın bu hasta popülasyonu için yeterli anti-koagülasyon sağladığını destekleyen verimevcut değildir. Bu hastalar için rivaroksaban ile tedavi önerilmemektedir. Antifosfolipid sendromlu hastalarAntifosfolipid sendromu tanısı ve tromboz öyküsü bulunan hastalarda, rivaroksaban dahil olmak üzere direkt etkili oral antikoagülan (DOAK) kullanımı önerilmez. Özellikle üçlü pozitif (lupusantikoagülan, antikardiyolipin antikorları ve anti-beta 2-glikoprotein I antikorları) hastalarda,DOAK tedavisi, K vitamin antagonisti tedavisine kıyasla, daha yüksek oranda rekürrentrombotik olaylarla ilişkili olabilir. Önceden inme ve/veya GİA'sı olan hastalar:AKS hastalarıONAXAN® 2,5 mg, önceden inme veya GİA öyküsü olan hastalarda AKS tedavisi için kontrendikedir (bkz. Bölüm 4.3 Kontrendikasyonlar). Önceden inme veya GİA geçirmiş olupAKS gelişen az sayıda hastayla çalışılmış olmakla birlikte, mevcut sınırlı etkililik verileri buhastaların tedaviden fayda görmeyeceğine işaret etmektedir. KAH/PAH hastalarıÖnceden hemorajik veya lakünar inme öyküsü olan veya önceki ay içinde laküner olmayan inme geçirmiş KAH/PAH hastaları ile ilgili çalışma yapılmamıştır (bkz. Bölüm 4.3Kontrendikasyonlar). 6 / 27Spinal/epidural anestezi veya ponksiyon:Nöraksiyal (epidural/spinal) anestezi ya da spinal/epidural ponksiyon uygulandığında, tromboembolik komplikasyonların önlenmesi için antitrombotik ajanlarla tedavi görenhastalarda, uzun dönemli veya kalıcı paraliz ile sonuçlanabilecek epidural ya da spinalhematom gelişme riski meydana gelir. Bu olaylara ilişkin risk, kalıcı epidural kateterlerinkullanılması sırasında ya da hemostazı etkileyen tıbbi ürünlerin eşzamanlı kullanımıylaartabilir. Bu risk, travmatik ya da tekrarlanan epidural veya spinal ponksiyon ile de artabilir. Hastaların nörolojik bozukluğa dair belirti ve semptomlar yönünden (örn. sersemlik hissi veya bacaklarda güçsüzlük, bağırsak ya da mesane işlev bozuklukları) karşı sık sık izlenmesigerekmektedir. Nörolojik bozukluk görülürse, acil tanı ve tedavi gereklidir.Antikoagüle olmuşhastalarda veya tromboprofilaksi için antikoagüle edilecek hastalarda nöraksiyal müdahaledenönce, doktorun riske karşı olası faydayı değerlendirmesi gerekir. Bu durum için rivaroksaban 2,5 mg'ın tek başına ASA ile birlikte veya ASA + klopidogrel veya tiklopidin ile kullanımınailişkin klinik deneyim mevcut değildir. Rivaroksaban ve nöraksiyal (epidural/spinal) anestezi veya spinal ponksiyonun eş zamanlı kullanımıyla ilişkilendirilen potansiyel kanama riskini azaltmak için rivaroksabanınfarmakokinetik profili göz önünde bulundurulmalıdır. Bir epidural kateter veya lombar ponksiyon, en iyi, rivaroksabanın antikoagülan etkisinin düşük olacağı tahmin edildiği zaman yerleştirilebilir veya (bkz. Bölüm 5.2 Farmakokinetiközellikler). Bununla birlikte, her hastada yeterli derecede düşük bir antikoagülan etkiye ulaşmakiçin kesin süre bilinmemektedir. Trombosit agregasyonu inhibitörleri, kısa ürün bilgilerinin belirttiği şekilde kesilmelidir. İnvazif prosedürler ve cerrahi müdahale öncesinde ve sonrasında doz önerileri:Bir invazif prosedür veya cerrahi müdahale gerekiyorsa, ONAXAN® 2,5 mg müdahaleden en az 12 saat önce ve doktorun klinik kararına göre kesilmelidir. Bir hasta elektif cerrahigeçirecekse ve antiplatelet etki istenmiyorsa, trombosit agregasyonu inhibitörleri ürünlerinkısa ürün bilgilerinin belirttiği şekilde kesilmelidir. Prosedür geciktirilemeyecekse, müdahalenin aciliyetine karşı artan kanama riski değerlendirilmelidir. ONAXAN®, klinik durumun izin vermesi ve tedavi eden doktorun belirlediği şekilde yeterli hemostaz gerçekleştirmesi koşuluyla, invazif prosedür veya cerrahi müdahaleden sonramümkün olan en kısa sürede başlatılmalıdır (bkz. Bölüm 5.2 Farmakokinetik özellikler). Geriatrik popülasyon:Yaş ilerledikçe hemorajik risk artabilir (bkz. Bölüm 5.1 Farmakodinamik özellikler ve Bölüm 5.2 Farmakokinetik özellikler). Dermatolojik reaksiyonlar:Rivaroksaban kullanımıyla ilişkili olarak pazarlama sonrası gözetim sırasında Stevens-Johnson sendromu/toksik epidermal nekroliz ve DRESS sendromu dahil olmak üzere, ciddi ciltreaksiyonları bildirilmiştir (bkz. Bölüm 4.8 İstenmeyen etkiler). Hastalar bu reaksiyonlarla ilgilien yüksek riski tedavi sürecinin başlarında taşımaktadır: reaksiyon başlangıcı vakaların büyükbölümünde tedavinin ilk haftalarında meydana gelir. Rivaroksaban, ciddi cilt döküntüsü (örn.yayılan, yoğun ve/veya kabarcıklanan) veya mukozal lezyonlarla bağlantılı başka birhipersensitivite emaresi ilk belirdiğinde kesilmelidir. 7 / 27Yardımcı maddeler ile ilgili bilgi:ONAXAN® laktoz içerir. Nadir kalıtımsal galaktoz intoleransı, Lapp laktaz yetmezliği ya da glukoz-galaktoz malabsorbsiyon problemi olan hastaların bu ilacı kullanmamaları gerekir. 4.5 Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleriCYP 3A4 ve P-gp inhibitörleri:Rivaroksaban'ın ketokonazol (günde bir kez 400 mg) veya ritonavir (günde iki kez 600 mg) ile eşzamanlı kullanımı, farmakodinamik etkilerde kanama riskinin artmasına yol açabilecekanlamlı artışlarla birlikte, ortalama rivaroksaban EAA'sında 2,6 kat / 2,5 kat artışa ve ortalamarivaroksaban Cmaks'ında 1,7 kat / 1,6 kat artışa yol açmıştır. Bu nedenle, rivaroksabanıneşzamanlı olarak ketokonazol, itrakonazol, vorikonazol ve posakonazol gibi azol-antimikotiklerya da HIV proteaz inhibitörleri ile sistemik tedavi gören hastalarda kullanılmasıönerilmemektedir. Bu etkin maddeler hem CYP3A4 hem de P-gp'nin güçlü inhibitörleridir(bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Rivaroksabanın eliminasyon yollarından (CYP 3A4 ya da P-gp) sadece birini kuvvetli şekilde inhibe eden diğer etkin maddelerin, rivaroksaban plazma