Vidaza 100 Mg Sc Enjeksiyonluk Süspansiyon İçin Toz İçeren Flakon Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADIVIDAZA® 100 mg SC enjeksiyonluk süspansiyon için toz içeren flakon Steril Sitotoksik 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir flakon 100 mg azasitidin içerir.Hazırlama sonrası elde edilen süspansiyon her mL'de 25 mg azasitidin içerir. Yardımcı maddeler:Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk süspansiyon için toz. Beyaz liyofilize toz. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarVIDAZA hematopoietik kök hücre transplantasyonuna uygun olmayan yetişkin hastalarda: Uluslararası Prognostik Skorlama Sistemi'ne (IPSS) göre intermediate 2 ve yüksek riskmiyelodisplastik sendrom (MDS) Miyeloproliferatif bozukluk olmaksızın kemik iliği blast ı %10-29 arasında olan kronikmiyelomonositer lösemi (KMML) Dünya Sağlık Örgütü (WHO) sınıflandırmasına göre %20-30 blast ve çoklu serilidisplazisi olan akut miyeloid lösemi (AML) Dünya Sağlık Örgütü (WHO) sınıflandırmasına göre % 30'dan fazla kemik iliği blastıolan 65 yaş ve üstü akut miyeloid lösemi (AML) tedavisinde endikedir. 4.2. Pozoloji ve uygulama şekliVIDAZA tedavisi kemoterapötik ajanlar konusunda tecrübeli bir hekim tarafından başlanmalı ve izlenmelidir. Hastalara, tedavi öncesinde bulantı ve kusmaya karşı anti-emetikpremedikasyonu uygulanmalıdır. Pozoloji/uygulama sıklığı ve süresi:İlk tedavi siklusunda tavsiye edilen başlangıç dozu tedavi öncesi hematoloji laboratuvar değerlerinden bağımsız olarak, tüm hastalar için vücut yüzey alanına göre 75 mg/m1 dozundaolmalı, subkutan olarak 7 gün boyunca yapılan enjeksiyonları takiben 21 günlük bir araverilmelidir (28 gün süren tedavi siklusu). Hastaların en az 6 siklus tedavi alması önerilir. Hasta tedaviden fayda gördüğü sürece ya da hastalıkta ilerleme görülünceye kadar tedavi devam ettirilmelidir. Hastalar hematolojik yanıt/toksisite ve renal toksisite açısından izlenmelidir (bakınız Bölüm 4.4); bir sonraki siklusa başlarken erteleme ya da aşağıda belirtildiği şekilde doz azaltımıgerekebilir. 2 Laboratuvar Testleri: Tedaviye başlamadan ve her tedavi siklusu öncesinde; karaciğer fonksiyon testleri, serum kreatinin ve serum bikarbonat seviyeleri ölçülmelidir. Tedaviye başlamadan ve en az hertedavi siklusundan önce, cevap ve toksisiteyi izlemek gerekli olduğu için tam kan sayımlar ıyapılmalıdır. Hematolojik Toksisite Nedeniyle Doz Ayarlaması:Hematolojik toksisiteye bağlı olarak doz ayarlamasında; Hematolojik toksisite, trombosit sayısı < 50,0 x 109/L ve/veya mutlak nötrofil sayısı (MNS) <1 x 109/L ise, siklus içerisindeulaşılan en düşük değer (en düşük) olarak tanımlanır. İyileşme ise hematolojik toksisite gözlenen hücre serilerinde başlangıç değerleri ile en düşük değer arasındaki mutlak farkın en az yarısı kadar bir artışın olma hali olarak tanımlanır.(iyileşme > En düşük sayım + (0,5 x [|Başlangıç sayım - En düşük sayım|]). Tedavi öncesi başlangıca göre kan sayımı değerleri düşmemiş hastalarda (örneğin beyaz kan hücresi - BKH - > 3,0 x 10 9/L ve MNS > 1,5 x 10 9/L ve trombosit > 75,0 x 10 9/L)VIDAZAtedavisine bağlı olarak hematolojik toksisite ortaya çıkarsa, bir sonraki tedavi siklusutrombosit sayısı ve MNS değerleri düzelene kadar ertelenmelidir. 14 Gün içerisinde değerlerdeiyileşme sağlanırsa herhangi bir doz değişikliğine gerek yoktur. Ancak 14 gün içerisindeiyileşme sağlanamazsa bu durumda aşağıdaki tabloya göre doz azaltılması yapılmalıdır. Dozayarlamalarını takiben siklus 28 güne döndürülmelidir.
*İyileşme = Sayım > En düşük sayım + (0,5 x [Başlangıç sayım - En düşük sayım]) Tedavi öncesi başlangıca göre kan sayımı değerleri düşmüş hastalarda (örneğin beyaz kan hücresi - BKH - < 3,0 x 10 9/Lveya MNS < 1,5 x 10 9/L veya trombosit < 75,0 x 10 9/L)VIDAZAtedavisini takiben BKH ya da MNS ya da trombosit sayısında, uygulama öncesine göre < %50bir azalma ya da %50'den fazla olmasına rağmen herhangi bir hücre seri farklılaşmasındaiyileşme görülmesi durumunda doz ayarlamasına ya da tedavinin ertelenmesine gerek yoktur.Eğer BKH ya da MNS ya da trombosit sayısındaki azalma uygulama öncesine göre %50'den fazla ise ve herhangi bir hücre seri farklılaşmasında iyileşme görülmemesi durumundaVIDAZA tedavisinin bir sonraki siklusu, trombosit sayısı ve MNS düzelene kadarertelenmelidir. 14 gün içerisinde iyileşme sağlanırsa herhangi bir doz ayarlamasına gerekyoktur. Ancak 14 gün süresinde bir düzelme gözlenmemesi durumunda kemik iliği hücreselyapısı değerlendirilmelidir. Eğer kemik iliği hücre düzeyi > %50 ise doz değişikliğine gerekyoktur. Kemik iliği hücre düzeyi <%50 ise tedavi ertelenmeli ve doz aşağıdaki tabloya göreazaltılmalıdır: 1
*İyileşme = Sayım > en düşük sayım + (0,5 x [Başlangıç sayım - en düşük sayım]) Doz ayarlamalarını takiben siklus 28 güne döndürülmelidir. Uygulama şekli:Sulandırılmış VIDAZA; üst kol bölgesi, uyluk ya da karına subkutan olarak enjekte edilmelidir. Enjeksiyon yerleri dönüşümlü olarak değiştirilmelidir. Yeni enjeksiyonlarbir öncekinden en az 2,5 cm uzağa yapılmalı ve kesinlikle hassasiyet, çürük, kızarıklıkya da sertleşme olan bölgelere uygulanmamalıdır. Sulandırıldıktan sonra süspansiyonfiltre edilmemelidir. VIDAZA için sulandırma ve uygulama prosedürü için detaylıtalimatlar bölüm 6.6'da verilmiştir. Özel popülasyonlara ilişkin ek bilgiler:Böbrek Yetmezliği:Azasitidin, başlangıç doz ayarlaması olmaksızın böbrek yetmezliği olan hastalara uygulanabilir (bakınız Bölüm 5.2). Eğer serum bikarbonat düzeyinde nedeni açıklanamayanbir şekilde 20 mmol/l'nin altında azalma ortaya çıkarsa, bir sonraki siklusta doz %50azaltılmalıdır. Eğer serum kreatinin ya da kan üre azot (BUN) değerleri açıklanamayan birşekilde başlangıç değerlerinin > 2 kat üzerine ve normal değerin en üst sınırı (ULN)'naçıkarsa, değerler normale ya da başlangıç düzeylerine dönene kadar bir sonraki siklusertelenmeli ve takip eden tedavi siklusunda doz % 50 azalt ılmalıdır (bakınız Bölüm 4.4). Karaciğer Yetmezliği:Karaciğer yetmezliği olan hastalarda yapılmış çalışma bulunmamaktadır (bakınız Bölüm 4.4). Ciddi karaciğer yetmezliği bulunan hastalar advers olaylar için dikkatlice izlenmelidir.Tedaviye başlamadan önce karaciğer yetmezliği olan hastalarda başlangıç dozu için spesifikbir doz değişikliği önerilmemektedir; takip eden doz değişiklikleri hematolojik laboratuvardeğerleri üzerinden yapılmalıdır. İleri evre malign hepatik tümörü olan hastalarda VIDAZAkontrendikedir (bakınız Bölüm 4.3 ve 4.4). Pediyatrik popülasyon:Yeterli güvenlilik ve etkililik verisi bulunmadığından 18 yaş altındaki çocuklar ve adölesanlarda VIDAZA kullanımı önerilmemektedir. Halihazırda mevcut veriler bölüm 4.8,5.1 ve 5.2'de açıklanmaktadır, ancak pozoloji konusunda herhangi bir tavsiyedebulunulmamaktadır. Geriyatrik popülasyon:3Yaşlı hastalar için spesifik bir doz ayarlaması önerilmemektedir. Yaşlılarda böbrek fonksiyonları zaten azaldığı için böbrek fonksiyonlarının izlenmesi yararlı olabilir. 