Arixtra 2,5 Mg /0,5 Ml Enjeksiyonluk Solüsyon Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADIARİXTRA® 2,5 mg/0,5 ml Enjeksiyonluk Çözelti Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:0,5 ml içinde: Fondaparinuks sodyum...............2,5 mg Yardımcı madde(ler):Sodyum klorür........4,2 mg Diğer yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti Subkutan ve intravenöz kullanım için berrak ve renksiz enjeksiyonluk çözelti 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarKalça kırığı, majör diz ameliyatı ya da kalça protezi ameliyatı gibi majör ortopedik cerrahi işlem geçiren yetişkinlerde Venöz Tromboembolik Olayların (VTE) önlenmesinde Abdominal kanser ameliyatına giren hastalar gibi, abdominal cerrahide derin ven trombozu (DVT) riski yüksek yetişkinlerde VTE'nin önlenmesinde (bkz. Bölüm 5.1) VTE açısından yüksek risk altında olduğu düşünülen ve kalp yetmezliği ve/veya akut solunum bozuklukları gibi akut hastalıklar ve/veya akut enfeksiyöz veya enflamatuvar hastalık nedeniyleimmobilize olan yetişkin medikal hastalarda VTE'nin önlenmesinde. Kararsız anjina ya da ST yükselmesi olmayan miyokard infarktüsünün (UA/NSTEMI) tedavisinde acil invazif girişim (<120 dk) endikasyonu olmayan yetişkinlerde (bkz. Bölüm 4.4 veBölüm 5.1). ST yükselmesi olan miyokard infarktüslü (STEMI) yetişkinlerde trombolitik tedavi ya da önceden reperfüzyon tedavisinin hiçbir formunu almamış yetişkinlerde Alt ekstremitelerde derin ven trombozunun eşlik etmediği akut semptomatik spontan yüzeyel ven trombozu görülen yetişkinlerin tedavisinde (bkz. Bölüm 4.2 ve 5.1) endikedir. 4.2. Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:Ortopedik ve abdominal cerrahi hastalarıÖnerilen ARİXTRA® dozu, postoperatif olarak subkutan enjeksiyonla günde bir kez uygulanan 2,5 mg'dır. İlk doz, cerrahi girişimin sonlanmasını takiben 6 saatten daha erken verilmemelidir ve ilaç mutlaka hemostazın sağlanmasını takiben uygulanmalıdır. Tedaviye ameliyat sonrası, venöz tromboemboli riski ortadan kalkıncaya ve genellikle hasta ayağa kalkıp yürüyebilir hale gelene kadar en az 5-9 gün devam edilmelidir. Deneyimler VTE 1 riskinin, kalça kırığı cerrahisinden 9 gün sonra hala devam ettiğini göstermiştir. Bu hastalarda, ek olarak 24 güne kadar ARIXTRA® ile uzatılmış profilaksi düşünülmelidir (bkz. Bölüm 5.1). Bireysel risk değerlendirmesine göre tromboembolik komplikasyonlar açısından yüksek risk altında olan medikal hastalardaÖnerilen fondaparinuks dozu günde bir defa subkutan enjeksiyonla uygulanan 2,5 mg'dir. Medikal hastalarda klinik olarak 6-14 günlük bir tedavi süresi incelenmiştir (bkz. Bölüm 5.1.). Kararsız anjina/ST yükselmesi olmayan miyokard infarktüsü (UA/NSTEMI) tedavisiÖnerilen ARİXTRA® dozu, subkutan enjeksiyonla günde bir kez uygulanan 2,5 mg'dır. Tedavi, tanı konduktan sonra mümkün olduğunca çabuk başlatılmalı ve 8 gün kadar ya da hasta taburcuolana kadar devam ettirilmelidir. Hastaya ARİXTRA® kullanımı sırasında perkütan koroner girişim (PCI) uygulanacak ise PCI sırasında standart uygulamaya göre heparin (unfraksiyone heparin (UFH)), son ARİXTRA®dozunun uygulanmasından sonra geçen zaman dahil, kanama gelişme riski göz önündebulundurularak uygulanmalıdır (bkz. Bölüm 4.4). Kılıf çekilmesi tamamlandıktan sonrasubkutan ARİXTRA® tedavisine tekrar ne zaman başlanacağı, klinik karara bağlıdır.UA/NSTEMI klinik çalışmasında ARİXTRA® tedavisine kılıf çekilmesi tamamlandıktan sonra,2 saat geçmeden tekrar başlanmamıştır. ST segment yükselmesi olan miyokard infarktüsü (STEMI) tedavisiÖnerilen