Novoeight 1500 Iu Enjeksiyonluk Çözelti Hazırlamak İçin Toz ve Çözücü Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ'VBu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADINovoEight® 1500 IU enjeksiyonluk çözelti hazırlamak için toz ve çözücü Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir toz flakonu nominal olarak 1500 IU turoktokog alfa (rekombinant insan koagülasyon faktörü VIII) içerir. NovoEight®, rekonstitüye edildikten sonra yaklaşık olarak 375 IU/mL turoktokog alfa (rekombinant insan koagülasyon faktörü VIII) içerir. Potens (IU), Avrupa Farmakopesi (Ph.Eur) kromojenik tayin kullanılarak belirlenir. NovoEight®'in spesifik aktivitesi yaklaşık 8,300 IU/mg proteindir. Turoktokog alfa (rekombinant insan koagülasyon faktörü VIII), 1,445 amino aside ve yaklaşık 166 kDA molekül kütlesine sahip saflaştırılmış bir proteindir. Çin hamsterı yumurtalık (CHO)hücrelerinde rekombinant DNA teknolojisi kullanılarak üretilir ve hücre kültürü prosesinde,saflaştırma ya da son formülasyon sırasında herhangi bir insan veya hayvan kaynaklı proteinilavesi olmaksızın hazırlanır. Turoktokog alfa, amino asit sekansında başka bir modifikasyonu olmayan bir B-domain trunkat rekombinant insan koagülasyon faktörü VIII'dir (B-domaini, yabanıl tip B-domaininin21 amino asidini içerir). Yardımcı maddelerToz36 mg/mL y.m. Sodyum klorür Sodyum hidroksit (pH ayarı için) ÇözücüSodyum klorür 9 mg/mL Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti için toz ve çözücü. Beyaz veya hafif sarı toz veya ufalanabilir kütle. Berrak ve renksiz enjeksiyonluk çözelti. 1 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarHemofili A (konjenital faktör VIII eksikliği) hastalarının kanama tedavisinde ve profılaksisinde kullanılır. NovoEight® tüm yaş gruplarında kullanılabilir. 4.2 Pozoloji ve uygulama şekliTedavi hemofili tedavisinde deneyimli bir hekimin gözetimi altında olmalıdır. Tedavinin izlenmesiTedavi süresince, uygulanacak dozun ve tekrar edilen infüzyon sıklığının belirlenmesinde, faktör VIII seviyelerinin uygun şekilde tespit edilmesi önerilir. Her bir hasta, faktör VIII'efarklı yanıt verebilir, farklı yarılanma ömrü ve geri kazanım (recovery) gösterebilir. Vücutağırlıkları normalden düşük veya fazla olan hastalarda vücut ağırlığına göre ayarlamayapılması gerekebilir. Yetişkin hastalardaki tek dozlu bir farmakokinetik çalışmada,maksimum maruz kalım (Cmaks) ve toplam maruz kalım (EAA), artan vücut kitle indeksi(VKİ) ile birlikte artmış olup, doz ayarlamasının gerekli olabileceğini göstermektedir. Vücutağırlıkları normalden az hastalarda (VKI <18,5 kg/m2) dozda artış ve obez hastalarda (VKİ>30 kg/m2) dozda azalma gerekebilir, ancak spesifik doz ayarlamaları önermek için yeterlibilgi bulunmamaktadır (bkz Bölüm 5.2). Özellikle majör cerrahi müdahaleler söz konusu olduğunda, ikame tedavinin koagülasyon analizi ile (plazma faktör VIII aktivitesi) kesin şekilde izlenmesi gerekmektedir. Hastaların kan numunelerinde faktör VIII tayini için in vitro tromboplastin zamanı (aPTT) esaslı tek aşamalı pıhtılaşma tayini kullanıldığında, plazma faktör VIII aktivitesi sonuçlarıhem aPTT reaktifi tipinden hem de tayinde kullanılan referans standarttan anlamlı derecedeetkilenebilir. Ayrıca, aPTT esaslı tek aşamalı pıhtılaşma tayini ve Avrupa Farmakopesi'neuygun olan kromojenik tayin ile elde edilen tayin bulguları arasında anlamlı çelişkiler olabilir.Bu durum, özellikle de laboratuarın ve/veya tayinde kullanılan reaktiflerin değiştirilmesihalinde önem taşımaktadır. Pozoloji/uygulama sıklığı ve süresi:Yerine koyma tedavisinin dozu ve süresi faktör