konsantrasyonlarını daha düşük birdüzeyde arttırması beklenmektedir. Örneğin,güçlü bir CYP 3A4 inhibitörü ve orta derecedebir P-gp inhibitörü olarak kabul edilen klaritromisin (günde iki kez 500 mg), ortalamarivaroksaban EAA'sında 1,5 kat, ve Cmaks'ında 1,4 kat artışa yol açar. Klaritromisin ile etkileşimin çoğu hastada klinik olarak anlamlı olması muhtemel olmamakla birlikte, yüksek risk taşıyan hastalarda anlamlı olabilir. (Böbrek yetmezliği olan hastalar için,bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). CYP 3A4 ve P-gp'yi orta derecede inhibe eden eritromisin (500 mg günde üç kez), ortalama rivaroksaban EAA'sı ve Cmaks'ında 1,3 kat artışa neden olmuştur. Eritromisin ile etkileşiminçoğu hastada klinik olarak anlamlı olması muhtemel olmamakla birlikte, yüksek risk taşıyanhastalarda anlamlı olabilir. Hafif böbrek yetmezliği olan hastalarda, eritromisin (günde üç kez 500 mg) böbrek fonksiyonu normal olan gönüllülere göre ortalama rivaroksaban EAA değerinde 1,8 kat artışa ve Cmaksdeğerinde 1,6 kat artışa yol açmıştır. Orta düzey böbrek yetmezliği olan hastalarda, eritromisinböbrek fonksiyonu normal olan gönüllülere göre ortalama rivaroksaban EAA değerinde 2,0 katartışa ve Cmaks değerinde 1,6 kat artışa yol açmıştır. Eritromisin böbrek yetmezliğine ek biretki yaratmıştır (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Orta derece kuvvetli CYP 3A4 inhibitörü olarak değerlendirilen flukonazol (günde bir kez 400 mg), ortalama rivaroksaban EAA değerinde 1,4 kat, ortalama Cmaks değerinde ise 1,3 kat artışaneden olmuştur. Flukonazol ile etkileşimin çoğu hastada klinik olarak anlamlı olmasımuhtemel olmamakla birlikte, yüksek risk taşıyan hastalarda anlamlı olabilir (Böbrekyetmezliği olan hastalar için, bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Dronedaron ile ilgili klinik veriler sınırlı olduğundan, rivaroksaban ile eşzamanlı kullanımından kaçınılmalıdır. Antikoagülanlar:Enoksaparin (40 mg tek doz) ile rivaroksabanın (10 mg tek doz) kombine kullanımından sonra, pıhtılaşma testlerine (protrombin zamanı [PTZ], aktive edilmiş parsiyel tromboplastin 8 / 27zamanı [aPTT]) ilave bir etkisi olmaksızın anti-faktör Xa aktivitesine ilave etki gözlenmiştir. Enoksaparin, rivaroksabanın farmakokinetiğini etkilemez. Artan kanama riski nedeniyle, diğer antikoagülanlarla eşzamanlı tedavi gören hastalarda dikkatli olunmalıdır. (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). NSAİİ/trombosit agregasyon inhibitörleri:Rivaroksaban (15 mg) ve 500 mg naproksenin eşzamanlı uygulanmasından sonra kanama zamanında klinik olarak önemli bir uzama gözlenmemiştir. Bununla beraber, daha belirginfarmakodinamik cevap veren bireyler olabilir (bkz. Bölüm 4.4 Özel kullanım uyarıları veönlemleri). Rivaroksaban 500 mg asetilsalisilik asit ile birlikte uygulandığında klinik olarak anlamlı farmakokinetik veya farmakodinamik etkileşimler gözlemlenmemiştir. Klopidogrel (300 mg yükleme dozu, ardından 75 mg idame dozu) rivaroksaban (15 mg) ile farmakokinetik bir etkileşim göstermez, ancak hastaların bir alt grubunda kanama zamanındatrombosit agregasyonu, P-selektin ya da GPIIb / IIIa reseptör seviyesi ile korele olmayan artışgözlenmiştir (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Hastalar NSAID'ler (asetilsalisilik asit dahil) ve trombosit agregasyon inhibitörleri ile eş zamanlı olarak tedavi görüyorsa dikkatli olunmalıdır, zira bu tıbbi ürünler tipik olarak kanama riskiniartırmaktadır (bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). SSRI'lar/SNRI'lar:Diğer antikoagülanlarda olduğu gibi, trombositler üzerindeki bildirilmiş etkileri nedeniyle SSRI veya SNRI'ların eş zamanlı kullanımı durumunda hastaların kanama riskinin yükselmesiolasılığı bulunmaktadır. Rivaroksaban klinik programında eş zamanlı kullanıldığında, tüm tedavigruplarında önemli veya önemli olmayan klinik olarak ilgili kanama oranlarının sayısal olarakdaha yüksek olduğu gözlenmiştir Varfarin:Tedavinin varfarinden (INR 2.0-3.0) rivaroksabana (20 mg) ya da rivaroksabandan (20 mg) varfarine (INR 2.0-3.0) değiştirilmesi PTZ/INR (Neoplastin) testinde beklenenden fazla artışa(12'ye varan münferit INR değerleri gözlenebilir) yol açmıştır; aPTT, Faktör Xa (Fxa) aktiviteinhibisyonu ve endojen trombin potansiyeli ise aditif olmuştur. Değişim periyodu sırasında rivaroksabanın farmakodinamik etkileri test edilmek istendiğinde, anti-faktör Xa aktivitesi, PiCT (Prothrombinase-induced Clotting Time) ve HepTest® kullanılabilir; bu testler varfarinden etkilenmez. Varfarin kesildikten sonra 4. günden itibaren tüm testler (PT,aPTT, FXa aktivitesinin inhibisyonu ve ETP [Endojen Trombin Potansiyeli] dahil) yalnızcarivaroksaban etkisini yansıtır (bkz. Bölüm 4.2 Pozoloji ve uygulama yöntemi). Değişim periyodu sırasında varfarinin farmakodinamik etkileri test edilmek istendiğinde, rivaroksaban Cvadi (önceki rivaroksaban dozundan 24 saat sonra) değerinde INR ölçümüyapılabilir; bu nokta bu testin rivaroksabandan en az etkilendiği noktadır. Varfarin verivaroksaban arasında farmakokinetik etkileşim gözlenmemiştir. CYP 3A4 indükleyicileri:Rivaroksabanın güçlü CYP3A4 ve P-gp indükleyicisi rifampisin ile beraber uygulanması, ortalama rivaroksaban EAA'sında yaklaşık %50 azalma ile farmakodinamik etkilerinde paralelbir azalmaya neden olur. Rivaroksabanın diğer güçlü CYP3A4 indükleyicileri (ör. fenitoin, 9 / 27karbamazepin, fenobarbital ya da St. John bitkisi (sarı kantaron otu, Hypericumperforatum))ile eşzamanlı kullanımı da rivaroksaban plazma konsantrasyonunda azalmaya yol açabilir. Bunedenle, hasta tromboz belirti ve semptomları açısından yakından izlenmediği sürece güçlüCYP3A4 indükleyicileriyle eşzamanlı uygulamadan kaçınılmalıdır.Diğer eş zamanlı tedavilerRivaroksaban ile midazolam (CYP3A4 substratı), digoksin (P-gp substratı) ya da atorvastatin (CYP3A4 ve P-gp substratı) veya omeprazol (proton pompası inhibitörü) eş zamanlıuygulandığında klinik olarak anlamlı farmakokinetik veya farmakodinamik etkileşimlergözlenmemiştir. Rivaroksaban, CYP3A4 gibi herhangi bir majör CYP izoformunu inhibe etmezve indüklemez. Klinik açıdan önemli herhangi bir gıda etkileşimi gözlenmemiştir (bkz. Bölüm4.2 Kullanım şekli ve dozu). Laboratuvar parametreleri:Rivaroksabanın etki mekanizmasına uygun olarak beklendiği üzere, pıhtılaşma parametresi testleri (PTZ, aPTT, HepTest®) etkilenmektedir (bkz Bölüm 5.1 Farmakodinamik özellikler). 