ARİXTRA® dozu günde bir kez 2,5 mg'dir. İlk ARİXTRA® dozu intarvenöz yolla, daha sonraki dozlar subkutan yolla uygulanır. Tedavi, tanı konduktan sonra mümkün olduğuncaçabuk başlatılmalı ve 8 gün kadar ya da hasta taburcu olana kadar devam ettirilmelidir. Hastaya ARİXTRA® kullanımı sırasında PCI uygulanacak ise PCI sırasında standart uygulamaya göre unfraksiyone heparin (UFH), son ARİXTRA® dozunun uygulanmasından sonra geçenzaman dahil, kanama gelişme riski göz önünde bulundurularak uygulanmalıdır ( bkz. Bölüm4.4).Kılıf çekilmesi tamamlandıktan sonra subkutan ARİXTRA® tedavisine tekrar ne zamanbaşlanacağı, klinik karara bağlıdır. STEMI klinik çalışmasında ARİXTRA® tedavisine kılıfçekilmesi tamamlanmasını takiben 3 saat geçmeden tekrar başlanmamıştır.Koroner arter bypass grefti (CABG) yapılacak hastalarKoroner arter bypass grefti (CABG) yapılacak STEMI veya UA/NSTEMI hastalarında mümkünse ameliyattan önceki 24 saat içinde ARİXTRA® verilmemelidir. Ameliyattan 48 saatsonra ARİXTRA®'ya tekrar başlanabilir. Yüzeyel ven trombozunun tedavisiÖnerilen fondaparinuks dozu günde bir defa subkutan enjeksiyonla uygulanan 2,5 mg'dir. Fondaparinuks 2,5 mg tedavisi için uygun olan hastalarda alt ekstremitelerde ultrason incelemesiveya başka bir objektif yöntemle belgelenmiş en az 5 cm uzunluğunda akut, semptomatik, izole,spontan yüzeyel ven trombozu bulunmalıdır. Tanı konmasını ve eşzamanlı DVT veya safeno-femoral bileşkenin 3 cm yakınında yüzeyel ven trombozu varlığının dışlanmasını takiben tedavimümkün olan en kısa süre içinde başlatılmalıdır. Tedavi en az 30 gün süresince vetromboembolik komplikasyonlar açısından yüksek risk altında olan hastalarda en fazla 45 günsüresince devam ettirilmelidir (bkz. Bölüm 4.4 ve 5.1). Hastaların kendi kendine enjeksiyonyapmak konusunda istekli ve yeterli olduğunun düşünülmesi durumunda hastalara bu yöndeöneride bulunulabilir. Doktorlar kendi kendine enjeksiyon konusunda açık talimatlar vermelidir. Cerrahi veya başka bir invazif prosedürün uygulanacak olan hastalarCerrahi veya başka bir invazif prosedürün uygulanacak olduğu yüzeyel ven trombozu hastalarında mümkün olduğunda fondaparinuks operasyondan önceki 24 saat içinde 2 uygulanmamalıdır. Fondaparinuks, hemostaz sağlanmış olması koşuluyla, operasyondan en az 6 saat sonra yeniden başlatılabilir. Uygulama şekli: Subkutan uygulama ARİXTRA®, hasta yatar pozisyondayken derin subkutan enjeksiyonla uygulanır. Subkutan enjeksiyon için sağ ve sol anterolateral ile sağ ve sol posterolateral karın duvarları dönüşümlükullanılmalıdır. Kullanıma hazır enjektör ile uygulama yapılırken tıbbi ürünün kaybını önlemekiçin enjeksiyondan önce enjektörün içinde bulunan hava kabarcığı dışarı çıkarılmamalıdır. İğne,başparmak ve işaret parmağı arasında tutulan deri boğumuna dikey olarak tümüyle batırılmalıdır.Deri boğumunun tutulması enjeksiyon boyunca sürdürülmelidir. İntravenöz uygulama (STEMI hastalarında sadece ilk doz) İntravenöz uygulama, mevcut bir intravenöz yolla doğrudan veya küçük hacimli (25 veya 50 ml'lik) % 0,9'luk serum fizyolojik seti kullanılarak yapılmalıdır. Tıbbi ürün kaybını önlemekamacıyla, kullanıma hazır enjektörün içindeki hava kabarcığını enjeksiyon öncesinde dışarıçıkarmayınız. Tıbbi ürünün tamamının verildiğinden emin olmak için enjeksiyontamamlandıktan sonra infüzyon kateteri serum fizyolojik ile yıkanmalıdır. Uygulama küçükhacimli serum fizyolojik seti ile yapılacaksa, infüzyon 1-2 dakikalık sürede yapılmalıdır. Kullanıma ve imhaya ilişkin ilave talimatlar için bölüm 6.6'ya bakınız. Özel