VIII eksikliğinin şiddetine, kanamanın yeri ve boyutuna ve hastanın klinik durumuna bağlıdır. Uygulanan faktör VIII ünitelerinin sayısı, faktör VIII ürünleri için mevcut WHO standardına ait Uluslararası Ünite (IU) ile ifade edilir. Faktör VIII'in plazmadaki aktivitesi yüzde (normaldüzey insan plazmasına göre) ya da Uluslararası Ünite (plazmada faktör VIII için UluslararasıStandarda göre) şeklinde ifade edilir. Bir Uluslararası Ünite (IU) faktör VIII aktivitesi, 1 mL normal insan plazmasında bulunan faktör VIII miktarına eşdeğerdir. Kanadıkça tedaviGereken faktör VIII dozunun hesaplanmasında, kg vücut ağırlığı başına 1 Uluslararası Ünite (IU) faktör VlII'in plazma faktör VIII aktivitesini 2 IU/dl artırdığı yönündeki gözlemsel/deneysel bilgi esas alınmaktadır. Gereken doz aşağıdaki formül kullanılarakhesaplanır: Gereken ünite sayısı = vücut ağırlığı (kg) x istenen faktör VIII yükselmesi (%) (IU/dL) x 0,5 (IU/kg / IU/dL). Uygulanacak miktar ve uygulama sıklığında her zaman söz konusu olgudaki klinik etkililik esas alınmalıdır. Aşağıdaki hemorajik olaylar söz konusu olduğunda faktör VIII aktivitesi, karşılık gelen periyottaki belirli plazma aktivite düzeyinin (IU/dl veya normalin yüzdesi olarak) altınadüşmemelidir. Kanama epizotlarında ve ameliyatlarda uygulanacak doza kılavuzluk etmesiiçin aşağıdaki tablo kullanılabilir: Tablo 1 Kanama epizotlarında ve ameliyatlarda uygulanacak doz için kılavuz
ProfilaksiAğır hemofili A hastalarında kanamaya karşı uzun süreli profilaksi. Normal şartlar altında 3 önerilen dozlar, iki günde bir verilen kg vücut ağırlığı başına 20-40 IU faktör VIII ya da haftada üç kez verilen kg vücut ağırlığı başına 20-50 IU faktör VIIFdirErişkin veadölesanlarda (>12 yaş) daha az sıklıkta dozaj (üç günde bir veya haftada 2 kez 40-60 IU/kg)uygulanabilir. Bazı durumlarda, özellikle daha genç hastalarda, daha kısa doz aralıkları veyadaha yüksek dozlar gerekli olabilir. CerrahiPediyatrik hastalarda cerrahi ile ilgili kısıtlı deneyim bulunmaktadır. Uygulama şekli:İntravenöz kullanım. NovoEight® için önerilen infüzyon hızı 1-2 mL/dakikadır. Hız, hastanın rahatlık düzeyine göre belirlenmelidir. Uygulama öncesinde tıbbi ürünün rekonstitüsyonu (karışım halinde hazırlanması) üzerine talimatlar için bölüm 6.6'ya bakınız. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karaciğer yetmezliği:Böbrek yetmezliği olan hastalarda deneyim bulunmamaktadır. Karaciğer yetmezliği olan hastalar klinik çalışmalara dahil edilmiştir. Bu hastalar ile karaciğer yetmezliği olmayan hastalar arasında etkililik ve güvenlilik açısından fark gözlenmemiştir. Pediyatrik popülasyon:12 yaşın altındaki hastalarda kanamaya karşı uzun süreli profilaksi için iki günde bir, kg vücut ağırlığı başına 25-50 IU faktör VIII ya da haftada 3 defa kg vücut ağırlığı başına 25-60 IUfaktör VIII önerilmektedir. 12 yaşın üzerindeki pediyatrik hastalarda doz önerileri,erişkinlerdeki ile aynıdır. Geriyatrik popülasyon:65 yaşın üzerindeki hastalar ile ilgili deneyim bulunmamaktadır. 4.3 KontrendikasyonlarEtkin maddeye ya da bölüm 6.1'de listesi bulunan yardımcı maddelerin herhangi birine karşı aşırı duyarlılık. Hamster proteinlerine karşı bilinen alerjik reaksiyon. 4.4 Özel kullanım uyarıları ve önlemleriAşırı duyarlılıkNovoEight® ile alerjik tip aşırı duyarlılık reaksiyonları olasıdır. Ürün, bazı hastalarda alerjik reaksiyonlara neden olabilen hamster proteinlerinden eser miktarlarda içerir. Aşırı duyarlılıksemptomlarının ortaya çıkması halinde hastalara bu tıbbi ürünün kullanımını derhaldurdurmaları ve hekimleri ile temasa geçmeleri tavsiye edilmelidir. Hastalar aşırı duyarlılığınkurdeşen, yaygın ürtiker, göğüs sıkışması, hırıltı, hipotansiyon ve anafilaksi gibi erkenişaretleri konusunda bilgilendirilmelidir. 