4.6 Gebelik ve laktasyonGenel tavsiye:Çocuk doğurma potansiyeli olan kadınlar / Doğum kontrolü (kontrasepsiyon):Çocuk doğurma potansiyeli olan kadınlar rivaroksaban ile tedavi süresince gebe kalmaktan kaçınmalıdır. Gebelik dönemi:Gebe kadınlarda rivaroksabanın güvenlilik ve etkililiği bilinmemektedir. Hayvanlar üzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. Bölüm 5.3 Klinik öncesi güvenlilik verileri). Olası üreme toksisitesi, doğal olarak meydanagelen kanama riski ve rivaroksabanın plasentaya geçtiği yönündeki veriler nedeniyleONAXAN® gebelikte kontrendikedir (bkz. Bölüm 4.3 Kontrendikasyonlar ve bölüm 5.3 Kliniköncesi güvenlilik verileri). Laktasyon dönemi:Emziren kadınlarda rivaroksabanın güvenlilik ve etkililiği bilinmemektedir. Hayvanlardan elde edilen veriler rivaroksabanın süte geçtiğini göstermektedir. Bu nedenle ONAXAN®emzirme döneminde kontrendikedir (bkz. B ölüm 4.3 Kontrendikasyonlar). Emzirmeyibırakma veya tedaviyi kesme/tedaviden kaçınma yönünde karar verilmelidir. Üreme yeteneği / Fertilite:Fertilite üzerindeki etkileri değerlendirmek için insanlarla özel rivaroksaban çalışmaları yapılmamıştır. Sıçanlarda erkek ve dişi fertilitesine ilişkin bir çalışmada, hiçbir etkigörülmemiştir (bkz. Bölüm 5.3 Klinik öncesi güvenlilik verileri). 4.7 Araç ve makine kullanımı üzerindeki etkilerRivaroksabanın araç ve makine kullanma yeteneği üzerinde önemsiz bir etkisi vardır. Senkop (sıklık: yaygın olmayan) ve baş dönmesi (sıklık: yaygın) gibi advers reaksiyonlar bildirilmiştir(bkz. Bölüm 4.8 İstenmeyen etkiler). Bu advers reaksiyonların görüldüğü hastalar araç ya damakine kullanmamalıdır. 10 / 274.8 İstenmeyen etkiler4.8.1 Güvenlilik profilinin özetiGüvenlilik profilinin özeti Rivaroksabanın güvenliliği, rivaroksabana maruz bırakılan 53.103 hastanın dahil olduğu on üç faz III çalışmada değerlendirilmiştir (bkz. Tablo 1).
11 / 27Rivaroksaban alan hastalarda en yaygın olarak bildirilen advers reaksiyon kanamadır (bkz. aşağıda Bölüm 4.4. ve 'Seçilen advers reaksiyonların açıklaması') (Tablo 2). En yaygın olarakbildirilen kanamalar epistaksis (%4,5) ve gastrointestinal kanal hemorajisidir (%3,8). Tablo 2: Tamamlanmış faz III çalışmaları boyunca rivaroksabana maruz kalan hastalarda kanama* ve anemi olaylarının oranı
* Tüm rivaroksaban çalışmaları için, bütün kanama olayları toplanır, bildirilir ve karara bağlanır. ** COMPASS çalışmasında, advers olay toplanmasına yönelik seçici bir yaklaşım uygulandığı için düşük bir anemi insidansı mevcuttur. Rivaroksaban kullanımında bildirilen advers ilaç reaksiyonlarının sıklıkları aşağıda özetlenmiştir. İstenmeyen etkiler, her bir sıklık grubunda azalan ciddiyet derecesine göresıralanmıştır. Çok yaygın (>1/10), yaygın (>1/100 ila <1/10), yaygın olmayan (>1/1000 ila<1/100), seyrek (>1/10000 ila <1/1000), çok seyrek (<1/10000) ve bilinmiyor (eldeki verilerdenhareketle tahmin edilemiyor) olarak tanımlanmıştır. Faz III klinik çalışmalarında veya pazarlama sonrası kullanımı boyunca hastalarda bildirilen tüm advers ilaç reaksiyonları*Kan ve lenf sistemi hastalıklarıYaygın: Anemi (ilgili laboratuvar parametreleri dahil) Yaygın olmayan: Trombositemi (trombosit sayısı artışı dahil)A, trombositopeni Bağışıklık sistemi hastalıklarıYaygın olmayan: Alerjik reaksiyon, alerjik dermatit, anjiyoödem ve alerjik ödem Çok seyrek: Anafilaktik şok da dahil olmak üzere anafilaktik reaksiyonlar 12 / 27Sinir sistemi hastalıklarıYaygın: Baş dönmesi, baş ağrısı, Yaygın olmayan: Serebral ve intrakranial kanama, senkop Göz hastalıklarıYaygın: Gözde kanama (Konjunktival kanama dahil) Kardiyak hastalıklarYaygın olmayan: Taşikardi Vasküler hastalıklarYaygın: Hipotansiyon, hematom Solunum, göğüs bozuklukları ve mediastinal hastalıklarYaygın: Epistaksis, hemoptizi Gastrointestinal hastalıklarYaygın: Jinjival kanama, gastrointestinal sistem kanaması (rektal kanama dahil), abdominal ve gastrointestinal ağrı, dispepsi, bulantı, konstipasyonA, diyare, kusmaAYaygın olmayan: Ağız kuruluğu Hepatobiliyer hastalıklarYaygın: Transaminaz artışıYaygın olmayan: Karaciğer yetmezliği, bilirubin artışı, kan alkalin fosfataz artışıA, GGT artışıA Seyrek: Sarılık, konjuge bilirubin artışı (eş zamanlı ALT artışı ile veya ALT artışı olmadan),kolestaz, hepatit (hepatoselüler yaralanma dahil) Deri ve deri altı dokusu hastalıklarıYaygın: Kaşıntı (nadiren jeneralize kaşıntı dahil), döküntü, ekimoz, deri ve deri altında kanama Yaygın olmayan: ÜrtikerÇok seyrek: Stevens-Johnson sendromu/ Toksik Epidermal Nekroliz, DRESS sendromu Kas-iskelet bozuklukları, bağ dokusu ve kemik hastalıklarıYaygın: Ekstremite ağrısıA Yaygın olmayan: HemartrozSeyrek: Kas kanamasıBilinmiyor: Kanamaya sekonder kompartman sendromu Böbrek ve idrar hastalıklarıYaygın: Ürogenital sistem kanaması (hematüri ve menoraji dahilB), renal bozukluk (kan kreatinin artışı, kan üre artışı dahil) Bilinmiyor: Böbrek yetmezliği/hipoperfüzyona neden olmaya yetecek ölçüde kanamaya sekonder akut böbrek