popülasyonlara ilişkin ek bilgiler:Cerrahiyi takiben VTE 'nin önlenmesiCerrahi operasyona giren hastalar söz konusu olduğunda >75 yaşındaki ve/veya vücut ağırlığı <50 kg olan ve/veya 20 ila 50 ml/dak arasında değişen kreatinin klirensiyle böbrek yetersizliğibulunan hastalarda ilk fondaparinuks enjeksiyonunun zamanlamasına katı şekilde uyulmasıgerekir. İlk fondaparinuks uygulaması cerrahi kapatmayı takiben 6 saat geçmeden önce yapılmamalıdır. Hemostaz sağlanmadığı takdirde enjeksiyon yapılmamalıdır (bkz. Bölüm 4.4). Böbrek yetmezliği: VTE önlenmesi -20 ml/dak'dan düşük kreatinin klerensi olan hastalarda fondaparinukskullanımı önerilmemektedir (bkz. Bölüm 4.3). Kreatinin klirensi 20 ila 50 ml/dak aralığında olanhastalarda doz, günde bir defa uygulanan 1,5 mg'ye düşürülmelidir (bkz. Bölüm 4.4 ve 5.2).Hafif düzeyde böbrek yetersizliği bulunan (kreatinin klirensi >50 ml/dak.) hastalarda herhangibir doz azaltımı gerekmemektedir. UA/NSTEMI ve STEMI tedavisi -ARİXTRA® kreatin klerensi 20 ml/dk'dan az olanhastalarda kullanılmamalıdır (bkz. Bölüm 4.3). Kreatin klerensi 20 ml/dk veya üzerinde olanhastalarda doz ayarlaması gerekli değildir. Yüzeyel ven trombozunun tedavisi- Fondaparinuks kreatinin klirensi <20 ml/dak olanhastalarda kullanılmamalıdır (bkz. Bölüm 4,3). Kreatinin klirensi 20 ila 50 ml/dak aralığındaolan hastalarda doz, günde bir defa uygulanan 1,5 mg'ye düşürülmelidir (bkz.Bölüm 4.4 ve 5.2).Hafif düzeyde böbrek yetersizliği bulunan (kreatinin klirensi >50 ml/dak.) hastalarda herhangibir doz azaltımı gerekmemektedir. 1,5 mg'nin güvenliliği ve etkililiği araştırılmamıştır (bkz.Bölüm 4.4).Karaciğer yetmezliği: VTE'nin Önlenmesi ve UA/NSTEMI ve STEMI tedavisi -Hafif veya orta seviyeli karaciğeryetersizliği olan hastalarda herhangi bir doz ayarlaması gerekli değildir. Şiddetli karaciğeryetersizliği olan hastalarda araştırma yapılmamış olduğundan, fondaparinuks bu hasta grubundadikkatli şekilde kullanılmalıdır (bkz. Bölüm 4.4 ve 5.2). 1 Yüzeyel ven trombozunun tedavisi-Pediyatrik popülasyon:17 yaşın altındaki hastalarda ARİXTRA®'nm güvenlilik ve etkililik verisi bulunmadığından bu hastalarda kullanılması önerilmez. Düşük vücut ağırlığı: VTE önlenmesi ve UA/NSTEMI ve STEMI tedavisi-Vücutağırlığı 50 kg'ın altında olan hastalarhastalar, artmış kanama riski altındadır Fondaparinuksun eliminasyonu kilo ile birlikte azalır.Fondaparinuks bu hastalarda dikkatli şekilde kullanılmalıdır (bkz Bölüm 4.4). Yüzeyel ven trombozunun tedavisiGeriyatrik popülasyon:Cerrahi operasyona giren hastalar söz konusu olduğunda >75 yaşındaki ve/veya vücut ağırlığı <50 kg olan ve/veya 20 ila 50 ml/dak arasında değişen kreatinin klirensiyle böbrek yetersizliğibulunan hastalarda ilk fondaparinuks enjeksiyonunun zamanlamasına katı şekilde uyulmasıgerekir. 4.3. Kontrendikasyonlar- ARİXTRA® veya bileşenlerinden herhangi birine karşı aşırı duyarlılığı olanlarda. - Klinik yönden anlamlı aktif kanama. - Akut bakteriyel endokardit. - Kreatinin klerensi < 20 ml/dak ile tanımlı şiddetli böbrek yetmezliği. 