4 Şok durumunda, şok tedavisine yönelik mevcut tıbbi standartlar izlenmelidir. İnhibitörlerFaktör VIII'e karşı nötralize edici antikor (inhibitörler) oluşumu, hemofili A hastalarının tedavisinde bilinen bir komplikasyondur. Bu inhibitörler genellikle faktör VIII prokoagülanaktiviteye yönelik olan IgG immünoglobülinleridir ve modifiye tetkik kullanılarak her mlplazmada Bethesda Ünitesi (BU) olarak ölçülür. İnhibitör gelişme riski, faktör VIII'emaruziyetin yanı sıra hastalığın şiddeti ile ilişkilidir ve bu risk ilk 50 maruziyet gününde enyüksek seviyededir; ancak risk yaygın görülmemesine rağmen yaşam boyu devam eder. İnhibitör gelişiminin klinik önemi inhibitör titresine bağlı olacaktır; düşük titrenin teşkil ettiği yetersiz klinik yanıt riski, yüksek titreli inhibitörlere kıyasla daha az olacaktır. Genel olarak,koagülasyon faktörü VIII ürünleri ile tedavi edilen tüm hastalar, uygun klinik gözlem velaboratuvar testleri ile inhibitörlerin gelişimi açısından dikkatle izlenmelidir. Eğer beklenenfaktör VIII aktivitesinin plazma düzeylerine ulaşılamazsa veya yeterli doz ile kanama kontrolaltına alınamazsa faktör VIII inhibitörü varlığı açısından test yapılmalıdır. İnhibitör düzeyleriyüksek olan hastalarda faktör VIII tedavisi etkili olmayabilir ve diğer tedavi seçenekleridikkate alınmalıdır. Böyle hastaların tedavisi hemofili ve faktör VIII inhibitörleri tedavisikonusunda deneyimli hekimler tarafından yönlendirilmelidir. Kardiyovasküler olayMevcut kardiyovasküler risk faktörleri olan hastalarda FVIII ile ikame tedavisi kardiyovasküler riski artırabilir. Katetere bağlı komplikasyonlarEğer bir santral venöz erişim cihazı (CVAD) gerekli ise, lokal enfeksiyonlar, bakteriyemi ve kateter yeri trombozu gibi CVAD ile ilişkili komplikasyonlar riski göz önündebulundurulmalıdır. Hasta ve tıbbi ürünün serisi arasındaki bağlantının devam etmesi için, hastaya her NovoEight uygulamasında ürünün adı ve seri numarasının kaydedilmesi kesinlikle önerilir. Pediyatrik popülasyonBelirtilen uyarılar ve önlemler hem erişkinler hem de çocuklar için geçerlidir. Yardımcı maddeler ile ilgili değerlendirmeBu tıbbi ürün, rekonstitüye edilmiş flakon başına 30,5 mg sodyum ihtiva etmekte olup, Dünya Sağlık Örgütü (WHO) tarafından önerilen yetişkinler için maksimum günlük 2 g'lık sodyumalımın %1,5'ine eşdeğerdir. İzlenebilirlikBiyolojik tıbbi ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına açıkça kaydedilmelidir. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriİnsan koagülasyon faktör VIII (rDNA) ürünleri ile diğer tıbbi ürünler arasında bilinen bir etkileşim bulunmamaktadır. 5 Özel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyon:NovoEight® ile etkileşim çalışmaları gerçekleştirilmemiştir. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)NovoEight®'in çocuk doğurma potansiyeli bulunan kadınlarda kullanımına ilişkin herhangi bir bilgi bulunmamaktadır. Gebelik dönemiHemofili genel olarak erkeklerde görüldüğünden, turoktokog alfanın gebe kadınlarda kullanımına ilişkin yeterli veri yoktur. Hayvanlar üzerinde yapılan çalısmalar, gebelik /ve-veya/ embriyonal / fetal gelisim /ve veya/ doğum /ve-veya/ doğum sonrası gelisim üzerindeki etkiler bakımından yetersizdir (bkz.Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. NovoEight® gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Laktasyon dönemiKadınlarda hemofili A'nın seyrek görülmesine bağlı olarak, emzirme süresince faktör VIII kullanımı ile ilgili deneyim bulunmamaktadır. Bu nedenle, faktör VIII laktasyon süresincesadece açıkça endike olduğunda kullanılmalıdır. Üreme yeteneği/FertiliteNovoEight®'in üreme yeteneği üzerindeki etkisine ilişkin bilgi bulunmamaktadır. 4.7 Araç ve makine kullanımı üzerindeki etkiler4.8 İstenmeyen etkilerGüvenlilik profilinin özetiAşırı duyarlılık ya da alerjik reaksiyonlar (bunlar arasında anjiyoödem, infüzyon bölgesinde yanma ve batma, titreme, sıcak basması, yaygın ürtiker, baş ağrısı, kurdeşen, kan basıncındadüşme, letarji, bulantı, huzursuzluk, taşikardi, göğüste sıkışma hissi, karıncalanma, kusma,hırıltılı solunum yer alabilir) nadiren gözlenmiştir ve bazı olgularda şiddetli anafilaksiye kadarilerleyebilir (şok dahil). İlgili aşırı duyarlılık reaksiyonları ile birlikte, hamster proteinine karşı antikor gelişimi çok nadiren gözlenmiştir. NovoEight® de dahil olmak üzere faktör VIII ile tedavi edilmiş hemofili A hastalarında nötralize edici antikorlar (inhibitörler) gelişebilir (bkz. Bölüm 5.1). Bu tür inhibitörler 6oluşursa, durum, yetersiz klinik yanıt şeklinde kendini gösterebilir. Bu gibi durumlarda uzmanhemofili merkezleriyle bağlantı kurulması önerilmektedir. Advers reaksiyonların tablolaştırılmış listesiAşağıda verilen tablo, MedDRA sistem organ sınıflandırmasına (SOC ve Tercihli Terim Düzeyi) uygundur. Görülme sıklıkları şu yaklaşıma göre değerlendirilmiştir: çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek(<1/10.000) ve bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık gruplamasında, istenmeyen etkiler, azalan ciddiyet sırasına göre sunulmaktadır. 7
b Sıklık, tüm FVIII ürünleriyle ilgili, ağır hemofili A hastalarının yer aldığı çalışmalara dayalıdır. c Enjeksiyon yeri reaksiyonları şunları içerir: enjeksiyon yerinde kızarıklık, enjeksiyon yerinde ekstravazasyon ve enjeksiyon yerine kaşıntı. d Hepatik enzim düzeylerinde yükselme şunları içerir: alanin aminotransferaz, aspartat aminotransferaz, gamma-glutamiltransferaz ve bilirubin. Seçilen advers reaksiyonların tanımıDaha önce tedavi görmüş hastalarla, NovoEight® ile yapılan tüm klinik çalışmalar sırasında, NovoEight® uygulanan 242 hastanın 23'ünde toplam 35 advers reaksiyon bildirilmiştir. En sıkbildirilen advers reaksiyonlar enjeksiyon yeri reaksiyonları, yanlış doz uygulanması vehepatik enzim düzeylerinde yükselme olmuştur. 35 advers reaksiyondan 2'si 6 yaşın altındaki31 hastanın 1'inde bildirilmiş, 6-12 yaş arası hastalarda hiçbir advers reaksiyonbildirilmemiş,12-18 yaş arasındaki 24 hastanın 1'inde bildirilmiş ve 155 erişkin hastanın (>18yaş) 21'inde 32 advers reaksiyon bildirilmiştir. Pediyatrik popülasyon0-12 yaş arası 63 önceden tedavi görmüş pediyatrik ağır hemofili A hastasını ve 12-18 yaş arası 24 adölesan ağır hemofili A hastasını içeren klinik çalışmalarda, NovoEight®'ingüvenlilik profili bakımından pediyatrik ile erişkin hastalar arasında herhangi bir farkgözlenmemiştir. Daha önce tedavi görmemiş hastalarla yapılan çalışmada, NovoEight® uygulanan yaşları 0-6 arasında olan 60 hastanın 33'ünde toplam 46 advers reaksiyon bildirilmiştir. En sık bildirilenadvers reaksiyon Faktör VIII inhibisyonudur (bakınız bölüm 4.4.). Yüksek riskli genetikmutasyonlar toplamın %92,3'ünde, yüksek titreli doğrulanmış inhibitörlerin %93,8'indetanımlanmıştır. Diğer faktörler inhibitör gelişimi ile anlamlı olarak ilişkili değildir. 