yetmezliği Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarYaygın: AteşA, periferik ödem, genel güç ve enerjide azalma (yorgunluk ve asteni dahil) Yaygın olmayan: İyi hissetmeme (malezi dahil) Seyrek: Lokalize ödemA AraştırmalarYaygın olmayan: LDH artışıA, lipaz artışıA, amilaz artışıA 13 / 27Yaralanma, Zehirlenme ve Prosedürel KomplikasyonlarYaygın: Prosedür sonrası kanama (postoperatif anemi ve yarada kanama da dahil), kontüzyon, yara yeri sızıntısıA Seyrek: Vasküler psödoanevrizmaC A: Elektif kalça veya diz replasman cerrahisi geçiren yetişkin hastalarda VTE'nin önlenmesinde gözlemlenmiştir. B: <55 yaş kadınlarda çok yaygın olarak DVT, PE tedavisi ve nüksün önlenmesinde gözlemlenmiştir C: AKS sonrası önlem tedavisinde (perkütan girişimin ardından) yaygın olmayan olarak gözlenmiştir. * Advers olay toplanmasında önceden belirtilen seçici bir yaklaşım uygulanmıştır. Advers reaksiyonların insidansı artmadığı ve yeni advers reaksiyon saptanmadığı için, bu tablodakifrekans hesaplamasına COMPASS çalışması verileri dahil edilmemiştir. Seçilmiş advers reaksiyonların tanımı Farmakolojik etki şekline bağlı olarak, ONAXAN® posthemorajik anemi ile sonuçlanabilecek, herhangi bir doku ve organda gelişebilecek açık ya da gizli kanama riskinde artışla ilişkili olabilir. Belirti, semptom ve şiddetli (ölümcül sonuç dahil) kanamave/veya aneminin yerleşimi ve derecesine göre değişir (bkz. Bölüm 4.9 Doz aşımı ve tedavisi). Klinik çalışmalarda, mukozal kanamalar (yani burun, dişeti, gastrointestinal, anormal vajinal veya artan adet kanaması dahil genito üriner kanama) ve anemi, VKA tedavisiylekıyaslandığında uzun süreli rivaroksaban tedavisinde daha sık görülmüştür. Bu yüzden, yeterliklinik gözetime ek olarak, hemoglobin/hematokrite yönelik laboratuvar testleri, o kültkanamayı tespit etmek ve uygun olduğu kararı verildiği üzere açık kanamanın klinik açıdananlamının miktarını tayin etmek için çok değerli olabilir. Kanama riski belirli hasta gruplarında artabilir, örn. kontrol altında olmayan şiddetli arteriyel hipertansiyonu olan ve/veya eş zamanlı olarak hemostazı etkileyen ilaçlar almakta olanhastalar (bkz. Bölüm 4.4 Kanama riski). Menstrüel kanama şiddetlenebilir ve/veya uzayabilir. Hemorajik komplikasyonlar, güçsüzlük, solukluk, baş dönmesi, baş ağrısı ya da açıklanamayan şişlikler, dispne ve açıklanamayan şok olarak görülebilir. Bazı olgularda, aneminin bir sonucuolarak göğüs ağrısı veya anjina pektoris gibi kardiyak iskemi semptomları gözlenmiştir. Rivaroksaban ile kompartman sendromu ve hipoperfüzyon nedeniyle renal yetmezlik gibi şiddetli kanamaya bağlı gelişen komplikasyonlar bildirilmiştir. Bu nedenle, antikoagülankullanan her hasta değerlendirilirken hemoraji olasılığı düşünülmelidir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; eposta:[email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9 Doz aşımı ve tedavisiKanama komplikasyonu ya da diğer advers reaksiyonlar olmaksızın 600 mg'a kadar nadir doz 14 / 27aşımı olguları bildirilmiştir. Sınırlı emilim nedeniyle >50 mg supraterapötik dozlarda ortalama plazma maruziyetinde daha fazla artış olmadan bir tavan etkisi beklenir. Rivaroksabanın farmakodinamik etkisini antagonize eden spesifik bir antidot bulunmamaktadır. Rivaroksaban doz aşımı durumunda emilimi azaltmak için aktif kömürkullanımı düşünülebilir. Kanamanın idaresiRivaroksaban kullanan bir hastada kanama meydana gelirse, bir sonraki rivaroksaban uygulaması geciktirilmeli veya uygun ise tedavi kesilmelidir. Rivaroksabanın yarı ömrüyaklaşık 5 ila 13 saattir (bkz. Bölüm 5.2). İdare kanamanın ciddiyetine ve yerine göre kişiyeözel hale getirilmelidir. Gerektiğinde mekanik kompresyon (örn. şiddetli epistaksisolgusunda), kanama kontrolü işlemleri ile birlikte cerrahi hemostaz, sıvı replasmanı vehemodinamik destek, kan ürünleri (anemi ya da koagülopatiye göre kırmızı kan hücresi, tazedonmuş plazma) ya da trombosit gibi uygun semptomatik tedavi uygulanmalıdır. Kanama yukarıdaki önlemlerle kontrol edilemediğinde, ya rivaroksabanın farmakodinamik etkilerini antagonize eden spesifik bir faktör Xa inhibitörü geri dönüştürücü ajanı (andexanetalfa) ya da protrombin kompleks konsantratı (PCC), aktive protrombin kompleks konsantratı(APCC) ya da rekombinant faktör VIIa (r- FVIIa) gibi özgün prokoagülan geri döndürücüajan kullanılması düşünülmelidir. Bununla birlikte, bugün için, bu ilaçların rivaroksabanalan kişilerde kullanımına dair klinik tecrübe oldukça kısıtlıdır. Tavsiye de sınırlı klinik dışı verilere dayanmaktadır. Rekombinant Faktör VIIa'nın yeniden doz ayarlaması düşünülebilir ve kanamanın gelişimine göre titre edilebilir. Majör kanamalaresnasında, lokal olarak uygun olma durumuna bağlı olarak, hematoloji uzmanı ilekonsültasyon düşünülmelidir (bkz. Bölüm 5.1 Farmakodinamik özellikler). Protamin sülfat ve K vitamininin rivaroksabanın antikoagülan aktivitesini etkilemesi beklenmez. Rivaroksaban alan hastalarda, traneksamik asit ile ilgili deneyim sınırlı olup, aminokaproik asit ve aprotinin ile deneyim bulunmamaktadır. Rivaroksaban kullanan hastalarda sistemikhemostatik olan desmopresin kullanımıyla ilgili olarak fayda açısından bilimsel gerekçe veyadeneyim bulunmamaktadır. Plazma proteinlerine bağlanma oranının yüksek olması nedeniyle,rivaroksabanın diyaliz ile uzaklaştırılması beklenmemektedir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: Antitrombotik ajanlar, direkt faktör Xa inhibitörleri ATC kodu: B01AF01 Etki mekanizması: Rivaroksaban oral biyoyararlanımı olan oldukça selektif direkt bir Faktör Xa (FXa) inhibitörüdür. Faktör Xa'nın inhibisyonu, hem trombin oluşumunu hem de tromboz gelişimini inhibe ederek kan koagülasyon kaskadının intrinsik ve ekstrinsik yolunu bozmaktadır. Rivaroksaban trombini(aktive faktör II) inhibe etmez ve trombositler üzerinde herhangi bir etkisi olmadığıkanıtlanmıştır. 