4.3. Kontrendikasyonlar- Azasitidine veya bölüm 6.1 'de listelenen herhangi bir bileşenine aşırı duyarlığı olan hastalarda, - İlerlemiş malign karaciğer tümörü olan hastalarda (bkz. Bölüm 4.4), - Laktasyonda (bkz. Bölüm 4.6). 4.4. Özel kullanım uyarıları ve önlemleriHematolojik toksisiteAzasitidin ile tedavi esnasında, özellikle ilk 2 siklus sırasında (bkz. Bölüm 4.8), anemi, nötropeni ve trombositopeni sık gözlenmektedir. Cevap ve toksisiteyi izlemek gerekli olduğuiçin, en az her tedavi siklusundan önce tam kan sayımları yapılmalıdır. İlk siklus içinönerilen dozun uygulanmasından sonra, en düşük sayımlara ve hematolojik cevaba dayanarak(bkz. Bölüm 4.2), daha sonraki sikluslar için doz azaltılabilir veya uygulama geciktirilebilir. Hastalara derhal febril ataklarını bildirmeleri tavsiye edilmelidir. Ayrıca hastalara ve doktorlara kanama belirtileri ve semptomları için dikkatli olmaları tavsiye edilir. Karaciğer yetmezliğiKaraciğer yetmezliği olan hastalarda herhangi bir çalışma yapılmamıştır. Metastatik hastalığa bağlı olarak büyük tümör yükü olan, özellikle albumin alt sınır değeri <30 g/L olanhastalarda, azasitidin tedavisi sırasında ilerleyen karaciğer koması ve ölüm seyrek olarakrapor edilmiştir. Azasitidin, ilerlemiş malign karaciğer tümörleri olan hastalardakontrendikedir (bkz. Bölüm 4.3). Böbrek yetmezliğiKemoterapötik ajanlarla birlikte i.v. azasitidin ile tedavi edilen hastalarda serum kreatinin düzeyi artışı, böbrek yetmezliği ve ölümle sonuçlanan böbrek fonksiyon bozukluklarıbildirilmiştir. Ek olarak, alkali idrar ve hipokalemi (serum potasyumu < 3mmol/L) ilebirlikte serum bikarbonatlarının <20 mmol/L'ye düşmesi olarak tanımlanan renal tübülerasidoz, azasitidin ve etoposid ile tedavi edilen 5 kronik miyeloid lösemi (KML) hastasındagelişmiştir. Serum kreatinin veya BUN seviyelerinde açıklanamayan artışlar veya serumbikarbonatta azalmalar (<20 mmol/L) oluşur ise, dozaj azaltılmalı veya uygulamageciktirilmelidir (bkz. Bölüm 4.2). Hastalar, oligüri ve anüri durumunda derhal doktorlarını bilgilendirmeleri konusunda uyarılmalıdırlar. Böbrek fonksiyonu normal olan hastalar ile böbrek yetmezliği olan hastalar arasında advers etkilerin sıklığı açısından klinik bir farklılık olmamasına rağmen, azasitidin ve/veyametabolitleri esas olarak böbrekten atıldığı için böbrek yetmezliği olan hastalar yakındanizlenmelidir (bkz. Bölüm 4.2) Laboratuvar Testleri:Tedaviye başlamadan ve her tedavi siklusundan önce karaciğer fonksiyon testleri, serum kreatinin ve serum bikarbonat düzeyleri belirlenmelidir. Tedaviye başlamadan ve en az her tedavi siklusundan önce, cevap ve toksisiteyi izlemek gerekli olduğu için tam kan sayımları yapılmalıdır (bkz. Bölüm 4.8). 4Kalp ve akciğer hastalığıCiddi konjestif kalp yetmezliği, klinik olarak stabil olmayan kalp hastalığı veya akciğer hastalığı olan hastalar Azasitidin endikasyon çalışmalarına (AZA PH GL 2003 CL 001 ve AZA-AML-001) alınmamıştır ve bu yüzden VIDAZA'nın bu hastalarda güvenliliği ve etkililiğisaptanamamıştır. Bilinen bir kalp veya akciğer hastalığı geçmişi olan hastalarda yapılan birklinik çalışmadan alınan yeni veriler, VIDAZA ile kardiyak olayların insidansında önemlibir artış olduğunu göstermiştir (bkz. Bölüm 4.8). Bu nedenle, bu hasta grubunda VIDAZAkullanırken dikkatli olunması önerilir. VIDAZA ile tedavi öncesinde ve tedavi sırasındakardiyopulmoner değerlendirme yapılması düşünülmelidir. Nekrotizan fasiitVIDAZA ile tedavi edilen hastalarda, ölümcül vakalar da dahil olmak üzere nekrotizan fasiit rapor edilmiştir. Nekrotizan fasiit gelişen hastalarda, VIDAZA tedavisi hemen durdurulmalıve acilen uygun bir tedaviye başlanmalıdır. Tümör lizis sendromu:Tedavi öncesinde yüksek tümör yükü olan hastalar tümör lizis sendromu açısından risk altındadır. Bu hastalar yakın takip edilmeli ve uygun önlemler alınmalıdır. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriİn vitroinvivoetkileşim olasılığının olmadığı düşünülmektedir.Azasitidinin sitokrom P450 enzimleri üzerinde klinik olarak önemli inhibitör veya indükleyici etkisi olası değildir (bkz Bölüm 5.2). Azasitidin ile klinik ilaç etkileşme çalışmaları yapılmamışt ır. Özel popülasyonlara ilişkin ek bilgilerHiçbir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyonHiçbir etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyon Genel tavsiyeGebelik kategorisi: D Çocuk doğurma potansiyeli bulunan kadınlar/ Doğum kontrolü (Kontrasepsiyon)Erkekler ve çocuk doğurma potansiyeli olan kadınlar tedavi sırasında ve tedaviden sonraki 3 aya kadar etkili kontrasepsiyon yöntemi kullanmalıdırlar. Gebelik dönemiAzasitidinin, gebe kadınlarda kullanımına ilişkin yeterli veri yoktur. Fareler üzerinde yapılan çalışmalar üreme toksisitesi olduğunu göstermiştir (bkz. Bölüm 5.3). VIDAZA'nın gebelikve/veya fetus/yeni doğan üzerinde zararlı farmakolojik etkileri bulunmaktadır. Azasitidin,hayvan çalışmalarından elde edilen sonuçlara ve mekanizmasına dayanarak gebeliksırasında, özellikle ilk trimesterde, kesinlikle gerekli olmadıkça kullanılmamalıdır.Tedavinin anne için avantajları fetus için olası risklerine karşı her vaka için tartışılarak kararverilmelidir. 5Laktasyon dönemiAzasitidin / metabolitlerinin anne sütüne geçip geçmediği bilinmemektedir. Emzirilen bebekte ciddi advers reaksiyon potansiyeli nedeniyle, azasitidin tedavisi sırasında emzirmekontrendikedir. Üreme yeteneği /Fertiliteİnsanlarda azasitidinin fertilite üzerindeki etkisine dair herhangi bir veri yoktur. Hayvanlarda azasitidin kullanımının erkek fertilitesi üzerinde advers reaksiyonları görülmüştür (bkz.Bölüm 5.3). Erkeklere tedavi alırken baba olmamaları ve tedavi sırasında ve tedaviden sonraki 3 aya kadar etkili kontrasepsiyon kullanmaları tavsiye edilmelidir. Tedaviye başlamadan önceerkek hastalara spermlerini saklamak üzere danışman aramaları tavsiye edilmelidir. 