4.4. Özel kullanım uyarıları ve önlemleriARİXTRA® intramüsküler yolla uygulanmamalıdır. KanamaARİXTRA®, konjenital ya da kazanılmış kanama bozuklukları (örn trombosit sayısı <50,000/mm1), aktif ülseratif gastrointestinal hastalık ve yakın zamanda geçirilmiş intrakranial hemoraji ya da beyin, omurilik ya da göz ameliyatı olan kanama riski artmışhastalarda ve aşağıda tanımlanan özel hasta gruplarında dikkatli kullanılmalıdır. VTE önlenmesiKanama riskini artırabilecek ilaçlar ARİXTRA® ile eşzamanlı kullanılmamalıdır. Bu ilaçlar desirudin, fibrinolitik ilaçlar, GP IIb/IIIa reseptör antagonistleri, heparin, heparinoidler ya daDüşük Moleküler Ağırlıklı Heparini (LMWH) içerir. Gerektiğinde K vitamini antagonistleri ilebölüm 4.5'teki bilgi doğrultusunda eşzamanlı tedavi uygulanabilir; diğer antitrombosit tıbbiürünler (asetilsalisilik asit, dipiridamol, sülfinpirazon, tiklopidin ya da klopidogrel) ve NSAİilaçlar dikkatli kullanılmalıdır. Eşzamanlı kullanılması gerektiğinde yakın izlem gerekir. UA/NSTEMI ve STEMI tedavisiFondaparinuks eşzamanlı olarak kanama riskini artıran diğer ilaçlar (GPIIb/IIIa inhibitörleri ya da trombolitikler gibi) kullanan hastalarda dikkatli kullanılmalıdır. Yüzeyel ven trombozunun tedavisi içinFondaparinuks kanama riskini arttıran başka tıbbi ürünlerle eşzamanlı şekilde tedavi uygulanan hastalarda dikkatli kullanılmalıdır. 4 PCI ve kılavuz kateter trombüs riskiSTEMI'de, primer PCI geçiren hastalarda, PCI öncesinde ve sırasında fondaparinuks kullanımı önerilmemektedir. Benzer şekilde, acil revaskülarizasyon gerektiren yaşamı tehdit edicidurumların görüldüğü UA/NSTEMI hastalarında PCI öncesinde ve sırasında fondaparinukskullanımı önerilmemektedir. Bunlar dinamik ST sapması, kalp yetmezliği, yaşamı tehdit ediciaritmiler veya hemodinamik dengesizlik ile ilişkili refrakter veya rekürren anjina hastalarıdır. Kılavuz kateter trombüsü riskinin artmış olmasından dolayı, primer dışı PCI uygulanan UA/NSTEMI ve STEMI hastalarında PCI sırasında tek antikoagülan olarak fondaparinukskullanımı önerilmemektedir (bkz. klinik çalışmalar, bölüm 5.1). Bu nedenle, primer olmayanPCI sırasında standart uygulamaya uygun şekilde ek tedavi olarak UFH kullanılmalıdır (bkz.pozoloji, bölüm 4.2). Yüzeyel ven trombozu görülen hastalarFondaparinuks tedavisi başlatılmadan önce safeno-femoral bileşkenin 3 cm'den daha fazla uzağında yüzeyel ven trombozu varlığı doğrulanmalıdır ve kompresyonlu ultrason veya objektifyöntemlerle eşzamanlı DVT dışlanmalıdır. Eşzamanlı DVT veya safeno-femoral bileşkenin 3 cmyakınında yüzeyel ven trombozu görülen yüzeyel ven trombozu hastalarında 2,5 mgfondaparinuks kullanımına ilişkin veri mevcut değildir (bkz. Bölüm 4.2 ve 5.1). Şu gruplarda 2,5 mg fondaparinuksun güvenliliği ve etkililiği araştırılmamıştır: skleroterapiyi takiben veya bir intravenöz yol komplikasyonu olarak ortaya çıkan yüzeyel ven trombozugörülen hastalar, önceki 3 ay içinde yüzeyel ven trombozu öyküsüne sahip hastalar, önceki 6 ayiçinde venöz tromboembolik hastalık öyküsüne sahip olan hastalar veya aktif kanser görülenhastalar (bkz. Bölüm 4.2 ve 5.1). Spinal / epidural anesteziParaliz ile sonuçlanabilecek epidural veya spinal hematomlara yol açabilir. Majör ortopedik cerrahi uygulanacak olan hastalarda eşzamanlı fondaparinuks ve spinal/epidural anestezi veyaspinal ponksiyon kullanımı ile birlikte uzun süreli veya kalıcı felce yol açabilecek epidural veyaspinal hematom olasılığı dışlanamaz. Seyrek görülen bu olayların gelişme riski, postoperatifindwelling epidural kateter kullanımı veya hemostazı etkileyen diğer tıbbi ürünlerin eşzamanlıkullanımıyla daha yüksek olabilir. Yaşlı hastalarYaşlı popülasyon kanama açısından artmış risk altındadır. Böbrek fonksiyonları genellikle yaşla azaldığı için, bu hastalarda eliminasyon azalır ve ARIXTRA® maruziyeti artabilir. ARIXTRA®yaşlı hastalarda dikkatli kullanılmalıdır (bkz.Pozoloji ve uygulama şekli).Düşük vücut ağırlığı VTE'nin Önlenmesi ve UA/NSTEMI ve STEMI tedavisi -(bkz.Pozoloji ve uygulama şekli). Yüzeyel ven trombozunun tedavisi -Vücut ağırlığı 50 kg'nin altında olan hastalarda yüzeyelven trombozu tedavisi için fondaparinuks kullanımına ilişkin klinik veri mevcut değildir. Bunedenle fondaparinuks bu hastalarda yüzeyel ven trombozu tedavisi için önerilmemektedir (bkz.Bölüm 4.2).Böbrek bozukluğuFondaparinuks başlıca böbrekler vasıtasıyla vücuttan uzaklaştırılır. VTE Profilaksisi -Kreatinin klirensi <50 ml/dak olan hastalarda kanama ve VTE riski 2artmıştır ve bu hastalara dikkatli şekilde tedavi uygulanmalıdır (bkz. Bölüm 4.2, 4.3 ve 5.2). Kreatinin klirensi 30 ml/dakikanın altında olan hastalarda sınırlı klinik veri mevcuttur. UA/NSTEMI ve STEMI tedavisi için -(bkz.Pozoloji ve uygulama şekli; Farmakokinetik özellikler). Yüzeyel ven trombozunun tedavisi -Fondaparinuks kreatinin klirensi <20 ml/dak olanhastalarda kullanılmamalıdır (bkz. Bölüm 4,3). Kreatinin klirensi 20 ila 50 ml/dak aralığındaolan hastalarda doz, günde bir defa uygulanan 1,5 mg'ye düşürülmelidir (bkz. Bölüm 4.2 ve 5.2).1,5 mg'nin güvenliliği ve etkililiği araştırılmamıştır.Şiddetli karaciğer bozukluğu VTE'nin Önlenmesi ve UA/NSTEMI ve STEMI tedavisi- Fondaparinuks dozunun ayarlamasıgerekli değildir. Bununla birlikte, şiddetli karaciğer bozukluğu bulunan hastalarda koagülasyonfaktörlerinin eksikliğinden kaynaklanan artmış kanama riski nedeniyle, fondaparinuks kullanımıdikkatle değerlendirilmelidir (bkz. Bölüm 4.2). Yüzeyel ven trombozunun tedavisi -Şiddetli karaciğer bozukluğu bulunan hastalarda yüzeyelven trombozu tedavisi için fondaparinuks kullanımına ilişkin klinik veri mevcut değildir. Bunedenle fondaparinuks bu hastalarda yüzeyel ven trombozu tedavisi için önerilmemektedir (bkz.Bölüm 4.2).Heparin ile indüklenen trombositopeniARlXTRA® HIT öyküsü olan hastalarda ilaç dikkatli kullanılmalıdır. ARIXTRA®'nın HIT tip II hastaları üzerindeki etkililik ve güvenliliği ile ilgili çalışmalar yapılmamıştır. ARIXTRA®trombosit faktör 4'e bağlanmaz ve heparin ile indüklenen trombositopeni (HIT) tip IIhastalarının serumları ile çapraz reaksiyon vermez. Bununla birlikte fondaparinuks ile tedaviedilen hastalarda seyrek HIT bildirimi mevcuttur. Lateks alerjisiKullanıma hazır enjektörün iğne koruyucusu, latekse duyarlılığı olan kişilerde ciddi alerjik reaksiyonlara neden olma potansiyeline sahip olan kuru doğal lateks lastik içerebilir. ARİXTRA®, her dozunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında sodyum içermez. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriFondaparinuks ve hemoraji riskini artırabilen ilaçlarla eşzamanlı kullanımda kanama riski artar (bkz. Bölüm 4.4). Oral antikoagülanlar (varfarin), asetilsalisilik asit (trombosit inhibitörü), NSAI (piroksikam) ve digoksin fondaparinuksun farmakokinetiğini etkilememiştir. Etkileşim çalışmalarındakullanılmış olan fondaparinuks dozu (10 mg) mevcut endikasyonlar için önerilen dozdanyüksektir. Fondaparinuks ne varfarinin Uluslararası Normalleştirilmiş Oran (INR) aktivitesini, neasetilsalisilik asit veya piroksikam tedavisi altında kanama zamanını, ne de kararlı durumdakidigoksin farmakokinetiğini etkilememiştir. Başka bir antikoagülan tıbbi ürünle takip tedavisiHeparin ya da düşük moleküler ağırlıklı heparin ile izlem tedavisine başlanırken, genel kural olarak ilk enjeksiyon son fondaparinuks enjeksiyonundan bir gün sonra yapılmalıdır. 