15 / 27Farmakodinamik etkiler: İnsanlarda FXa aktivitesinin doza bağımlı şekilde inhibe olduğu gözlenmiştir. Eğer testte Neoplastin® kullanılırsa, PTZ, plazma konsantrasyonları (r değeri 0,98'e eşit) ile yakın ilişkiliolarak doza bağımlı şekilde rivaroksabandan etkilenir. Diğer reaktifler farklı sonuçlarsağlayacaktır. INR sadece kumarinler için kalibre edildiği ve onaylandığından ve herhangi diğerbir antikoagülan için kullanılamadığından, PTZ okuması saniyeler içinde yapılmalıdır. Sağlıklı yetişkin gönüllülerde (n=22) rivaroksabanın farmakodinamik özelliklerinin geri çevrilmesine ilişkin bir klinik farmakoloji çalışmasında, 3 faktörlü PCC (Faktör II, IX ve X) ve4 faktörlü PCC'nin (Faktör II, VII, IX ve X) olmak üzere iki farklı PCC tipinin tek dozlarınınetkileri (50 IU/kg) değerlendirilmiştir. 3 faktörlü PCC ortalama Neoplastin PTZ değerlerini 30dakika içinde yaklaşık 1,0 saniye düşürürken, 4 faktörlü PCC ile yaklaşık 3,5 saniyelikdüşüşler gözlemlenmiştir. Buna karşın, 3 faktörlü PCC, 4 faktörlü PCC'ye kıyasla, endojentrombin oluşumunda değişiklikleri geri çevirme üzerinde daha büyük ve daha hızlı bir geneletki yapmıştır (bkz. Bölüm 4.9 Doz aşımı ve tedavisi). Aktive parsiyel tromboplastin zamanı (aPTT) ve HepTest® de doza bağımlı şekilde uzarlar; ancak rivaroksabanın farmakodinamik etkisini değerlendirmek için önerilmezler. Klinik rutinderivaroksaban ile tedavi sırasında koagülasyon parametrelerinin izlenmesine gerek yoktur. Ancak,klinik olarak belirtilirse rivaroksaban seviyeleri kalibre edilmiş nicel anti-faktör Xa testleriyleölçülebilir (bkz. bölüm 5.2 Farmakokinetik özellikler). Klinik etkililik ve güvenlilik: AKSRivaroksaban klinik programı, yakın dönemde AKS (ST-elevasyonlu miyokard infarktüsü [STEMI], ST-elevasyonsuz miyokard infarktüsü [NSTEMI] ya da unstabil angina [UA])geçirmiş bireylerde KV ölüm, MI ya da inmenin önlenmesinde rivaroksabanın etkililiğinigöstermek için tasarlanmıştır. Pivotal çift kör ATLAS AKS 2 TIMI 51 çalışmasında yer alan15.526 hasta 3 tedavi grubundan birine 1:1:1 oranında randomize edilmiştir: oral olarak gündeiki kez 2,5 mg rivaroksaban grubu, oral olarak günde iki kez 5 mg rivaroksaban grubu ya da gündeiki kez tek başına ASA veya ASA+bir tiyenopiridin (klopidogrel veya tiklopidin) ile birlikte verilen plasebogrubu. 55 yaşın altındaki AKS hastlannda diyabet veya önceden MI öyküsü olması gerekmiştir. Medyantedavi süresi 13 ay ve genel tedavi süresi yaklaşık olarak 3 yıla kadardır. Hastaların %93,2'si eşzamanlı olarak ASA ile birlikte tiyenopiridin tedavisi ve %6,8'i yalnızca ASA almıştır. İkili antitrombosit tedavisi gören hastaların %98,8'i klopidogrel, %0,9'u tiklopidin ve %0,3'ü prasugrel almıştır. Hastalar, hastaneye kabulden en az 24 saat sonra ve 7 güne kadar (ortalama 4,7 gün), AKS olay indeksinin (revaskülarizasyon prosedürleri dahil) stabilizasyonundan sonra mümkün olan enkısa sürede ve parenteral antikoagülasyon tedavisinin normalde kesileceği zaman ilkrivaroksaban dozunu almıştır. Günde iki kez 2,5 mg ve günde iki kez 5 mg rivaroksaban rejimlerinin her ikisi de standart antitrombosit tedavinin yanı sıra KV olayların insidansının daha fazla düşürülmesinde de etkiliolmuştur. Günde iki kez 2,5 mg rejimi mortaliteyi azaltmıştır ve düşük dozun daha düşükkanama riski oluşturduğuna dair kanıtlar mevcuttur; dolayısıyla kardiyak biyogöstergelerinyüksek olduğu AKS sonrası hastalarda aterotrombotik olayların önlenmesi için tek başınaasetilsalisilik asit (ASA) ya da ASA ve klopidogrel veya tiklopidin ile kombinasyon şeklindegünde iki kez 2,5 mg rivaroksaban kullanılması önerilmektedir. 16 / 27Rivaroksaban, plasebo ile karşılaştırıldığında KV ölüm, MI ya da inmenin primer birleşik sonlanım noktasını anlamlı oranda azaltmıştır. Fayda KV ölüm ve MI'deki azalma ile elde edilmiş ve tüm tedavi periyodu boyunca sabit tedavi etkisi ile erken meydana gelmiştir (bkz. Tablo 3 ve Şekil 1). Ayrıca ilk sekonder sonlanımnoktası (her nedene bağlı ölüm, MI veya inme) anlamlı derecede azalmıştır. İlave bir retrospektifanaliz, plaseboya kıyasla stent tromboz insidans oranlarında nominal olarak anlamlı bir azalmagöstermiştir (bkz. Tablo 3). Temel güvenlilik sonucuna (koroner arter bypass greft [CABG] ileilişkili olmayan TIMI majör kanama olayları) ait insidans oranları, rivaroksaban ile tedavi edilenhastalarda plasebo alan hastalara kıyasla yüksektir (bkz. Tablo 5). Bununla birlikte insidansoranları, ölümcül kanama olayları intravenöz inotropik ilaçlar ile tedavi gerektiren hipotansiyonve devam eden kanamaya yönelik cerrahi müdahale bileşenleri için rivaroksaban ve plaseboarasında dengelenmiştir. Tablo 4'te, perkutanöz koroner girişim (PCI) geçiren hastaların etkililik sonuçları sunulmaktadır. Bu PCI geçiren hasta alt grubundaki güvenlilik sonuçları genel güvenlilik sonuçlarına benzerdir.Biyogöstergelerinde (troponin veya CK-MB) yükselme olan ve önceden inme/GİA geçirmemişhastalar çalışma popülasyonunun %80'ini oluşturmuştur. Bu hasta popülasyonunun sonuçları dagenel etkililik ve güvenlilik sonuçları ile tutarlıdır.
17 / 27Tablo 4: Faz III ATLAS AKS 2 TIMI 51'den elde edilen PCI uygulanan hastalarda güvenlilik sonuçları
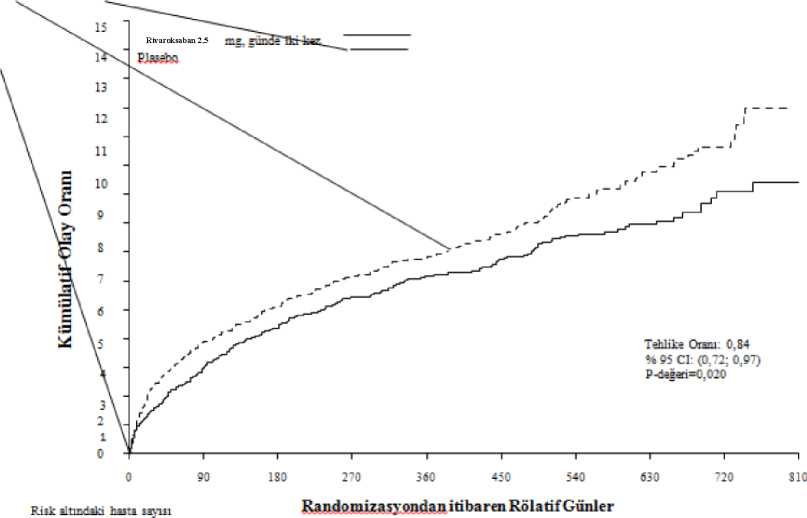
18 / 27Şekil 1: Primer etkililik sonlanım noktasının (KV ölüm, MI veya inme) ilk ortaya çıkışına kadar geçen süre
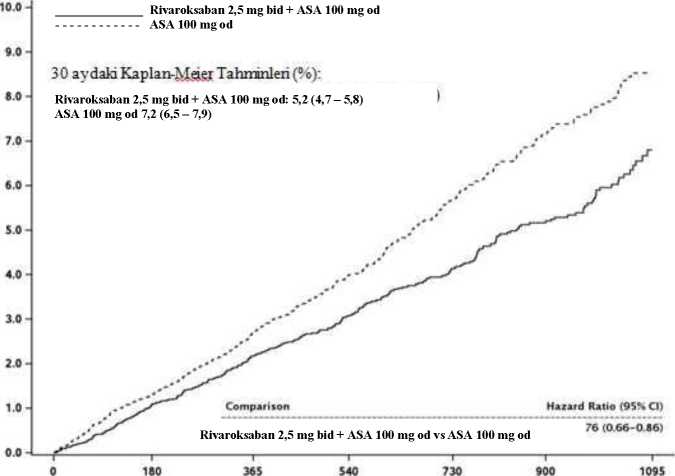