4.7. Araç ve makine kullanımı üzerindeki etkilerAzasitidinin araç ve makina kullanımına hafif ve orta derecede etkisi vardır. Ayrıca, azasitidin tedavisi sırasında yorgunluk gibi istenmeyen etkilerin oluşabileceği konusundahastalar bilgilendirilmelidir. Bu nedenle, araç ve makine kullanırken dikkatli olunmasıönerilmelidir. 4.8. İstenmeyen etkilerGüvenlilik profili özetiMDS, KMML ve % 20-30 kemik iliği blastlı AML'si olan yetişkinlerde: Hastaların %97'sinde VIDAZA uygulaması ile ilişkili advers reaksiyonlar oluşmuştur. Azasitidin tedavisi ile çok yaygın görülen advers reaksiyonlar trombositopeni, nötropeni ve lökopeniyi (genellikle Derece 3-4) içeren hematolojik reaksiyonlar (% 71,4), bulantı,kusmayı (genellikle Derece1-2) içeren gastrointestinal olaylar (%60,6) veya enjeksiyonbölgesi reaksiyonlarıdır ( %77,1; genellikle Derece 1-2). Azasitidin endikasyon çalışmasında (AZA PH GL 2003 CL 001) en sık görülen ciddi advers reaksiyonlar febril nötropeni (%8,0) ve anemi (%2,3) olup bu çalışmayı destekleyençalışmalarda da (CALGB 9221 ve CALGB 8921) benzer ciddi advers reaksiyonlarraporlanmıştır. Daha az sıklıkta bildirilen diğer ciddi advers reaksiyonlar nötropenik sepsis(% 0 ,8 ) ve bazen ölümcül sonuçları olabilen pnömoni (%2,5) gibi enfeksiyonları,trombositopeni (%3,5), aşırı duyarlılık reaksiyonları (% 0,25) ve kanama olaylarını [örneğinserebral kanama (%0,5), gastrointestinal kanama (%0,8) ve intrakraniyal kanama (%0,5)]içermektedir. % 30'dan fazla kemik iliği blastı olan 65 yaş ve üstü hastalarda: Azasitidin tedavi kolunda AZA-AML-001 çalışması ile belirlenmiş olan çok yaygın ciddi advers reaksiyonlar (> % 10) arasında febril nötropeni (%25), pnömoni (%20,3) ve pireksi(%10,6) bulunmaktadır. Ayrıca daha az sıklıkla raporlanmış olan ciddi advers reaksiyonlararasında sepsis (%5,1), anemi (%4,2), nötropenik sepsis (%3,0), idrar yolu enfeksiyonu (%3,0),trombositopeni (%2,5), nötropeni (%2,1), selülit (%2,1), baş dönmesi (% 2,1) ve dispne (%2,1)bulunmaktadır. Azasitidin tedavisi ile en sık raporlanan advers reaksiyonlar (çalışmadaki hastaların %30'unda görülen), kabızlık (%41,9), mide bulantısı (%39,8) ve ishali de içeren sindirim sistemi olayları(%36,9; genellikle Derece 1-2), pireksiyi de içeren genel bozukluklar ve uygulama bölgesineilişkin durumlar (%37,7; genellikle Derece 1-2) ve febril nötropeni (%32,2) ve nötropeniyi(%30,1; genellikle Derece 3-4) de içeren hematolojik olaylardır 6Aşağıdaki tablo azasitidin tedavisi ile ilişkili olabilecek advers reaksiyonları içermektedir. Sıklıklar, MDS ve AML üzerine yapılmış temel klinik çalışmalara ve pazarlama sonrasıgözlemlere dayanmaktadır. Sıklıklar şu şekilde tanımlanmıştır: çok yaygın (>1/10), yaygın (>1/100- <1/10), yaygın olmayan (>1/1000- <1/100), seyrek (>1/10.000- <1/1000), çok seyrek (<1/10.000),bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubu içindeadvers reaksiyonlar azalan ciddiyet sırasına göre verilmiştir.
8
9
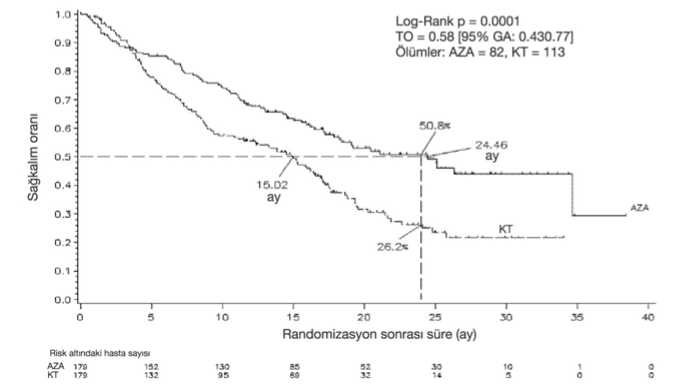
Hematolojik advers reaksiyonlar Azasitidin tedavisi ile ilişkili olarak çok yaygın rapor edilen (>%10) hematolojik advers reaksiyonlar, genellikle 3. veya 4. dereceden anemi, trombositopeni, nötropeni, febrilnötropeni ve lökopenidir. Bu olayların olma riski daha çok ilk 2 siklus sırasındadır, daha sonra hematolojik fonksiyonun normale döndüğü hastalarda daha az sıklıkta oluşur. Çoğu hematolojik adversreaksiyonlar, tam kan sayımlarının rutin olarak izlenmesi ve bir sonraki siklusta azasitidinuygulamasının geciktirilmesi, nötropeni için profilaktik antibiyotikler ve/veya büyümefaktörü desteği (örneğin G-CSF) ve anemi veya trombositopeni için transfüzyonlar ilegerektiği gibi tedavi edilmektedir. Enfeksiyonlar Miyelosupresyon nötropeniye ve enfeksiyon riskinin artmasına neden olabilir. Azasitidin alan hastalarda nötropenik sepsisi de içeren sepsis ve pnömoni gibi ve bazıları ölümcülsonuçlara neden olan ciddi advers reaksiyonlar rapor edilmiştir. Enfeksiyonlar, nötropeni içinanti-enfektif ajanlar ve büyüme faktör desteği (örneğin G-CSF) kullanımı ile kontrol altınaalınabilir. Kanama Azasitidin alan hastalarda kanama görülebilir. Gastrointestinal kanama ve intrakraniyal kanama gibi ciddi advers reaksiyonlar rapor edilmiştir. Özellikle daha öncedentrombositopenisi olan veya tedaviye bağlı trombositopenisi gelişen hastalar, kanama 10belirtileri ve semptomları için izlenmelidir. Aşırı duyarlılık Azasitidin alan hastalarda ciddi aşırı duyarlılık reaksiyonları rapor edilmiştir. Anafilaktik benzeri reaksiyon durumunda azasitidin tedavisi derhal kesilmelidir ve uygun semptomatiktedavi başlatılmalıdır. Deri ve deri altı doku hastalıkları Deri ve deri altı advers reaksiyonlarının çoğunluğu enjeksiyon bölgesi ile ilgilidir. Bu advers reaksiyonların hiçbiri azasitidinin kesilmesine veya ana çalışmalarda azasitidin dozununazaltılmasına neden olmamıştır. Advers reaksiyonlarının çoğunluğu tedavinin ilk 2 siklususırasında olmuştur ve sonraki sikluslar ile azalmaya yönelmiştir. Enjeksiyon bölgesindedöküntü/enflamasyon/pruritus, döküntü, eritem ve deri lezyonu gibi subkutan adversreaksiyonlar, antihistaminikler, kortikosteroidler ve non-steroidal anti-enflamatuarlar(NSAIIler) gibi ilaçların birlikte kullanımını gerektirebilir. Bu kutanöz reaksiyonlar, bazenenjeksiyon bölgesinde oluşan yumuşak doku enfeksiyonlarından ayırt edilmelidirler.Pazarlama sonrasındaki gözlemlerde; azasitidin ile birlikte nadir vakalarda, ölüme yol açanselülit ve nekrotizan fasiit gibi yumuşak doku enfeksiyonları rapor edilmiştir. Enfeksiyözadvers reaksiyonların klinik yönetimi için 4.8 Enfeksiyonlar bölümüne bakınız. Gastrointestinal advers reaksiyonlar Azasitidin tedavisi ile çok yaygın rapor edilen advers reaksiyonlar kabızlık, ishal, bulantı ve kusmadır. Bu advers reaksiyonlar, bulantı ve kusma için anti-emetikler, ishal için anti-diyaretikler ve kabızlık için laksatif ve/veya feçes yumuşatıcıları ile semptomatik olaraktedavi edilmelidirler. Renal advers reaksiyonlar Azasitidin ile tedavi edilen hastalarda, serum kreatinin değerlerinde artış ve hematüriden renal tübüler asidoz, renal yetmezlik ve ölüme kadar giden derecelerde böbrek bozukluklarırapor edilmiştir (bkz. Bölüm 4.4). Hepatik advers reaksiyonlar Azasitidin tedavisi sırasında, metastatik hastalığa bağlı olarak tümör yükü çok olan hastalarda hepatik yetmezlik, ilerleyen hepatik koma ve ölüm gözlenmiştir (bkz. Bölüm 4.4). Kardiyak olaylar Kardiyovasküler veya pulmoner hastalık geçmişi olduğu bilinen hastaların dahil edildiği bir klinik çalışmadan alınan veriler, VIDAZA ile tedavi edilen yeni AML teşhisi konmuşhastalarda kardiyak olaylarda bir artış olduğunu göstermiştir (bkz. Bölüm 4.4). Yaşlı hastalar 85 yaş ve üstü hastalarda azasitidinin güvenliliği ile ilgili sınırlı bilgi bulunmaktadır (AZA-AML-001 çalışmasında tedavi edilen 85 yaş ve üstü hastalarda 14 hasta [% 5,9] bulunmaktadır.) Pedivatrik _ popülasvonAZA-JMML-001 çalışmasında, 28 pediyatrik hasta (1 aydan 18 yaşına kadar) MDS (n = 10) veya juvenil miyelomonositik lösemi (JMML) (n = 18) için VIDAZA ile tedavi edilmiştir(bkz.Bölüm 5.1). 