8 Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr;e-posta: [email protected]; tel: 00 800 314 00 08; faks: 0 312 218 35 99) 4.9 Doz aşımı ve tedavisiRekombinant koagülasyon faktörü VIII ile herhangi bir doz aşımı semptomu bildirilmemiştir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: Antihemorajikler, kan koagülasyon faktörü VIII ATC kodu: B02BD02 Etki mekanizmasıNovoEight®, kesilmiş B-domainli bir rekombinant insan koagülasyon faktörü VIII olan turoktokog alfa içerir. Bu glikoprotein aktive edildiğinde insan faktörü VIII ile aynı yapıyasahiptir ve post-translasyonel değişiklikler plazma türevli molekülün değişiklikleri ilebenzerdir. von Willebrand faktörünün bağlanması için önemli olan Tyr1680 (doğuştan tamuzunlukta) noktasında mevcut olan tirozin sülfasyon bölgesinin turoktokog alfa molekülündetamamen sülfatlanmış olduğu bulunmuştur. Hemofili hastasına infüze edildiğinde, faktör VIIIhastanın dolaşımındaki endojen von Willebrand faktörüne bağlanır. Faktör VIII/vonWillebrand faktörü kompleksi, farklı fizyolojik fonksiyonlara sahip iki molekülden oluşur(faktör VIII ve von Willebrand faktör). Aktive faktör VIII, aktive faktör IX için ko-faktörişlevi görerek faktör X'un aktive faktör X'a dönüşümünü hızlandırır. Aktive faktör X,protrombini trombine dönüştürür. Trombin daha sonra fibrinojeni fibrine dönüştürür ve pıhtıoluşabilir. Hemofili A, düşük faktör VIII:C düzeylerine bağlı olan ve eklemlerde, kaslardaveya iç organlarda spontan oluşan ya da kaza veya cerrahi travma sonucu ortaya çıkabilenciddi kanamalara neden olan cinsiyet kromozomuna bağlı kalıtsal bir kan pıhtılaşmabozukluğudur. Replasman tedavisi ile faktör VIII'in plazma düzeyleri artırılır, böyleliklefaktör eksikliği geçici olarak düzeltilir ve kanama eğilimleri giderilir. Ayrıca, yıllık kanama oranı (ABR), farklı faktör konsantreleri ve farklı klinik çalışmalar arasında kıyaslanabilir değildir. Klinik etkililikNovoEight®'in ağır hemofili A (FVIII aktivitesi <%1) hastalarında kanamaların önlenmesi ve tedavisinde ve cerrahi müdahaleler sırasında güvenliliği ve etkililiğini değerlendirmekamacıyla dört çok merkezli, açık etiketli, kontrol grubu olmayan çalışma gerçekleştirilmiştir.Bu çalışmalardan üçü daha önce tedavi görmüş hastalarda, dördüncüsü daha önce tedavigörmemiş hastalarda gerçekleştirilmiştir. Bu çalışmalarda 298 hastaya ilaç uygulanmıştır;inhibitörü olmayan 12 yaş ve üzeri 175 adölesan ve erişkin hasta (>150 maruziyet günü),inhibitörü olmayan 12 yaş altı daha önceden tedavi görmüş 63 pediyatrik hasta (>50maruziyet günü) ve daha önceden tedavi görmemiş 6 yaşın altındaki 60 hasta. 9 238 daha önceden tedavi görmüş hastanın 188'si güvenlilik uzatma çalışmasına devam etmiştir. NovoEight® ile tedavinin güvenli olduğu ve amaçlanan hemostatik ve önleyici etkiyesahip olduğu gösterilmiştir. Hastaların 298'inde gözlenen 3.293 kanamadan 2.902'si (%88,1)NovoEight®'in 1-2 infüzyonu ile düzelmiştir. Tablo 3 Daha önceden tedavi edilmemiş hastalarda (HTGH) ve daha önceden tedavi edilmiş hastalarda (TGH) NovoEight tüketimi ve hemostatik başarı oranları
Ruhsatlandırma öncesi klinik veriler, rutin klinik uygulamada NovoEight®'in immunojenesitesi, etkililiği ve güvenliliğine ilişkin ek dokümantasyon sağlamak amacıylayürütülen, girişimsel olmayan, ruhsatlandırma sonrası bir güvenlilik çalışmasıyladesteklenmiştir. 14'ü 12 yaşın altında ve 54'ü 12 yaş ve üzerinde olan toplam 68 dahaönceden tedavi görmüş hasta (> 150 maruziyet günü), toplamda 87,8 hasta yılı ve 8967maruziyet günü süresince kanadıkça (N = 5) ve profilaktik (N = 63) tedavi almıştır. Cerrahi 25 hastada toplam 30 cerrahi operasyon gerçekleştirilmiş olup bunların 26'sı majör ve 4'ü minör cerrahidir. Tüm cerrahi operasyonlarda hemostaz başarılı olmuştur ve tedavibaşarısızlığı bildirilmemiştir. Faktör VIII'e karşı inhibitör geliştiren hemofili A hastalarında İmmün Tolerans İndüksiyonu (ITI) hakkında veriler toplanmıştır. Daha önceden tedavi edilmemiş hastalardaki (HTGH)klinik çalışmalar sırasında, 21 hasta ITI ile tedavi edilmiş ve 18 hasta (%86) ITI'yı negatif 10 inhibitör test sonucu ile tamamlamıştır. 