KAH veya PAHFaz III COMPASS çalışması (27.395 hasta, %78 erkek, %22 kadın), yüksek iskemik olay riski taşıyan KAH'li veya semptomatik PAH'li hastalarda KV ölüm, MI, inme bileşimininönlenmesinde rivaroksabanın etkililiğini ve güvenliliğini göstermiştir. Hastalar ortalama 23ay ve en fazla 3,9 yıl izlenmiştir. Bir proton pompası inhibitörü ile sürekli tedaviye ihtiyaç duymayan gönüllüler pantoprazol veya plaseboya randomize edilmiştir. Tüm hastalar daha sonra 1:1:1 olarak günde iki kez rivaroksaban 2,5 mg/günde bir kez ASA 100 mg'a, günde iki kez rivaroksaban 5 mg'a veya tek başına günde bir kere ASA 100 mg'ave bunlara karşılık gelen plasebolara randomize edilmiştir. KAH hastalarında çok damarlı KAH ve/veya önceden MI öyküsü vardır. 65 yaş üstü hastalar için, en az iki vasküler yatak içeren ateroskleroz veya en az iki ilave kardiyovasküler riskfaktörü gerekmiştir. PAH hastaları, ayak bileği/kol kan basınç oranı <0,90 olan arteriyel vasküler hastalık ya da intermittan klaudikasyo ve/veya anlamlı periferik arter stenozu veya önceden karotidrevaskülarizasyonu öyküsü ya da %50 ve üzerinde asemptomatik karotid arter stenozunedeniyle önceden bypass ameliyatı veya perkutanöz transluminal anjiyoplasti veya bacakyada ayak ampütasyonu gibi müdahaleler geçirmiştir. Hariç bırakma kriterleri arasında, çift trombosit veya diğer ASA olmayan antitrombosit veya oral antikoagülan tedavisi gerekliliğinin yanı sıra, yüksek kanama riski taşıyan, veyaejeksiyon fraksiyonu %30'un altında ya da New York Kalp Derneği sınıf II veya IV olan kalpyetmezliği olan ya da 1 ay içinde herhangi bir iskemik, laküner olmayan inme geçiren veyaherhangi bir kanama veya laküner inme öyküsü olan hastalar bulunmaktadır. 19 / 27Günde bir kez ASA 100 mg ile kombine olarak günde iki kez 2,5 mg rivaroksaban KV ölüm, MI, inme primer birleşik sonucunun azaltılmasında ASA 100 mg'dan daha üstün olmuştur(bkz. Tablo 6 ve Şekil 2). ASA 100 mg alan hastalara kıyasla günde bir kez ASA 100 mg ile kombine olarak günde iki kez 2,5 mg rivaroksaban alan hastalarda primer güvenlilik sonucunda (değiştirilmiş ISTHmajör kanama olayları) anlamlı bir artış olmuştur (bkz. Tablo 7). Primer etkililik sonucu için, günde bir kez ASA 100 mg'a kıyasla günde iki kez 2,5 mg rivaroksaban + günde bir kez ASA 100 mg'ın gözlemlenen faydası 75 yaş ve üzeri hastalardaHR=0,89 (%95 GA 0,7-1,1) (insidans: %7,0'a karşı %6,3) ve 75 yaş altı hastalardaHR=0,70'tir (%95 GA 0,6-0,8) (%'a karşı %3,6). Değiştirilmiş ISTH majör kanama için,gözlemlenen risk artışı 75 yaş ve üzeri hastalarda HR=2,12 (%95 GA 1,5-3,0) (%2,5'e karşı%5,2) ve 75 yaş altı hastalarda HR=1,53'tür (%95 GA 1,2-1,9) (%1,7'ye karşı %2,6). Proton pompası inhibitörüne klinik gereksinimi olmayan hastalarda antitrombotik çalışma ilacına ek olarak günde bir kez 40 mg pantoprazol kullanımı, üst gastrointestinal olayların (yaniüst gastrointestinal kanama, üst gastrointestinal ülserasyon veya üst gastrointestinalobstrüksiyon veya perforasyon bileşiminin) önlenmesinde fayda göstermemiştir; üstgastrointestinal olayların görülme oranı günde bir kez 40 mg pantoprazol grubunda 0,39/100hasta-yıl ve günde bir kez plasebo grubunda 0,44/100 hasta-yıl'dır. Tablo 6: Faz III COMPASS çalışmasından elde edilen etkililik sonuçları
* Primer etkililik sonucundaki azalma istatistiksel olarak üstün olmuştur. CI: güven aralığı, % KM: 900 günde hesaplanan kümülatif insidans riske yönelik Kaplan-Meier tahminleri; KV: kardiyovasküler; MI: miyokard enfarktüsü
21 / 27Şekil 2: COMPASS'ta primer etkililik sonucunun (inme, miyokard enfarktüsü, kardiyovasküler ölüm) ilk ortaya çıkmasına kadar geçen süre
CI: güven aralığı Kalp yetmezliğini içeren KAHCOMMANDER HF çalışması, dekompanse kalp yetmezliğinin (KY) hastanede tedavisinin ardından kalp yetmezliği ve önemli koroner arter hastalığı (KAH) olan, şu iki tedavigrubundan birine rastgele atanan 5.022 hastayı içermiştir: sırasıyla, rivaroksaban günde ikikez 2,5 mg (n = 2.507) veya karşılık gelen plasebo (n = 2.515). Genel ortalama çalışma tedavisüresi 504 gündür. Hastalarda en az 3 aydır semptomatik KY ve kayıttan sonraki bir yıl içinde < %40 sol ventrikül ejeksiyon fraksiyonu (SVEF) olmalıdır. Başlangıçta, medyan ejeksiyon fraksiyonu%34 olmuştur (IQR: %28-38) ve gönüllülerin %53'ü NYHA Sınıf III veya IV'tür. Birincil etkililik analizi (yani, tüm nedenlere bağlı mortalite, MI veya inme bileşimi), HR = 0.94 (%95 GA 0,84-1,05), p=0,270 olmak üzere, günde iki kez 2,5 mg rivaroksaban grubuile plasebo grubu arasında istatistiksel olarak anlamlı bir fark ortaya koymamıştır. Tümnedenlere bağlı mortalite için, rivaroksaban ve plasebo arasında olay sayısı açısından farkyoktur (100 hasta-yıl başına olay oranı; 11.63'e karşı 11.41, HR: 0,98; %95 GA: 0,87 ila1,10; p=0,743). MI için 100 hasta-yıl başına olay oranları (plaseboya karşı rivaroksaban)2.52'ye karşı 2.08 (HR 0,83; %95 GA: 0,63 ila 1.08; p = 0,165) ve inme için 100 hasta-yılbaşına olay oranları 1.62'ye karşı 1.08'dir (HR: 0,66; %95 GA: 0,47 ila 0,95; p=0,023).Temel güvenlilik sonuçları (yani ölümcül kanama veya kalıcı sakatlanma potansiyeli olankritik bir alana kanamanın bileşimi), sırasıyla, 2,5 mg rivaroksaban günde iki kez tedavigrubunda 18 (%0,7) hastada ve plasebo grubunda 23 (%0,9) hastada meydana gelmiştir(HR=0,80; %95 GA 0,43-1,49; p=0.484). Rivaroksaban grubunda ISTH majör kanamasında plaseboya kıyasla istatistiksel olarak anlamlı bir artış olmuştur (100 hasta-yıl başına olay oranı: 1,21'e karşı 2,04, HR 1,68; %95GA: 1,18 ila 2,39; p=0,003). Hafif ve orta derecede kalp yetmezliği olan hastalarda, COMPASS çalışma alt grubunun tedavi etkileri tüm çalışma popülasyonunun tedavi etkilerine benzer olmuştur (bkz. KAHveya PAHbölümü).22 / 27Yüksek risk taşıyan üçlü pozitif antifosfolipid sendromlu hastalarAraştırmacı tarafından desteklenen randomize, açık etiketli, çok merkezli bir çalışmada körlenmiş sonlanım noktası karar verme yaklaşımı kullanılarak tromboz öyküsü ileantifosfolipid sendromu tanısı olan ve tromboembolik olaylar bakımından yüksek risktaşıyan (lupus antikoagülan, antikardiyolipin antikorları ve anti-beta 2-glikoprotein Iantikorları şeklindeki 3 fosfolipid testi pozitif olan) hastalarda rivaroksaban ve varfarinkarşılaştırılmıştır. Bu çalışma, rivaroksaban kolundaki hastalarda fazla sayıda olay görülmesisebebiyle, çalışmaya 120 hasta alındıktan sonra erken dönemde sonlandırılmıştır. Ortalama takip süresi 569 gündür. 