28 hastanın tümü en az 1 advers olay yaşadı ve 17'si (%60,7) en az 1 tedaviyle ilişkili olay 11yaşadı. Genel pediyatrik popülasyonda en sık bildirilen advers olaylar ateş, anemi, trombositopeni ve febril nötropeni dahil hematolojik olaylar ile kabızlık ve kusma dahilgastrointestinal olaylardır. Klinik çalışmadaki üç (3) hasta, ilacın kesilmesine neden olan tedavi ilişkili olay yaşadı (ateş, hastalığın ilerlemesi ve karın ağrısı). AZA-AML-004 çalışmasında, moleküler relapslı 7 pediyatrik hasta (2-12 yaş arası), ilk tam remisyondan [CR1] VIDAZA ile tedavi edilmiştir (bkz. Bölüm 5.1). 7 hastanın tümü, tedaviye bağlı en az 1 advers olay yaşamıştır. En sık bildirilen yan etkiler nötropeni, bulantı, lökopeni, trombositopeni, diyare ve alanin aminotransferaz (ALT) artışıdır.İki hasta, dozun kesilmesine yol açan (ateşli nötropeni, nötropeni) tedaviyle ilişkili bir olayyaşamıştır. Klinik çalışma sırasında VIDAZA ile tedavi edilen sınırlı sayıda pediyatrik hastada yeni güvenlilik sinyali tespit edilmemiştir. Genel güvenlik profili, yetişkin popülasyonun güvenlilikprofiliyle tutarlıdır. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9. Doz aşımı ve tedavisiKlinik çalışmalar sırasında azasitidin ile doz aşımı bir vakada rapor edilmiştir. Hasta, önerilen başlangıç dozunun neredeyse 4 katı olan, yaklaşık 290 mg/m2 tek bir i.v. dozualdıktan sonra, hastada ishal, bulantı ve kusma görülmüştür. Doz aşımı durumunda, hasta uygun kan sayımları yapılarak izlenmeli ve gerekli olduğu şekilde destekleyici tedavi almalıdır. Azasitidinin doz aşımı için bilinen spesifik bir antidotyoktur. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antineoplastik maddeler. Pirimidin analogları. Azasitidinin antineoplastik etkilerini, kemik iliğindeki anormal hematopoietik hücreler üzerinde sitotoksisite ve DNA'nın hipometilasyonu da dahil olmak üzere çoklu mekanizmalarile gösterdiğine inanılmaktadır. Azasitidinin sitotoksik etkileri şu mekanizmalardankaynaklanıyor olabilir: DNA, RNA ve protein sentezinin inhibisyonu, RNA ve DNA'ylabirleşme ve DNA yıkım yolaklarının aktivasyonu. Non-proliferatif hücreler azasitidinegöreceli olarak dirençlidir. Azasitidinin DNA'ya katılımı DNA metiltransferazlarınıninaktivasyonu ve DNA'nın hipometilasyonu ile sonuçlanır. Normal hücre siklusu kontrolü,diferansiyasyonu ve ölüm yolaklarında görev alan anormal derecede metillenmiş genlerin 12DNA hipometilasyonu, genlerinyenidenekspresyonu ve kanser- baskılayıcı fonksiyonların tamiri ile sonuçlanabilir. DNA hipometilasyonu ile azasitidinin sitotoksik veya diğer aktivitelerinin klinik sonuçlar üzerindeki göreceli önemleri henüzbilinmemektedir. Klinik etkililik ve güvenlilik:MDS, KMML ve kemik iliğinde % 20-30 blast olan AML tanılı yetişkinlerde VIDAZA'nın etkililiği ve güvenliliği uluslararası, çok merkezli, kontrollü, açık-uçlu, randomize, paralel gruplu, Faz 3 karşılaştırmalı araştırmada (AZA PH GL 2003 CL 001)incelenmiştir. Araştırmaya Uluslararası Prognostik Skorlama Sistemine (UPSS) göreintermediate-2 ile yüksek riskli MDS ve Fransız Amerikan İngiliz (FAB) sınıflandırmasistemine göre ise RAEB, RAEB-T (%21-30 blast) ile mKMML olan MDS hastaları dahiledilmiş, sekonder MDS'si olan hastalar araştırmaya dahil edilmemiştir. Azasitidin (n= 179)konvansiyonel tedavi rejimleri (n=179) ile karşılaştırılmıştır. Konvansiyonel tedavi rejimleri,tek başına destek tedavi (n= 105), düşük doz sitarabin ve beraberinde destek tedavi (n= 49)veya standart indüksiyon kemoterapi ile destek tedaviden (n= 25) oluşmuştur. Hastalarrandomizasyondan önce doktorları tarafından 3 konvansiyonel tedavi rejiminden bir tanesineseçilmişlerdir. Hasta VIDAZA grubuna randomize olmamışsa, bu önceden seçilen rejimialmıştır. Hastanın araştırmaya dahil edilmesi için gereken kriterlerden bir tanesi de EasternCooperative Oncology Group (ECOG) performansının 0-2 arasında olmasıdır. SekonderMDS'si olan hastalar araştırmaya dahil edilmemiştir. Araştırmanın primer sonlanım noktasıtoplam sağ kalım süresidir. VIDAZA medyan 9 siklus (1-39 siklus aralığında) ve ortalama 10,2siklus olacak şekilde 7 gün boyunca günlük 75 mg/m2 subkutan dozda uygulanmış ve 21 günara verilmiştir (28 günden oluşan tedavi siklusu). Tedavi Amaçlı Popülasyonda (ITT) yaşortalaması 69'dur (38-88 yaş arası). 358 hasta (179 azasitidin ve 179 konvansiyonel tedavi rejimleri üzerinde yapılan ITT analizinde, VIDAZA ile medyan 24,46 aylık bir sağ kalıma karşı, konvansiyonel tedavi rejimitedavisinde 15,02 aylık sağ kalım olduğu saptanmıştır. Aradaki fark 9,4 aydır. (p<0,0001).Azasitidin kullanan hastalarda iki yıllık sağ kalım oranı %50,8 iken; konvansiyonel tedavirejimi hastalarında %26,2'dir (p< 0,0001). VIDAZA'nın sağkalım faydaları, kontrol kolunda kullanılan konvansiyonel tedavi rejimi seçeneğinden (tek başına en iyi destek tedavi, düşük doz sitarabin ve beraberinde en iyi destek 13tedavi veya standart indüksiyon kemoterapisi ve beraberinde en iyi destek tedavi) bağımsız olarak tutarlıdır. UPSS (Uluslararası Prognostik Skorlama Sistemi) sitogenetik alt grup analiz edildiğinde, tüm gruplarda (iyi, orta, kötü sitogenetikli, monozomi 7 dahil) medyan genel sağ kalım açısındanbenzer sonuçlar. Yaş alt grupları analiz edildiğinde, tüm gruplarda medyan genel sağ kalımdabir artış gözlendi (<65 yaş, >65 yaş ve >75 yaş). ANAHTAR: AZA= azasitidin; KT= konvansiyonel tedavi; GA= güvenlilik aralığı; TO= tehlike oranı VIDAZA grubunda ölüm veya AML'ye dönüşüm için geçen medyan süre 13,0 ay iken; bu süre konvansiyonel rejim tedavisi alan grupta 7,6 aydır. VIDAZA 5.4 aylık avantaj sağlamışolup, p-değeri 0,0025'dir. Ayrıca, VIDAZA tedavisi sitopeni ve semptomlarında azalma ilebirliktelik göstermiştir. Azasitidin grubunda, araştırmanın başında eritrosit transfüzyonunabağımlı olan hastaların %45'i eritrosit transfüzyonundan bağımsız hale gelmiştir, bu orankonvansiyonel tedavi rejimi gruplarında %11,4'dir (p< 0,0001) eritrosit transfüzyonundanbağımsız kalma süreleri ise medyan 13 aydır. Azasitidin grubunda elde edilen