5.2 Farmakokinetik özelliklerGenel özelliklerNovoEight ile yapılan tüm farmakokinetik (FK) çalışmalar, 50 IU/kg NovoEight' in i.v uygulamasından sonra önceden tedavi edilmiş ağırhemofili A (FVIII <%1) hastaları ilegerçekleştirilmiştir. Plazma örneklerinin analizi hem tek aşamalı pıhtılaşma testi hem dekromojenik yöntem ile yürütülmüştür. NovoEight®'in FVIII:C tayinlerinde tayin performansı değerlendirilmiş ve piyasalarda yer alan tam uzunlukta bir rekombinant FVIII ürünü ile karşılaştırılmıştır. Bu çalışma, bu ikiürünle karşılaştırılabilir ve tutarlı sonuçlar alındığını ve NovoEight®'in plazmada, ayrı birNovoEight® standardına gerek olmadan güvenilir bir şekilde ölçülebileceğini göstermiştir. NovoEight®'in tek doz farmakokinetik parametreleri, pıhtılaşma testi için Tablo 4'te, kromojenik test için Tablo 5'te gösterilmektedir. Emilim:NovoEight intravenöz olarak, doğrudan damar içine uygulanır. Bu nedenle, emilim çalışmaları yapılmamıştır. Dağılım:Plazma FVIII aktivitesi, dağılım fazı belirgin olmayan bir mono-üstel bozunma ile azalır. Tablo 4 Yaşa göre NovoEight'in (50 IU/kg) tek doz farmakokinetik parametreleri - tek aşamalı pıhtılaşma testi - Ortalama (SS)
Biyotransformasyon:Uygulanabilir değil. Eliminasyon:Plazma FVIII aktivitesi, mono-üstel bir düşüş ile elimine edilir. 11 Tablo 5 Yaşa göre NovoEight'in (50 IU/kg) tek doz farmakokinetik parametreleri -Kromojenik test - Ortalama (SS)
Farmakokinetik parametreler 6 yaşın altındaki pediyatrik hastalar ile 6 ila 12 yaş arası pediyatrik hastalar arasında benzer olmuştur. Pediyatrik hastalar ile erişkin hastalar arasındaNovoEight®'in farmakokinetik parametrelerinde bir miktar değişkenlik gözlenmiştir.Hemofili A'lı erişkin hastalar ile karşılaştırıldığında pediyatrik hastalarda görülen dahayüksek KL ve daha kısa t/ , kısmen, daha genç hastalarda kilogram vücut ağırlığı başına dahayüksek olduğu bilinen plazma hacmine bağlı olabilir. Farklı VKİ kategorilerindeki 35 hemofili hastasında (>18 yaş) tek dozlu bir farmakokinetik çalışma (50 IU/kg) yürütülmüştür. Artan VKİ ile maksimum maruz kalım (Cmaks) ve toplammaruz kalım (EAA) artmış olup, vücut ağırlıkları normalden düşük hastalarda (VKI <18,5kg/m2) ve obez hastalarda (VKİ >30 kg/m2) doz ayarlaması gerekebileceğini göstermektedir,bkz Bölüm 4.2.
12 a VKİ grupları: Normalden az kilolu: VKİ <18,5 kg/m2, Normal kilolu: VKİ 18,5-24,9 kg/m2, Normalden fazla kilolu: VKİ 25-29,9 kg/m2, Obez Sınıf I: VKİ 30-34,9 kg/m2, Obez Sınıfn/III: VKİ >35 kg/m2.b Sadece 6 hastaya dayanmaktadır. Tablo 7 VKI sınıflarına3 göre NovoEight'in (50 IU/kg) tek doz farmakokinetik parametreleri - Kromojenik test - Ortalama (SS) göre NovoEight'in (50 IU/kg) tek doz farmakokinetik parametreleri - Kromojenik test - Ortalama (SS)
n/III: VKİ >35 kg/m2. Doğrusallık/doğrusal olmayan durum:Tüm farmakokinetik çalışmalar, 50 IU/kg dozundan sonra yapılmıştır. Bu nedenle, doz doğrusallığı araştırılmamıştır. 5.3 Klinik öncesi güvenlilik verileriGüvenlilik farmakolojisi ve tekrarlı doz toksisitesinden oluşan konvansiyonel çalışmalar bazında klinik dışı veriler, insanlar için endişeye neden olabilecek bir durum meydanagetirmemektedir. 