59 hasta 20 mg rivaroksaban (kreatinin klerensi < 50 ml/dak olan hastalar için 15 mg), 61 hasta ise varfarin (INR 2.0-3.0) almak üzere randomizeedilmiştir. Rivaroksaban tedavisine randomize edilen hastaların %12'sinde tromboembolikolaylar görülmüştür (4 iskemik inme ve 3 miyokard enfraktüsü). Varfarin tedavisinerandomize edilen hastalarda olay bildirilmemiştir. Rivaroksaban grubundaki 4 hastada (%7)ve varfarin grubundaki 2 hastada (%3) majör kanama gelişmiştir. 5.2 Farmakokinetik özelliklerGenel özelliklerEmilim:Tablet alımından sonra rivaroksaban hızla emilir ve Cmaks 2-4 saatte görülür. Rivaroksabanın oral emilimi neredeyse tamdır ve oral biyoyararlanımı açlık/tokluk durumundan bağımsız olarak 2,5 mg ve 10 mg tablet dozu için yüksektir (%80-100).Yiyeceklerle birlikte alınması, 10 mg dozda rivaroksabanın EAA ya da Cmaks değerlerinietkilemez. ONAXAN® 2,5 mg ve 10 mg tabletler yemeklerle birlikte ya da ayrı alınabilir(bkz. bölüm 4.2 Pozoloji ve uygulama şekli). Rivaroksaban farmakokinetiğindeki değişkenlik, bireyler arası %30-40 değişkenlik (%CV) ile orta derecededir. Rivaroksabanın emilimi ilaç serbest bırakılmasının gastrointestinal kanalın hangi bölgesinde gerçekleştiğine bağlıdır. Rivaroksaban granülü ince bağırsak proksimalindesalındığı zaman EAA ve Cmaks bakımından tablete kıyasla %29 ve %56 azalma olduğubildirilmiştir. İlaç salınımı ince bağırsak distalinde veya çıkan kolonda gerçekleştiğindemaruziyet daha da azalmaktadır. Bundan dolayı, rivaroksabanın midenin distalineuygulanmasından kaçınılmalıdır, bu durum emilimin ve ilgili rivaroksaban maruziyetininazalmasına neden olabilir. Biyoyararlanım (EAA ve Cmaks), tam bir tablete kıyasla, elma püresine karıştırılmış veya suda süspansiyon haline getirilmiş ezilmiş bir tablet halinde oral yoldan uygulanan ve birsulu yemeğin ardından gastrik sonda ile uygulanan 20 mg rivaroksaban için benzerdir.Rivaroksabanın öngörülebilen, dozla orantılı farmakokinetik profili dikkate alındığında, buçalışmada elde edilen biyoyararlanım bulgularının daha düşük rivaroksaban dozları için degeçerli olma ihtimali bulunmaktadır. Dağılım:İnsanlarda plazma proteinlerine bağlanma yaklaşık % 92-% 95 ile yüksek orandadır ve serum albumini temel bağlayıcı bileşendir. Dağılım hacmi Vss yaklaşık 50 L ile olmaküzere, orta düzeydedir. 23 / 27Biyotransformasyon:Uygulanan rivaroksaban dozunun yaklaşık 2/3'ü metabolik degradasyona uğrar. Eliminasyon:Degradasyondan uğrayan kısmın yarısı renal, diğer yarısı da fekal yoldan elimine edilir. Uygulanan dozun kalan 1/3'ü, başlıca aktif renal sekresyon yoluyla olmak üzere,direkt renal ekskresyona uğrar ve idrarda değişmemiş etkin madde şeklinde bulunur. Rivaroksaban, CYP 3A4, CYP 2J2 ve CYP'den bağımsız mekanizmalarla metabolize edilir. Morfolinonun oksidatif degradasyonu ve amid bağlarının hidrolizi majörbiyotransformasyon alanlarıdır. In vitroaraştırmalara dayanarak, rivaroksaban P-gp (P-glikoprotein) ve Bcrp (meme kanseri direnç proteini) taşıyıcı proteinlerinin bir substratıdır.Değişmemiş rivaroksaban insan plazmasındaki en önemli bileşiktir, dolaşımda majör ya da aktif metaboliti bulunmaz. Yaklaşık 10 l/saat'lik sistemik klerensi ile rivaroksabandüşük klerensli bir ilaç olarak sınıflandırılabilir. 1 mg dozun intravenöz uygulanmasındansonra, eliminasyon yarılanma ömrü yaklaşık 4,5 saattir. Oral uygulamadan sonraeliminasyon sınırlı emilim hızı haline gelir. Rivaroksabanın plazmadan eliminasyonu gençbireylerde 5-9 saatlik, yaşlılarda 11-13 saatlik terminal yarılanma ömrü ile gerçekleşir. Hastalardaki farmakokinetik veriler Akut koroner sendromlu hastalarda aterotrombotik olayların önlenmesi için günde iki kez 2,5 mg rivaroksaban alan hastalarda, doz sonrası 2. - 4. saatte ve yaklaşık 12. saatte (yaklaşıkolarak doz aralığı sırasındaki maksimum ve minimum konsantrasyonları temsil eder)geometrik ortalama konsantrasyon (%90 tahmin aralığı) (kabaca doz aralığı sırasındamaksimum ve minimum konsantrasyonları temsil eder), sırasıyla 47 (13-123) ve 9,2 (4,418) mcg/l olarak saptanmıştır. Farmakokinetik/farmakodinamik ilişki Rivaroksaban plazma konsantrasyonu ile birkaç FD sonlanım noktası (faktör-Xa inhibisyonu, PTZ, aPTT, Heptest) arasındaki farmakokinetik/farmakodinamik (FK/FD)ilişki, geniş bir doz aralığının (günde iki kez 5-30 mg) uygulanmasından sonradeğerlendirilmiştir. Rivaroksaban konsantrasyonu ve Faktör Xa aktivitesi arasındaki ilişkiyien iyi açıklayan yöntem Emaks modeli olmuştur. PTZ için genellikle doğrusal kesiştirmemodeli verileri daha iyi açıklamaktadır. Kullanılan farklı PTZ reaktiflerine bağlı olarak, eğimönemli oranda değişiklik sergilemiştir. Neoplastin PTZ kullanıldığında başlangıç PTZ değeriyaklaşık 13 saniye, eğim ise 3 ila 4 s/(100 mcg/l) olmuştur. Faz II ve III çalışmalarındakiFK/FD analizlerinin sonuçları sağlıklı kişilerde elde edilen verilerle tutarlılık sergilemiştir. Doğrusallık/ Doğrusal olmayan durum:Rivaroksabanın farmakokinetiği günde bir kez uygulanan 15 mg'lık dozuna kadar doğrusaldır. Daha yüksek dozlarda rivaroksaban, artan dozla azalan emilim oranı veazalmış biyoyararlanımın görüldüğü çözünmeyle sınırlı emilim gösterir. Bu durum toklukdurumuna kıyasla aç karnına daha belirgindir. Hastalardaki karakteristik özelliklerYaşlı hastalar:Yaşlı hastalar, temel olarak düşük total ve renal klerense (belirgin) bağlı olarak yaklaşık 1,5 kat yüksek ortalama EAA değerleri ile genç hastalardan daha yüksek plazmakonsantrasyonları sergilemiştir. Doz ayarlamasına gerek yoktur. 