toplam yanıt (tam remisyon [TR] + parsiyel remisyon [PR]) %29 iken kombine konvansiyonel tedavi rejimleri grubunda ise %12'dir (p = 0,0001).Bağımsız İnceleme Komitesi'nin AZA PH GL 2003 CL1 çalışmasında elde ettiği genel yanıt(TR + PR), azasitidin grubunda %7 (12/179) olup bu oran kombine konvansiyonel tedavigruplarında %1 (2/179)'dur (p=0,0113). Bağımsız İnceleme Komitesi ve araştırmacıdeğerlendirmeleri yanıtları arasındaki farklar periferik kan sayımlarının iyileştirilmesini ve enaz 56 gün bu iyileştirmenin idamesini gerektiren Uluslararası Çalışma Grubu (IWG)kriterlerinin bir sonucudur. Azasitidin tedavisini takiben TR ve PR elde edilemeyen hastalardada sağ kalımda avantaj gözlenmiştir. Bağımsız İnceleme Komitesinin yaptığıdeğerlendirmeye göre azasitidin alan hastaların %49'unda hematolojik iyileşme (major veyaminör) tespit edilmiş olup bu oran kombine konvansiyonel tedavi rejimleri ile tedavi edilenhastalarda %29'dur (p< 0,0001). Başlangıçta bir veya daha fazla sitogenetik anormalliği olan hastalarda, major sitogenetik yanıt görülen hastaların oranı azasitidin ve kombine konvansiyonel tedavi rejimi gruplarındabirbirine benzerdir. Minör sitogenetik yanıt, kombine konvansiyonel tedavi rejimi grubu ilekarşılaştırıldığında (%10), azasitidin grubunda (%34) istatistiksel olarak anlamlı düzeydedaha yüksektir (p = 0,0015). %30'dan fazla kemik iliği blastı olan 65 yaş ve üstü akut miyeloid lösemi (AML) hastaları AZA-AML-001 klinik araştırmasında yer alan tedavi amaçlı hasta popülasyonuna ait sonuçlar aşağıda sunulmuştur ( Bkz. 4.1- Terapötik Endikasyonlar). VİDAZA'nın etkililik ve güvenliliği hematopoietik kök hücre transplantasyonuna uygun olmayan, Dünya Sağlık Örgütü sınıflandırmasına göre 65 yaş ve üstü yeni teşhiş konmuş veya% 30'dan fazla kemik iliği blastlı ikincil AML'si olan hastalarda uluslararası, çok merkezli,kontrollü, açık-uçlu, paralel grup Faz 3 çalışması yapılmıştır. VİDAZA ile birlikte en iyidestek tedavileri (n = 241) konvansiyonel tedavi rejimleri ile karşılaştırılmıştır.Konvansiyonel tedavi rejimleri, tek başına destek tedavileri (n = 45), düşük doz sitarabin veberaberinde destek tedavileri (n = 158) veya sitarabin ve antrasiklin ile birlikte standart 14yoğunlaştırılmış kemoterapi ile beraber destek tedaviden (n = 44) oluşmaktadır. Randomizasyondan önce konvansiyonel tedavi rejimi alan 3 hastadan 1'i doktorlarıtarafından seçilmişlerdir. Hastalar eğer VİDAZA grubuna randomize edilmediyse öncedenseçilmiş tedavi rejimini almaya devam etmiştir. Çalışmaya alınma kriterleri, hastaların ECOGperformans durumlarının 0 ila 2 arasında olması ve orta dereceli veya düşük riskli sitogenetikanormalliği olmasıydı. Çalışmanın birincil sonlanım noktası genel sağkalım olarakbelirlenmiştir. VIDAZA alanlar için, 21 gün dinlenme periyodunu takiben 7 gün boyunca (28 günlük tedavi siklusu) 75 mg/m2 subkutan medyan 6 siklus (1-28 siklus) olacak şekilde uygulanırken,sadece en iyi destek tedavisi alanlarda medyan 3 siklus (1-20 siklus), düşük doz sitarabinalanlarda medyan 4 siklus (1-25 siklus) ve standart yoğunlaştırılmış kemoterapi alanlardamedyan 2 siklus (1-3 indüksiyon siklusu artı 1 veya 2 konsolidasyon siklusu) olacak şekildeuygulanmıştır. Bireysel başlangıç parametreleri açısından VIDAZA ile konvansiyonel tedavi rejimindeki gruplar karşılaştırılabilirdir. Hastalardaki medyan yaş 75'tir (64 ile 91 yaş aralığı). %%60,7'si tek başına AML, %32,4'ü miylodisplaziye bağlıdeğişiklikler ile AML, %4,1'i terapiye bağlı miyeloid neoplazma ve % 2,9'u tekrar edengenetik anormallikleri ile birlikte AML olarak kategorize edilmiştir.488 hastanın ITT analizinde (241 hasta VIDAZAve 247 hasta konvansiyonel tedavi rejimi ile tedavi edilmiştir.), VIDAZA tedavisi alan hastalar ile konvansiyonel tedavi rejimi alanhastalar da medyan sağkalım oranı sırasıyla 10,4 ay ve 6,5 aydır. Aradaki fark 3,8 aydır(p=0,1009). Tedavi etkisinin risk oranı 0,85'tir (%95 Cl=0,69; 1,03). Bir yıllık sağkalımoranları VIDAZA alan hastalarda % 46,5, konvansiyonel tedavi rejimi alan hastalarda %34,3'tür. Âz a s ıtı din ¦ KTe davs: ;¦2E~mıniMMT>*miMTMi Loa rarA p = 0.0629 ,S«rfinanin*}Loç fank p = 0 1009 0 1009feacitHS»* = 104 0. 12 7. kt
£b TauiMM^ (%) = 4&19 9). kt = 4*118 6)SManaaommamş HR = 0 84 0 69 - 1 021 S»rfiaBdrtın«ş HR = O 85 |95*CI C 69 - 1 03)ra O6-OE 05-o i-0.2 -0 ı -Risk altındaki hasta sayısıRandomizasyon sonrası süre (ay)AzasitidinÖnceden tanımlanmış başlangıçtaki prognostik faktörler için Cox PH modele uyarlanarak 15VIDAZA'nın konvansiyonel tedavi rejimleri karşılaştırması için risk oranı 0,80 (%95 Cl=0,66;0,99; p=0,0355) olarak belirlenmiştir. Buna ek olarak, azasitidin ile önceden seçilmiş konvansiyonel tedavi rejimi alan hastalar karşılaştırıldığında çalışma istatistiksel olarak belirli bir fark göstermemesine rağmen,VIDAZA kullanan hastaların sağkalım oranı konvansiyonel tedavi rejimi seçeneklerindendestek tedavisi ve düşük doz sitarabin artı destek tedavisi alan hastalardan daha uzundur.Yoğun kemoterapi ile destek tedavisi alan hastalar ile karşılaştırıldığında ise sağkalım oranıbenzerlik göstermektedir. VIDAZA'nın lehine toplam sağkalım yararı yönünden, bütün önceden seçilmiş alt gruplarda yaş [-75 yaş altı ve 75 yaş ve üstü], cinsiyet, ırk, ECOG performans durumu [0 veya 1 ve 2],temel sitogenetik risk [orta veya düşük], coğrafik bölge, AML'nin DSÖ sınıflandırması(miyelodisplaziye bağlı değişiklikler ile birlikte AML'yi de içeren), başlangıçtaki lökositsayısı [< 5 x109/L ve >5 x 109/L], başlangıçtaki kemik iliği blastı [% 50 ve daha az ve > %50'den çok], önceki MDS geçmişi] bir eğilim bulunmaktadır. Yalnızca çok küçük bir gruptatoplam sağkalım risk oranı istatiksel anlamlılığa ulaşmıştır. Bu gruplar arasında zayıfsitogenetik riski olan hastalar, miyelodisplaziye bağlı değişiklikler olan AML hastaları, 75yaş altı hastalar, kadın hastalar ve beyaz ırktan hastalar yer almaktadır. Hematolojik ve sitogenetik cevaplar araştırmacılar ve IRC tarafından benzer sonuçlar ile değerlendirilmişlerdir. IRC tarafından tam yanıtların oranı (tam remisyon [CR] ve kan sayımıdüzelmesiz tam remisyon [CRi]) VIDAZA grubu için %27,8, ve birleştirilmiş konvansiyoneltedavi rejimi için % 25,1 olarak belirlenmiştir (p=0,5384). CR ve Cri'ye ulaşan hastalarda,remisyon için medyan süre VIDAZA kullanan hastalarda 10,4 ay (% 95 CI =7,5; 15,2) olup ,konvansiyonel tedavi rejimi alan hastalarda ise 12,3 aydır (% 95 CI =9,0; 17,0). VIDAZA iletedavi edilen ve tam yanıt sağlanamayan