3 Bir K Vitamini antagonisti ile izlem tedavisi gerektiğinde, fondaparinuks tedavisi hedef INR değerlerine ulaşılana dek devam ettirilmelidir. Özel popülasyonlara ilişkin ek bilgiler Böbrek / Karaciğer yetmezliği:Böbrek / karaciğer yetmezliği olan hastalarda etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon :Pediyatrik popülasyonda etkileşim çalışması yapılmamıştır. Geriyatrik popülasyon:Geriyatrik popülasyonda etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiye Gebelik kategorisi:BÇocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)ARİXTRA® tedavisinin oral kontraseptifler üzerindeki etkisi çalışılmamıştır. Gebelik dönemiFondaparinuksun gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fötal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir. Fondaparinuks açık biçimde gerekli olmadıkça gebe kadınlarda kullanılmamalıdır ( bkz.Laktasyon dönemiFondaparinuksun sıçanda sütle atıldığı, ancak insanda anne sütü ile atılıp atılmadığı bilinmemektedir. ARİXTRA® tedavisi süresince emzirme tavsiye edilmemektedir. Bununlabirlikte çocuk tarafından oral absorbsiyonun gerçekleştirilmesi muhtemel değildir. Üreme yeteneği /FertiliteFondaparinuksun insanlarda fertilite üzerindeki etkilerine ilişkin herhangi bir veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar fertilite üzerinde herhangi bir etki ortayakoymamaktadır. 4.7. Araç ve makine kullanımı üzerindeki etkilerAraç veya makine kullanma yeteneği üzerindeki etkisi ile ilgili herhangi bir çalışma yapılmamıştır. 4.8. İstenmeyen etkilerFondaparinuksla en yaygın şekilde bildirilen ciddi advers reaksiyonlar kanama komplikasyonları (seyrek intrakraniyal/ intraserebral ve retroperitoneal kanama vakalarını içeren çeşitli durumlar)ve anemidir. Fondaparinuks kanama riskinin artmış olduğu hastalarda dikkatli şekildekullanılmalıdır (bkz. Bölüm 4.4). VTE'nin önlenmesine yönelik uygulamada, Aadvers reaksiyonlar sistem organ sınıflamasına, sıklığına ve endikasyona göre aşağıda sıralanmıştır. Advers reaksiyonlar sıklığa göre şöylesınıflandırılmıştır: Çok yaygın (> 1/10), yaygın (> 1/100, < 1/10), yaygın olmayan (> 1/1000, <1/100), seyrek (> 1/10000, < 1/1000), çok seyrek (<1/10000), bilinmiyor (eldeki verilerdenhareketle tahmin edilemiyor olarak tanımlanmıştır. Bu advers etkiler, endikasyonların cerrahi yada tıbbi içeriği ile bağlantılı olarak yorumlanmalıdır. 7 Fondaparinuks 2,5 mg güvenliliği 9 güne kadar alt ekstremite majör ortopedik cerrahisi uygulanan 3.595 hastada, bir haftalık başlangıç profilaksisi ardından 3 hafta tedavi edilen 327kalça kırığı ameliyatı geçirmiş hastada, 9 güne kadar tedavi edilen 1.407 abdominal cerrahihastasında ve 14 güne kadar tedavi edilen tromboembolik komplikasyon riski olan 425 medikalhastada, UA veya NSTEMI ACS tedavisi görmekte olan 10.057 hastada ve STEMI ACS tedavisigörmekte olan 6.036 hastada değerlendirilmiştir. Enfeksiyonlar ve enfestasyonlarSeyrek: Post-operatif yara enfeksiyonları.(A) Kan ve lenf sistemi hastalıklarıYaygın: Anemi, post operatif kanama, (A) Yaygın: Kanama (hematom, hematüri, hemoptizi, dişeti kanaması) (B) Yaygın olmayan: Anemi (B) Bağışıklık sistemi hastalıklarıSeyrek: Alerjik reaksiyon (anjiyoödem ve anaflaktoid/anaflaktik reaksiyon üzerine çok seyrek olarak bildirilenler dahil) (A) Seyrek: Alerjik reaksiyon (anjiyoödem ve anaflaktoid/anaflaktik reaksiyon üzerine çok seyrek olarak bildirilenler dahil) (B) Metabolizma ve beslenme hastalıklarıSeyrek: Hipokalemi (A) Sinir sistemi hastalıklarıSeyrek: Anksiyete, konfüzyon, sersemlik, uyku hali, baş dönmesi, baş ağrısı (A) Vasküler hastalıklarSeyrek: Hipotansiyon (A) Solunum, göğüs bozuklukları ve mediastinal hastalıklarSeyrek: Dispne, öksürük (A) Yaygın olmayan: Dispne (B) Gastrointestinal hastalıklarYaygın olmayan: Bulantı, kusma (A) Seyrek: Karın ağrısı, dispepsi, gastrit, konstipasyon, diyare (A) Hepatobiliyer hastalıklarYaygın olmayan: Karaciğer fonksiyon testlerinde anormallik, karaciğer enzimlerinde artış (A) Seyrek: Bilirubinemi (A) Deri ve deri altı doku hastalıklarıYaygın olmayan: Döküntü, kaşıntı (A) (B) Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarYaygın olmayan: Ateş, ödem, periferik ödem, ödem, yara sekresyonu (A) Seyrek: Göğüs ağrısı, bacak ağrısı, bitkinlik, yüzde ani kızarma, sıcak basması, senkop, genital ödem. (A) Yaygın olmayan: Göğüs ağrısı (B) 4 (A) : Alt ekstremitelerde majör ortopedik cerrahi ve/veya abdominal cerrahi uygulanan hastalardagözlenen advers reaksiyonlar (B) : Medikal hastalarda gözlenen advers reaksiyonlar Diğer çalışmalarda veya pazarlama sonrası deneyimde, seyrek intrakraniyal / intraserebral ve retroperitoneal kanama vakaları bildirilmiştir. ACS programında bildirilen advers olay profili, VTE profilaksisi için tanımlanan advers ilaç reaksiyonları ile tutarlıdır. Kanama, UA/NSTEMI ve STEMI hastalarında sık bildirilen bir olay olmuştur. Karara bağlanmış majör kanama insidansı Faz III UA/NSTEMI çalışmasında 9. güne kadar (9. gün dahil) %2,1'e(fondaparinuks) karşılık %4,1 (enoksaparin) olmuştur ve modifiye TIMI kriterlerine göre birleşikşiddetli hemoraji insidansı Faz III STEMI çalışmasında 9. güne kadar (9. gün dahil) %1,1'e(fondaparinuks) karşılık %1,4 (kontrol [UFH/plasebo]) olmuştur. Faz III UA/NSTEMI çalışmasında en yaygın şekilde bildirilmiş olan kanama dışı advers olaylar (fondaparinuks alan gönüllülerin en az %1'inde bildirilmiş) baş ağrısı, göğüs ağrısı ve atriyalfibrilasyondur. STEMI hastaları üzerinde gerçekleştirilen Faz III çalışmada en yaygın şekilde bildirilmiş olan kanama dışı advers olaylar (fondaparinuks alan gönüllülerin en az %1'inde bildirilmiş) atriyalfibrilasyon, pireksi, göğüs ağrısı, baş ağrısı, ventriküler taşikardi, kusma ve hipotansiyondur. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr;[email protected]. Doz aşımı ve tedavisiBelirti ve bulgularTavsiye edilenin üzerindeki ARIXTRA® dozları kanama riskinde artışa yol açabilir. TedaviFondaparinuksun bilinen bir antidotu yoktur. Doz aşımına bağlı kanama komplikasyonu görüldüğünde tedavi durdurulmalı ve birincil neden araştırılmalıdır. Cerrahi hemostaz, kantransfüzyonu, taze plazma transfüzyonu, plazmaferez gibi uygun bir tedaviye başlanmasıdüşünülmelidir. 5. FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antitrombotik ilaçlar ATC Kodu: B01AX05 Farmakodinamik özellikler Fondaparinuks aktif faktör X'un (Xa) sentetik ve selektif bir inhibitörüdür. Fondaparinuksun antitrombotik aktivitesi, Faktör Xa'nın antitrombin III (ATIII) aracılı selektif inhibisyonu ilesağlanır. ATIII'e selektif olarak bağlanan fondaparinuks, Faktör Xa'nın ATIII ilenötralizasyonunu artırır (yaklaşık 300 kat). Faktör Xa'nın nötralizasyonu, koagülasyon yolağınıbloke eder ve hem trombin oluşumunu hem de trombüs gelişimini önler. Fondaparinuks 9 trombini (aktif Faktör II) inaktive etmez ve trombosit fonksiyonları üzerinde bilinen etkisi yoktur. 2,5 mg dozunda fondaparinuks, plazmada aktif parsiyel tromboplastin zamanı (aPTT), aktif pıhtılaşma zamanı (aPT)veya protrombin zamanı (PT)/(INR) testleri gibi rutin koagülasyontestlerini, kanama zamanını veya fibrinolitik aktiviteyi etkilemez. Bununla birlikte, 2,5 mg dozdayükselmiş aPPT seyrek spontan raporları alınmıştır. Fondaparinuks, heparinin indüklediği trombositopeni (HIT) hastalarının serumlarıyla çapraz reaksiyona girmez. Bununla birlikte, fondaparinuks ile tedavi gören hastalarda HIT seyrekspontan raporları alınmıştır. 