6. FARMASOTIK ÖZELLİKLER6.1 Yardımcı maddelerin listesiToz:Sodyum klorür L-histidin Sukroz Polisorbat 80 L-metiyonin Kalsiyum klorür dihidrat Sodyum hidroksit (pH ayarı için) Hidroklorik asit (pH ayarı için) 13 Çözücü:Sodyum klorür Enjeksiyonluk su 6.2 GeçimsizliklerGeçimlilik çalışmaları bulunmadığından, bu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3 Raf ömrüAçılmamış flakon:30 ay buzdolabında (2°C - 8°C) saklandığı sürece. Raf ömrü boyunca, 9 ayı geçmeyecek kesintisiz bir süre için 30°C ve altındaki oda sıcaklığında (< 30°C) veya3 ayı geçmeyecek kesintisiz bir süre için oda sıcaklığı üzerinde(30°C ila 40°C arasında) saklanabilir.Ancak buzdolabından bir kere çıkarıldıktan sonra tekrar buzdolabına koyulmamalıdır. Saklamanın başladığı zaman ve saklama sıcaklığı ürün kutusu üzerine kaydedilmelidir. Rekonstitüsyon sonrasında (kullanıma hazırlandıktan sonra):Kullanıma hazırlandıktan sonraki kimyasal ve fiziksel stabilite aşağıdakiler için gösterilmiştir: - 24 saat süreyle 2°C- 8°C'de saklanır. - 4 saat süreyle 30°C'de saklanır; 9 ayı geçmeyecek kesintisiz bir süre için odasıcaklığında (< 30°C) saklanan ürün için geçerlidir. - 4 saat süreyle 40°C'ye kadar saklanır; 3 ayı geçmeyecek kesintisiz bir süre için odasıcaklığı üzerinde (30°C ila 40°C arasında) saklanan ürün için geçerlidir. Mikrobiyolojik açıdan bu ürün, rekonstitüsyon sonrasında hemen kullanılmalıdır. Hemen kullanılmadığı takdirde, kullanıma hazırlanmış ürünün saklama süreleri ve koşullarıkullanıcının sorumluluğundadır ve normal şartlar altında, rekonstitüsyon kontrollü ve valideedilmiş aseptik koşullar altında gerçekleşmediği sürece, yukarıda bahsedilen sürelerigeçmemelidir. Oda sıcaklığında (<30°C) veya 40°C'ye kadar 4 saatten fazla saklanan kullanılmamış rekonstitüye ürün atılmalıdır. 6.4 Saklamaya yönelik özel tedbirlerBuzdolabında saklanmalıdır (2°C - 8°C). Dondurulmamalıdır. Işıktan korumak için, flakon orijinal kutusunda saklanmalıdır. Oda sıcaklığında (<30°C) veya 40°C'ye kadar saklama ve tıbbi ürünün rekonstitüsyon sonrası saklama koşulları için bkz. bölüm 6.3. 14 6.5 Ambalajın niteliği ve içeriğiHer NovoEight® 1500 IU enjeksiyonluk çözelti için toz ve çözücü ambalajı şunları içerir: - 1 adet kauçuk (klorobütil) tıpalı flakon (tip I cam) içinde toz - Rekonstitüsyon için 1 adet steril flakon adaptörü - 1 adet bromobütil tıpalı enjektör başlıklı, kauçuk (bromobütil) pistonlu, geri kaçış kilidi (polipropilen) bulunan kullanıma hazır enjektör içinde 4 mL çözücü - 1 adet piston kolu (polipropilen) 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliği gereklerine uygun olarak imha edilmelidir. NovoEight®, enjektör içinde verilen çözücü ile toz karıştırılarak hazırlandıktan sonra (rekonstitüye edilerek) intravenöz yolla uygulanır. Rekonstitüsyon sonrası çözelti berrak veyahafif opelesan görünümdedir. Bulanık görünen veya tortu içeren çözeltileri kullanmayınız. Ayrıca bir infüzyon setine (ince tüp ve kelebek iğne), steril alkollü bezlere, gazlı bezlere ve flasterlere ihtiyacınız olacaktır. Bunlar, NovoEight® ambalajında yer almaz. Her zaman aseptik teknik kullanınız. Rekonstitüsyon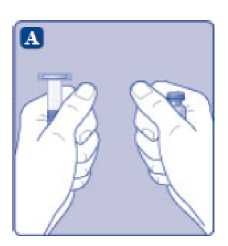 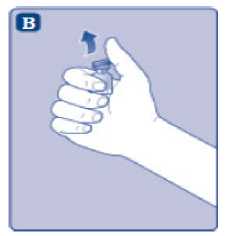 A) Flakonu, flakon adaptörünü ve kullanıma hazır enjektörü ambalajından çıkarınız. Piston koluna dokunmayınız vekutu içinde bırakınız. Flakonun ve kullanıma hazırenjektörün oda sıcaklığına gelmesini sağlayınız. Bunu,elleriniz kadar ılık olana kadar elleriniz içinde tutarakyapabilirsiniz. Flakonu ve kullanıma hazır enjektörüısıtmak için başka bir yol kullanmayınız. B) Plastik kapağı flakondan çıkarınız. Eğer plastik kapak gevşekse veya yoksa bu flakonu kullanmayınız.Flakondaki lastik tıpayı steril alkollü bez ile siliniz vekullanmadan önce birkaç saniye kurumasını bekleyiniz. 