hastalarda konvansiyonel tedavi rejimlerine göre sağkalım avantajı gösterilmiştir.VIDAZA tedavisi periferik kan değerlerini iyileştirmiş ve eritrosit ve trombosit transfüzyonu ihtiyacını azaltmıştır. Eğer hasta sırasıyla 56 gün (8 hafta) boyunca veya randomizasyonöncesi bir veya daha fazla eritrosit veya trombosit transfüzyonu almışsa, başlangıçta eritrositveya trombosit transfüzyonuna bağımlı kabul edilmiştir. Eğer hasta sırasıyla tedavi süresiboyunca ve raporlama periyodunda ardışık gelen herhangi 56 gün boyunca eritrosit veyatrombosit transfüzyonu almıyorsa, eritrosit veya trobosit transfüzyonuna bağımlı olmadığıdüşünülmektedir. Başlangıçta eritrosit transfüzyonuna bağımlı o lan VIDAZA gurubundaki hastalardan % 38,5'inin (% 95 Cl=31,1; 46,2) tedavi periyodu süresince eritrosit transfüzyonuna bağımlılığıkalmamıştır. Birleştirilmiş konvasiyonel tedavi rejimi alan hastalarda bu oran % 27,6'dır (%95 Cl=20,9; 35,1). Başlangıçta eritrosit transfüzyonuna bağımlı olan ve tedavi iletransfüzyona bağımsız hale gelen hastalar için , transfüzyona bağımsız hale gelmek için geçenmedyan süre VIDAZA gurubunda 13,9 ay iken konvansiyonel tedavi rejimi alan hastalardaise bu süreye ulaşılamamıştır. Çalışma başlangıcında trombosit transfüzyonuna bağımlı olan VIDAZA gurubundaki hastalardan % 40,6'sının (% 95 Cl=30,9; 50,8) tedavi periyodu süresince trombosittransfüzyonuna bağımlılığı kalmamıştır. Birleştirilmiş konvasiyonel tedavi rejimi alanhastalarda bu oran % 29,3'tür (% 95 Cl=19,7; 40,4). Başlangıçta trombosit transfüzyonuna 16bağımlı olan ve tedavi ile transfüzyona bağımsız hale gelen hastalar için, transfüzyona bağımsız hale gelmek için geçen medyan süre VİDAZA gurubunda 10,8 ay ikenkonvansiyonel tedavi rejimi alan hastalarda ise bu süre 19,2 aydır. Sağlığa Bağlı Yaşam Kalitesi (HRQoL), Avrupa Organizasyonu Kanser Araştırma ve Tedavi Çekirdek Yaşam Kalitesi (EORTC QLQ-C30) anketi kullanılarak belirlenmiştir. HRQoLverileri test çalışmasındaki bütün popülasyonun alt kümesi için analiz edilebilir. Analizdebazı sınırlamalar olmasına rağmen, elde bulunan veriler VIDAZA tedavisi sırasındahastaların yaşam kalitesinde anlamlı bir kayıp yaşamadıklarını göstermektedir. Pediyatrik popülasyonAZA-JMML-001 çalışması, yeni tanı almış ileri MDS veya JMML'li pediyatrik hastalarda HSCT'den önce VIDAZA'nın farmakokinetiğini, farmakodinamiğini, güvenliğini veaktivitesini değerlendirmek için gerçekleştirilmiş; Faz 2, uluslararası, çok merkezli, açıketiketli bir çalışmadır. Klinik çalışmanın birincil amacı VIDAZA'nın 3. siklus, 28. günde yanıtoranı üzerindeki etkisini değerlendirmektir. Hastalar (MDS, n = 10; JMML, n = 18, 3 aylık - 15 yaş arası; %71 erkek), minimum 3 siklus ve maksimum 6 siklus boyunca 28 günlük bir siklusun ilk 7 günü boyunca, günlük 75 mg/m2intravenöz VIDAZA dozu ile tedavi edilmiştir. MDS koluna hasta alımı, 10 MDS hastasından sonra etkililik gözlenmemesi nedeniyle durdurulmuştur: bu 10 hastada doğrulanmış yanıt kaydedilmemiştir. JMML çalışma kolunda, 18 hasta (13 PTPN11, 3 NRAS, 1 KRAS somatik mutasyonu ve 1 nörofibromatozis tip 1 klinik tanılı [NF 1]) kaydedildi. On altı hasta 3 siklus, 5 hasta 6 siklustedaviyi tamamladı. Toplam 11 JMML hastasında, 3. siklusun 28. gününde klinik yanıtalınmıştır. Bu 11 hastanın 9'unda (%50) doğrulanmış bir klinik yanıt gözlenmiştir (3 hastadacCR - doğrulanmış tam yanıt ve 6 hastada cPR - doğrulanmış kısmi yanıt). VIDAZA ile tedaviedilen hasta kohortunda, 7 (%43,8) hastada sürekli trombosit yanıtı (sayımlar > 100 x 109/L)gözlenmiş ve HSCT'de 7 (%43,8) hasta transfüzyona ihtiyaç duymuştur. 18 hastadan 17'siHSCT'ye geçmiştir. Çalışma tasarımı nedeniyle (az hasta sayısı ve karışıklığa neden olan çeşitli faktörler), bu klinik çalışmadan HSCT öncesi VIDAZA'nın JMML hastalarında sağkalımı arttırdığı veyaarttırmadığı sonucu çıkarılamaz. AZA-AML-004 çalışması, ilk tam remisyondan sonraki moleküler relaps gelişen AML tanılı pediyatrik hastalarda ve moleküler relapstaki AML'li çocuklarda ve genç erişkinlerde anti-kanser tedavisine kıyasla VIDAZA'nın güvenliğini, farmakodinamiğini ve etkinliğinideğerlendirmek için bir Faz 2, çok merkezli, açık etiketli bir çalışmadır. VIDAZA, 7 hastada (yaşları 2 ila 12 arasında, ortanca 6, 7 yıl ve %71,4'ü erkek olan), her 28 günlük siklusun, ilk 7 gününde 100 mg/m2 olacak şekilde en fazla 3 siklus kullanılmıştır. 84. günde, yani 3 siklus sonundaki değerlendirmede; 5 hastada minimal rezidüel hastalık (MRD) saptandı; bunların birinde klinik relaps, kalan 4 hastanın üçünde (n=3) molekülerstabilizasyon, birinde de (n = 1) moleküler ilerleme tespit edildi. Azasitidin ile tedavi edilen7 hastanın altısına (% 90 [% 95 GA = 0,4; 1,0]) HSCT uygulandı. 17Bu küçük örneklem büyüklüğü nedeniyle, VIDAZA'nın pediyatrik AML'deki etkililiği belirlenemez. Güvenlilik bilgileri için bölüm 4.8'e bakınız. 5.2. Farmakokinetik özelliklerGenel özelliklerEmilim:Azasitidin tek 75 mg/m2 subkutan doz uygulamasından sonra, azasitidin 0.5 saatte oluşan (ilk numune alma noktası) 750±403 ng/mL'lik doruk plazma konsantrasyonlarıyla hızlaabsorbe edilmiştir. Eğri altındaki alana (EAA) dayanarak subkutan uygulama sonrası azasitidinin I.V. azasitidine (tek 75 mg/m2 doz) göre biyoyararlanımı eğri altındaki alan (EAA) olarakyaklaşık %89'dur. Azasitidinin subkutan uygulamasının eğri altındaki alanı ve maksimum plazma konsantrasyonu (Cmaks) yaklaşık 25-100 mg/m2 doz aralığı içinde orantılıdır. Dağılım:IV uygulamanın ardından ortalama dağılım hacmi 76±26 L ve sistemik klirensi 147±47 L/saattir. Biyotransformasyon:İn vitroverilere göre sitokrom P450 izoenzimleri (CYPler), UDP-glukuronoziltransferazlar (UGTler), sulfotransferazlar (SULTlar) ve glutatyon transferazların (GSTler) azasitidinmetabolizmasında yer almadığı görülmektedir.Azasitidin metabolizması, sitidin deaminaz aracılığı ile oluşan deaminasyon ve spontan olarak gelişen hidroliz ile gerçekleşmektedir. İnsan karaciğeri S9 fraksiyonlarında metabolitoluşumunun NADPH'dan bağımsız olduğu gözlenmiştir, bu durum metabolik basamaklarınsitozolik enzimler tarafından katalizlendiğine işaret etmektedir. İnsan hepatosit kültürleriüzerinde yapılan in vitro araştırmalar 1,0-100 ^M azasitidin konsantrasyonlarının (yaniklinik olarak elde edilebilecek konsantrasyonlardan yaklaşık 30 kat daha yüksekkonsantrasyonlarda) sitokrom P450 izoenzimleri (CYP) olan 1A2, 2C19 veya 3A4 veya3A5'i indüklemediğini göstermektedir. 