24 / 27CinsiyetErkek ve kadın hastalar arasında farmakokinetik özelliklerde klinik olarak belirgin farklılıklar yoktur. Vücut ağırlığı:Vücut ağırlığındaki uç noktaların (<50 kg vs >120 kg) rivaroksaban plazma konsantrasyonları üzerinde sadece küçük bir etkisi vardır (%25'ten daha az). Dozayarlamasına gerek yoktur. Çocuklar ve ergenler:Güvenlilik ve etkililik, çocuklarda ve 18 yaşına kadar adolesanlarda belirlenmemiştir. Etnik farklılıklar:Beyaz, Afrikalı-Amerikalı, Latin kökenli, Japon ya da Çinli hastalar arasında rivaroksabanın farmakokinetik ve farmakodinamik özellikleri ile ilgili klinik olarak belirginetnik farklılıklar gözlenmemiştir. Karaciğer yetmezliği:Hafif karaciğer yetmezliği izlenen sirozlu hastalar (Child Pugh A olarak sınıflandırılanlar), rivaroksabanın farmakokinetik özelliklerinde neredeyse uygun sağlıklıkontrol gruplarına benzer düzeyde, yalnızca minör değişiklikler sergilemiştir (ortalamaolarak rivaroksaban EAA değerinde 1,2 kat artış). Orta derecede karaciğer yetmezliğiizlenen sirozlu hastalarda (Child Pugh B olarak sınıflandırılanlar), rivaroksabanınortalama EAA değeri sağlıklı gönüllülere kıyasla 2,3 kat olmak üzere anlamlı derecedeartmıştır. Serbest ilaç EAA değeri 2,6 kat artmıştır. Bu hastalarda, orta derecedeböbrek yetmezliği hastalarına benzer şekilde, renal eliminasyon da azalmıştır. Şiddetli karaciğer yetmezliği izlenen hastalarla ilgili veri bulunmamaktadır. Faktör XA inhibisyonu, orta derecede karaciğer yetmezliği izlenen hastalarda sağlıklı gönüllülere kıyasla 2,6 kat artmıştır; PT uzamasında da benzer şekilde 2,1 kat artışgözlenmiştir. Orta derecede karaciğer yetmezliği izlenen hastalar rivaroksabana karşıdaha duyarlı olduğundan konsantrasyon ve PT arasında daha dik bir FK/FD ilişkisi sözkonusu olmuştur. ONAXAN®, Child Pugh B ve C derecesinde sirozlu olan hastalar dahil olmak üzere koagülopati ve klinik açıdan anlamlı kanama riski ile ilişkili karaciğer hastalığı izlenenhastalarda kontrendikedir (bkz. Bölüm 4.3 Kontrendikasyonlar). Böbrek yetmezliği:Kreatinin klerensi ölçümü yoluyla değerlendirilen renal fonksiyonda azalmayla ilişkili olarak, rivaroksaban maruziyetinde artış vardır. Hafif (kreatinin klerensi: 50-80 mL/dk), orta (kreatinin klerensi 30-49 mL/dk) ya da ciddi (kreatinin klerensi 15-29 mL/dk) renal yetmezliği olan bireylerde, rivaroksaban plazmakonsantrasyonları (EAA) sırasıyla 1.4, 1.5 ve 1.6 kat artmıştır. Kreatinin klerensi <15 ml/dak olan hastalar için veri bulunmamaktadır. Plazma proteinlerine bağlanma oranının yüksek olması nedeniyle, rivaroksabanın diyaliz ile uzaklaştırılması beklenmemektedir. 25 / 27Kreatinin klerensi <15 ml/dak olan hastalarda kullanımı önerilmemektedir. ONAXAN ®, kreatinin klerensi 15-29 ml/dak olan hastalarda dikkatle kullanılmalıdır (bkz. Bölüm 4.4Özel kullanım uyarıları ve önlemleri). Hafif, orta ya da ciddi böbrek yetmezliği olan bireylerde, FXa aktivitesinin genel inhibisyonu sağlıklı gönüllülerle karşılaştırıldığında sırasıyla 1.5, 1.9 ve 2.0 kat artmıştır;PTZ uzaması benzer şekilde sırasıyla 1.3, 2.2 ve 2.4 kat artmıştır. 5.3 Klinik öncesi güvenlilik verileriRivaroksabanın konvansiyonel güvenlilik farmakolojisi, tekli ve tekrarlı doz toksisitesi, fototoksisite, genotoksisite, karsinojenisite ve üreme toksisitesi çalışmaları iledeğerlendirilen klinik dışı güvenlilik verisi insanlar için özel bir tehlike ortayaçıkarmamaktadır. Tekrarlanan doz toksisitesi çalışmalarında gözlemlenen etkiler, esasen rivaroksabanın aşırı farmakodinamik aktivitesinden kaynaklanmıştır. Sıçanlarda, artan IgG ve IgA plazmaseviyeleri, klinik olarak önemli maruziyet seviyelerinde görülmüştür. Sıçanlarda, erkek veya dişi fertilitesi üzerinde herhangi bir etki görülmemiştir. Hayvan çalışmaları, rivaroksabanın farmakolojik etki mekanizmasıyla ilgili üreme toksisitesi (örn.hemorajik komplikasyonlar) göstermiştir. Klinik olarak anlamlı plazmakonsantrasyonlarında, embriyo-fetal toksisite (implantasyon sonrası kayıp,gerilemiş/ilerlemiş osifikasyon, karaciğerde çoklu açık renkli noktalar) ve yaygın şekilbozuklukları insidansının artmasının yanı sıra plasentada değişiklikler gözlemlenmiştir.Sıçanlarla prenatal ve postnatal çalışmalarda, dişiler için toksik olan dozlarda yavrucanlılığında azalma gözlemlenmiştir. 6. FARMASÖTİK ÖZELLİKLER6.1 Yardımcı maddelerin listesiLaktoz monohidrat (inek sütü kaynaklı) Kroskarmelloz sodyum Hiproksipropil metil selülozSodyum lauril sülfatMikrokristalin selülozMagnezyum stearat Film kaplama içeriği; Hipromelloz Titanyum dioksitPolietilen GlikolSarı demir oksit 6.2 GeçimsizliklerBilinen herhangi bir geçimsizliği bulunmamaktadır. 6.3 Raf ömrü24 ay 26 / 276.4 Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriği56 tabletlik kutularda PVC / PVDC Şeffaf- Alüminyum blisterler şeklindedir. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürün ya da atık materyaller, Tıbbi Atıkların Kontrolü ve Ambalaj ve Ambalaj Atıklarının Kontrolü yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİAli Raif İlaç Sanayi Yeşilce Mah. Doğa Sok. No:4 Kağıthane/İstanbul 8. RUHSAT NUMARASI2021/313 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 17.09.2021 Ruhsat yenileme tarihi: 10. KUB'UN YENİLENME TARİHİ27 / 27 |
İlaç BilgileriOnaxan® 2,5 Mg Film Kaplı TabletEtken Maddesi: Rivaroksaban Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.