15 C) 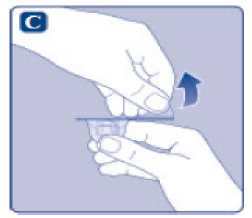 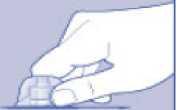 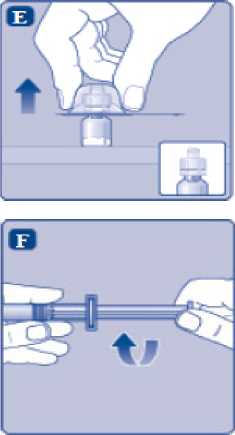 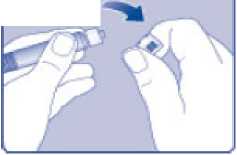 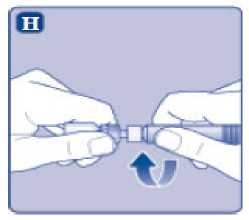 Koruyucu kağıdı flakon adaptöründen çıkarınız. Koruyucu kağıt tam kapalı değilse veya yırtıksa, bu flakon adaptörünü kullanmayınız. Flakon adaptörünü koruyucu kapaktan parmaklarınızla çıkarmayınız. D) Koruyucu kapağı ters çeviriniz ve flakon adaptörünü flakona takınız. Taktıktan sonra flakon adaptörünüflakondan çıkarmayınız. E) Koruyucu kapağı baş ve işaret parmağınızla şekilde gösterildiği gibi hafifçe sıkınız. Koruyucu kapağı flakonadaptöründen çıkarınız. F) Piston kolunu geniş üst kısmından kavrayınız ve piston kolunu hemen kullanıma hazır enjektör içindeki dalıcıpistona doğru, direnç hissedilene kadar saat yönündeçevirerek enjektöre bağlayınız. G) Delikli kısımdan kırılana kadar aşağı yönde eğmek suretiyle enjektör kapağını kullanıma hazır enjektördençıkarınız. Enjektör kapağının altında bulunan enjektörünucuna dokunmayınız. H) Kullanıma hazır enjektörü direnç hissedilene kadar çevirerek flakon adaptörüne sıkıca takınız. 16 I) 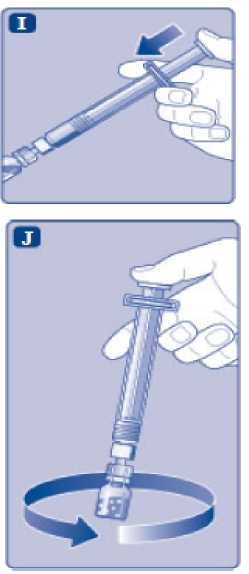 Kullanıma hazır enjektörü hafif eğik ve flakon aşağı bakacak şekilde tutunuz. Piston kolunu flakonun içinetamamen girene kadar itiniz. J) Piston kolunu aşağıya doğru basılı tutunuz ve toz çözülene kadar flakonu dairesel hareketlerle döndürünüz.Köpüklenmeye yol açacağından flakonu çalkalamayınız. Rekonstitüsyondan hemen sonra NovoEight®'in kullanılması önerilir. Rekonstitüye tıbbi ürünün saklama koşulları için bkz. bölüm 6.3. Daha büyük bir doz gerekirse, A ile J arası adımları ek flakonlarla, flakon adaptörleri ve kullanıma hazır enjektörler ile tekrarlayınız. Rekonstitüye çözeltinin uygulanması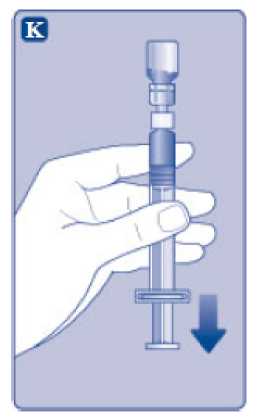 K) Piston kolunu tamamen içe basılı halde tutunuz. Flakon baş aşağı bakacak şekilde enjektörü çeviriniz. Piston kolunu itmeyi durdurunuz ve kendiliğinden gerihareket etmesine izin veriniz; bu esnada rekonstitüyeçözelti enjektörü dolduracaktır. Rekonstitüye çözeltiyienjektöre çekmek için piston kolunu hafifçe aşağıçekiniz. Eğer dolu flakonun sadece bir kısmına ihtiyacınız varsa, doktorunuz veya hemşireniz tarafından size anlatıldığıgibi, rekonstitüye çözeltiden ne kadar çektiğinizi görmekiçin enjektör üzerindeki ölçeğe bakınız. Hava kabarcıkları varsa bunların üst kısma yükselmesini sağlamak için, flakonu baş aşağı tutarken enjektöreparmaklarınızın ucuyla hafifçe vurunuz. Tüm havakabarcıkları kaybolana kadar piston kolunu itiniz. 17 L) 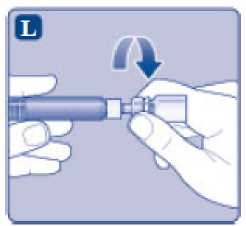 Flakon adaptörünü flakon ile çevirerek açınız. NovoEight® artık enjeksiyon için hazırdır. Uygun bir yer belirleyiniz ve NovoEight®'i damar içine, 2-5 dakikalık bir sürede yavaşça enjekte ediniz. İmhaEnjeksiyondan sonra kullanılmamış tüm NovoEight® çözeltisini, infüzyon setiyle birlikte enjektörü, flakon adaptörü ile birlikte flakonu ve diğer atık maddeleri eczacınızın tarif ettiğişekilde atınız. Olağan ev atıkları ile birlikte atmayınız. 7. RUHSAT SAHİBİNovo Nordisk Sağlık Ürünleri Tic. Ltd. Şti. Faks: 0 212 282 21 20 8. RUHSAT NUMARASI/NUMARALARI2018/155 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 15.03.2018 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ18
|
İlaç BilgileriNovoeight 1500 Iu Enjeksiyonluk Çözelti Hazırlamak İçin Toz ve ÇözücüEtken Maddesi: Turoktokog Alfa (rekombinant İnsan Koagülasyon Faktörü Viii) Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.