100 |iM azasitidin ile inkübe edilen bir seri P450izoenziminde (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ve 3A4) inhibisyonoluşturmamıştır. Bu nedenle klinik olarak elde edilebilir azasitidin plazmakonsantrasyonlarında enzim inhibisyonu olasılığı düşünülmemektedir. Eliminasyon:Azasitidin s.c. uygulamadan sonra 41±8 dakikalık ortalama eliminasyon yarılanma ömrü t( 12) ile hızlı bir şekilde plazmadan atılır. Günde 1 defa 7 gün boyunca subkutan 75 mg/m2azasitidin uygulamasından sonra herhangi bir birikme oluşmaz.Azasitidin ve/veya metabolitleri başlıca idrarla atılır. 14C-azasitidinin s.c. ve i.v. uygulamasının ardından, uygulanan radyoaktivitenin <%1'i feçes ile atılırken, % 50-85'i idrar ile atılır. 18Hastalardaki karakteristik özelliklerÖzel popülasyonlar:Karaciğer yetmezliğinin (bkz Bölüm 4.2), cinsiyetin, yaşın veya ırkın azasitidinin farmakokinetiği üzerine olan etkileri incelenmemiştir. Pediyatrik popülasyonAZA-JMML-001 çalışmasında, Farmakokinetik analiz, 1. siklusun 7. gününde 10 MDS ve 18 JMML pediatrik hasta üzerinden gerçekleştirilmiştir. (Bkz. Bölüm 5.1). Ortalama yaş MDShastaları için 13,3 (yaş aralığı 1,9-15) ve JMML hastaları için de 2,1 (yaş aralığı 0,2-6,9) idi. 75 mg/ m2'lik bir dozun intravenöz uygulanmasını takiben VIDAZA, hem MDS hem de JMML popülasyonlarında 0,083 saat içinde Cmax değerine hızlı bir şekilde ulaşmıştır. MDS ve JMMLhastaları için Cmax'ın geometrik ortalaması sırasıyla 1797,5 ve 1066,3 ng/mL iken AU^Vıngeometrik ortalaması ise 606,9 ve 240,2 ng saat/mL'dir. MDS ve JMML hastalarında geometrikortalama dağılım hacmi sırasıyla 103,9 ve 61,1 L'dir. VTDAZA'nın toplam plazma maruziyetininMDS hastalarında daha yüksek olduğu görülmüş; bununla birlikte, hem AUC hem de Cmaxdeğerleri için hastalar arasında orta ila yüksek değerli değişkenlik kaydedilmiştir. MDS ve JMML için tAnin geometrik ortalaması sırasıyla 0,4 ve 0,3 saat ve klerenslerin geometrik ortalaması ise sırasıyla 166,4 ve 148,3 L /saat'tir. AZA-JMML-001 çalışmasından elde edilen farmakokinetik veriler birlikte toplanmış ve AZA-2002-BA-002 çalışmasında intravenöz yolla 75 mg/m2 dozluk VIDAZA uygulanan MDS'li 6 yetişkin hastadan alınan farmakokinetik verilerle karşılaştırılmıştır. VTDAZAnın Cmax ve AUC 0-t'nin ortalaması, intravenöz uygulamadan sonra yetişkin hastalar ve pediatrik hastalar arasındabenzerdir (sırasıyla, 2750 ng/mL'ye karşı 2841 ng/mL ve 1025 ng-saat/mL'ye karşılık 882,1ng-saat/mL).AZA-AML-004 Çalışmasında farmakokinetik analiz, yedi hastanın doz sonrası en az bir ölçülebilir farmakokinetik konsantrasyon saptanabilmiş altısının verileriyle yapılmıştır. (bkz.Bölüm 5.1). AML hastalarının medyan yaşı 6,7 ve yaş aralığı ise 2-12 idi. 100 mg/m2'lik çoklu dozun bir çok kez verilmesi sonrasında 1.siklusun 7. günü Cmax ve AUC0-tau geometrik ortalamaları sırasıyla 1557 ng/mL ve 899,6 ng-saat/mL olmuştur.Hastalar arası Cmax ve AUC0-tau değerlerinde yüksek değişkenlik olduğu gözlenmiştir (CVyüzdesi Cmax ve AUC0-tau için sırasıyla %201,6 ve %87,8 olmuştur). Azasitidin, intravenözuygulamadan sonra ortalama 0,090 saatlik bir medyan sürede hızla Cmax'a ulaşmış ve 0,380saatlik bir geometrik ortalama yarılanma ömrü (L/ 2) ile azalmıştır. Klirens ve dağılma hacmiiçin geometrik ortalama sırasıyla 127,2 L/sa ve 70,2 L'dir.AML'li çocuklarda ilk tam remisyon (CR1)'dan sonra moleküler relapsda gözlenen farmakokinetik (azasitidin) maruziyet, MDS'li 10 çocuk ve JMML'li 18 çocuğun havuzlanmışverilerinden elde edilen maruziyet ile karşılaştırılabilir ve ayrıca MDS'li yetişkinlerdekiazasitidin maruziyeti ile karşılaştırılabilir seviyededir. Böbrek yetmezliğiBöbrek yetmezliğinin, tek ve çoklu subkutan uygulamalardan sonra azasitidinin farmakokinetik maruziyetinde herhangi bir önemli etkisi yoktur. Tek 75 mg/m2 subkutan 19doz uygulamasından sonra, normal böbrek fonksiyonu olan hastalara kıyasla hafif, orta ve ciddi böbrek yetmezliği olan hastaların ortalama maruziyet değerleri (EAA veCmaks),sırasıyla %11-21, %15-27 ve %41-66 oranında artmıştır. Bununla birlikte, maruziyet,normal böbrek fonksiyonu olan hastalar için gözlenen aynı genel maruziyet aralığındadır.Azasitidin ve/veya metabolitleri esas olarak böbrekten atıldığı için böbrek yetmezliği olanhastaların yakından izlenmesi koşulu ile, azasitidin, başlangıç doz ayarlaması olmaksızınböbrek yetmezliği olan hastalara uygulanabilir. Farmakogenomikler:Azasitidin metabolizması üzerinde bilinen sitidin deaminaz polimorfizmlerinin etkisi incelenmemiştir. 5.3. Klinik öncesi güvenlilik verileriAzasitidin in vitrobakteriyel ve memeli hücre sistemlerinde hem gen mutasyonlarını hem de kromozomal anomalileri indükler. Azasitidinin potansiyel karsinojenitesi farelerde vesıçanlarda incelenmiştir. Azasitidin 52 hafta boyunca haftada 3 defa intraperitonal (i.p.)uygulandığında, dişi farelerde hematopoetik sistem tümörlerini indüklemiştir. 50 haftasüreyle i.p. olarak uygulanan azasitidin ile tedavi edilen farelerde lenforetiküler sistem,akciğer, süt bezi ve deri tümörlerinin insidansının arttığı görülmüştür. Sıçanlarda bir tümöroluşturma çalışmasında testiküler tümörlerin insidansı artmıştır.Farelerde yapılan ilk embriyotoksisite çalışmalarında, organogenezis sırasında azasitidinin tek bir i.p. enjeksiyonundan sonra, intrauterin embriyonal ölüm %44 sıklıkta (artanrezorpsiyon) görülmüştür. Azasitidin verilen farelerde, sert damağın kapanması sırasında veya kapanmasından önce beyinde gelişimsel anormallikler görülmüştür. Sıçanlara preimplantasyon sürecindeverildiğinde, azasitidin herhangi bir advers reaksiyon göstermemiştir; fakat organogenezissırasında verildiğinde açıkça embriyotoksiktir. Organogenezis sırasında sıçanlarda meydanagelen fetal anomaliler şunlardır: MSS anomalileri (eksensefali, ensefalosel), kol-bacakanomalileri (mikromeli, yumru ayak, sindaktili, oligodaktili) ve diğerleri (mikroftalmi,mikrognazi, gastroşizis, ödem ve kaburga anormallikleri). Azasitidinin, tedavi edilmemiş dişi fare ile çiftleşmeden önce erkek fareye uygulanması, fertilite azalması ve embriyonik ve postnatal gelişim sırasında yavrunun kaybı ilesonuçlanmıştır. Erkek sıçanlara verilmesi, testis ve epididimislerin ağırlığının azalması,sperm sayısının azalması, gebelik oranlarının azalması, çiftleşen dişilerde embriyolarınkaybı ve anormal embriyo artışı ile sonuçlanmışt ır (bkz. Bölüm 4.4). 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiMannitol (E421) 6.2. GeçimsizliklerBu ürün, Bölüm 6.6'da b6.3. Raf ömrüAçılmamış toz flakonu:48 ayHazırlandıktan sonra: VIDAZA, buzdolabında saklanmayan enjeksiyonluk su ile hazırlandığında, hazırlanan tıbbi ürün 25°C'de 45 dakika ve 2-8°C'de 8 saat süre ile kimyasal ve fiziksel stabilitesini korur. 20Hazırlanan tıbbi ürünün raf ömrü buzdolabında (2-8°C) saklanan enjeksiyonluk su ile uzatılabilir. VIDAZA, buzdolabında (2-8°C) saklanan enjeksiyonluk21iltere hazırlandığında,hazırlanan tıbbi ürün 2-8°C'de 22 saat süre ile kimyasal ve fiziksel stabilitesini korur. Mikrobiyolojik açıdan hazırlanan ürün derhal kullanılmalıdır. Hemen kullanılmayacak ise, kullanım öncesi saklama süresi ve koşulları kullanıcının sorumluluğundadır ve buzdolabındasaklanmayan enjeksiyonluk su ile hazırlandığında 2-8°C'de 8 saatten fazla ve buzdolabında(2-8°C) saklanan enjeksiyonluk su ile hazırlandığında 2-8°C'de 22 saatten fazlaolmamalıdır. 6.4. Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklığında saklayınız. Hazırlanan tıbbi ürünün saklama koşulları için Bölüm 6.3'e bakınız. 6.5. Ambalajın niteliği ve içeriğiButil kauçuk tıpa ve aluminyum kapak ile kapatılan, polipropilen plastik düğmesi olan renksiz Tip I 30 mL cam flakon Ambalaj büyüklüğü:1 flakon içinde 100 mg azasitidin. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanma talimatıGüvenlik için öneriler:VIDAZA sitotoksik bir ilaçtır ve diğer potansiyel toksik bileşiklerde olduğu gibi, azasitidin süspansiyonlarını hazırlarken ve taşırken dikkatliolunmalıdır.Antikanser ilaçların imhası ve doğru şekilde tutulma prosedürleri uygulanmalıdır. Hazırlanan azasitidin süspansiyonu cilt ile temas ederse, derhal ve iyice su ve sabun ile yıkanmalıdır. Mukus membranlarla temas eder ise, su ile iyice yıkanmalıdır. Hazırlama prosedürü:1. Aşağıdaki malzemeler hazırlanmalıdır: Azasitidin flakonu: enjeksiyonluk su flakonu(ları); steril olmayancerrahi eldiven; Alkollü bezler; 5 mL'lik, iğneli enjeksiyon şırıngası(ları). 2. Şırıngaya 4 mL enjeksiyonluk su çekilmeli, şırıngada hiç hava olmamalıdır. 3. 4 mL enjeksiyonluk su içeren şırınganın iğnesi plastik kapaklı azasitidin flakonunabatırılmalı ve enjeksiyonluk su flakona enjekte edilmelidir. 4. İğne ve şırınga, azasitidin flakonundan çıkarıldıktan sonra azasitidin flakonu kuvvetleçalkalanarak bulanık, homojen bir süspansiyon elde edilmelidir. Bu noktada süspansiyonunher mL'sinde 25 mg azasitidin (100 mg/4 mL) bulunur. Oluşan ilaç homojen, bulanık birsüspansiyondur, herhangi bir topak içermemelidir. Eğer büyük partikül veya topak mevcutsaürün atılmalıdır. Etkin maddeyi uzaklaştırabileceği için süspansiyonu filtre etmeyiniz. Bazıadaptörlerde, şırıngalarda ve doz sistemlerinde filtrelerin bulunduğu dikkate alınmalıdır.Bu nedenle, bu tip sistemler ilaç hazırlandıktan sonra uygulama için kullanılmamalıdır.5. Azasitidin flakonunun plastik kapağı temizlenmeli ve yeni bir şırınga batırılmalıdır. 21Flakon ters döndürülmek, iğne ucunun sıvı seviyesinin alt ında olduğundan emin olunmalıdır. Şırınganın pistonu çekilerek doz için gerekli miktarda ilaç çekilmeli ve şırıngada havaolmamasına dikkat edilmelidir. Daha sonra şırınga ve iğnesi flakondan çıkarılmalı veşırınganın iğnesi atılmalıdır. 6. Şırıngaya yeni bir subkutan iğne ucu (25 ölçek önerilmektedir) takılır. Enjeksiyonbölgesinde lokal reaksiyon insidansını azaltmak için iğne ucu enjeksiyondan öncetemizlenmemelidir. 7. 1 flakondan fazla gerektiği zaman yukarıdaki basamaklar takip edilerek yeni ilaçsüspansiyonu hazırlanır. 1 flakondan fazla gereken dozlarda doz eşit bölünmelidir (örneğindoz 150 mg= 6 mL ise 2 şırınganın her biri 3 mL süspansiyon içermelidir). Flakon ve iğneiçindeki gecikmeden dolayı, flakondan bütün süspansiyonu çekmek mümkün olmayabilir. 8. Dozlama yapılan şırınganın içerikleri hastaya uygulanmadan önce tekrarçalkalanmalıdır. Enjeksiyon sırasında süspansiyonun ısısı yaklaşık 20°C-25°C olmalıdır.Süspansiyon, bulanık bir görünüm elde edilene kadar iki el arasında kuvvetle yuvarlanarakçalkalanır. Büyük partikül veya topak mevcutsa ürün atılmalıdır.VIDAZA süspansiyonu kullanılmadan hemen önce hazırlanmalı, oluşan süspansiyon 45 dakika içinde kullanılmalıdır. Süspansiyonun hazırlanmasından sonra 45 dakikadan dahauzun süre geçmesi halinde ilaç uygun şekilde atılmalı ve yeni bir doz hazırlanmalıdır.Alternatif olarak, süspansiyonun hastaya uygulanmadan önce hazırlanması gerektiğidurumlarda hazır ilaç, hazırlandıktan hemen sonra buzdolabına (2-8°C) konulmalıdır.Süspansiyon bu şekilde buzdolabında maksimum 8 saat bekleyebilir. İlacın buzdolabında 8saatten uzun süre kalması durumunda süspansiyon uygun şekilde atılmalı ve yeni bir dozhazırlanmalıdır. Buzdolabında (2-8°C) saklanan enjeksiyonluk su ile hazırlandığında, hazırlandıktan sonra hemen buzdolabına (2-8°C) konulmalıdır. Süspansiyon buzdolabında en fazla 22 saatbekleyebilir. İlacın buzdolabında 22 saatten uzun süre kalması durumunda süspansiyonuygun şekilde atılmalı ve yeni bir doz hazırlanmalıdır. Süspansiyonu içeren şırınga hastaya uygulanmadan önce 30 dakikaya varan sürelerde buzdolabı dışında bekletilerek ısısının yaklaşık 20-25°C'ye ulaşması sağlanmalıdır. Eğerbuzdolabı dışında geçen bu süre 30 dakikayı geçerse süspansiyon uygun şekilde atılmalı veyeni bir doz hazırlanmalıdır. Tek dozun hesaplanmasıVücut yüzey alanına (VYA) göre toplam doz aşağıdaki şekilde hesaplanabilir: Toplam doz (mg) = Doz (mg/m2) x VYA (m2) Aşağıda 1.8 m2'lik ortalama VYA değerine göre azasitidin dozlarının nasıl olması gerektiğine dair örnek bir tablo verilmiştir.
Uygulama şekli:Süspansiyonu hazırladıktan sonra filtre etmeyiniz! 22Hazırlanan VIDAZA subkutan olarak üst kola, uyluğa veya karna 25 ölçekli iğne kullanarak enjekte edilmelidir (45-90° açı ile iğneyi sokunuz). 4 mL'den büyük dozlar iki ayrı bölgeye enjekte edilmelidir. Enjeksiyon yapılan alan değiştirilmelidir. Yeni enjeksiyonlar, eski enjeksiyon bölgesine en az 2.5 cm uzaklıkta yapılmalıdır ve asla yumuşak, morarmış, kırmızı ve sert olan yerlereenjeksiyon yapılmamalıdır. Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imhaedilmelidir. 7. RUHSAT SAHİBİEr-Kim İlaç Sanayi ve Ticaret A.Ş. Zorlu Center, Levazım Mah. Koru Sk. No:2 D-Blok 342-345 34340, Beşiktaş - İSTANBUL Tel: (0212) 275 39 69 Faks: (0212) 211 29 77 e-mail: 8. RUHSAT NUMARASI123/18 9. İLK RUHSAT TARİHİ/ RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 10.10.2007 Ruhsat yenileme tarihi: 17.02.2014 10. KÜB'ÜN YENİLENME TARİHİ231
|
İlaç BilgileriVidaza 100 Mg Sc Enjeksiyonluk Süspansiyon İçin Toz İçeren FlakonEtken Maddesi: Azasitidin Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.