5.2. Farmakokinetik özelliklerGenel özelliklerEmilim:Subkutan uygulamadan sonra fondaparinuks tamamen ve hızlıca absorbe edilir (mutlak biyoyararlanım %100). Sağlıklı genç bireylerde 2,5 mg fondaparinuksun tek subkutanuygulamasını takiben, doruk plazma konsantrasyonuna, ortalama Cmaks 0,34 mg/L yaklaşık 2 saatsonra ulaşılmıştır. Ortalama Cmaks değerinin yarısı düzeyindeki plazma konsantrasyonlarınauygulamadan 25 dakika sonra ulaşılır.Günde bir kez yapılan subkutan uygulamayı takiben kararlı durum plazma seviyesi, Cmaks ve EAA'da 1,3 kat artışla 3 - 4 günde elde edilmiştir. Subkutan yolla günde bir kez 2,5 mg fondaparinuks kullanan, kalça protezi cerrahisi geçiren hastalarda: Cmaks (mg/l)- 0,39 (%31), Tmaks (saat) - 2,8 (%18) ve Cmin (mg/l) -0,14 (%56)bulunmuştur. Kalça kırığı hastalarında ileri yaşları ile ilişkili olarak fondaparinuks kararlı durumplazma konsantrasyonu Cmaks (mg/l) - 0,50 (%32) ve Cmin (mg/l) - 0,19 (%58) bulunmuştur. Dağılım:Fondaparinuksun dağılım hacmi sınırlıdır (7 - 11 litre). Fondaparinuks in vitrokoşullarda antitrombin proteine yüksek oranda (0,5 ila 2 mg/l konsantrasyon aralığından %98,6 ila %97,0) ve spesifik olarak bağlanır vet. Trombosit Faktör 4'ü (PF4) ya da diğer plazmaproteinlerine önemli düzeyde bağlanmaz.Fondaparinuks ATIII'den başka plazma proteinlerine önemli düzeyde bağlanmadığından, proteinlere bağlanan diğer tıbbi ürünler ile bir etkileşme beklenmez. Biyotransformasyon:Tam olarak değerlendirilmemiş olmakla birlikte, fondaparinuksun metabolize olduğuna ya da aktif metabolit oluşumuna ilişkin kanıt bulunmamaktadır. Fondaparinuks in vitro koşullarda CYP450 (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ya da CYP3A4) inhibisyonu yapmaz. Bu nedenle fondaparinuksun in vivo koşullardaCYP aracılı metabolizma inhibisyonu nedeniyle diğer tıbbi ürünlerle etkileşmesi beklenmez. Eliminasyon:Sağlıklı genç bireylerde eliminasyon yarı ömrü yaklaşık 17 saat ve sağlıklı yaşlı bireylerde ise yaklaşık 21 saattir. Fondaparinuksun % 64 ila %77'si böbreklerden değişmemiş olarak atılır. Doğrusallık/Doğrusal olmayan durum:Yaşlı sağlıklı bireylerde, subkutan yolla uygulanan 2-8 mg fondaparinuksun farmakokinetiği doğrusaldır. 10 Hastalardaki karakteristik özelliklerPediyatrik hastalar:Fondaparinuks bu popülasyonda VTE'nin önlenmesi veya yüzeyel ven trombozu ya da akut koroner sendrom (ACS) tedavisi açısından araştırılmamıştır. Geriyatrik hastalar:Böbrek fonksiyonu yaşla birlikte azalabilir ve dolayısıyla yaşlılarda fondaparinuksa ilişkin eliminasyon kapasitesi düşebilir. Ortopedik cerrahi uygulanan 75 yaşından büyük hastalardahesaplanan plazma klirensi, 65 yaş altı hastalardakinden 1,2 ila 1,4 kat daha düşük bulunmuştur. Böbrek bozukluğu:Böbrek işlevleri normal (kreatinin klerensi > 80 ml/dak.) olan hastalarla karşılaştırıldığında, plazma klerensi hafif böbrek bozukluğu (kreatinin klerensi 50 - 80 ml/dak.) olan hastalarda 1,2 -1,4; orta derecede böbrek bozukluğu (kreatinin klerensi 30 - 50 ml/dak.) olan hastalarda ortalama2 kat düşmüştür. Şiddetli böbrek bozukluğu olan (kreatinin klerensi < 30 ml/dak.) hastalardaplazma klerensi normal böbrek işlevine sahip hastalara göre yaklaşık 5 kat düşmüştür. İlişkiliterminal yarı ömür orta dereceli böbrek bozukluğu olan hastalarda 29 saat, şiddetli böbrekbozukluğu olan hastalarda 72 saat olmuştur. Cinsiyet:Vücut ağırlığı için doz ayarlaması yapıldıktan sonra cinsiyet ile ilgili herhangi bir farklılık gözlenmemiştir. Irk:Irka bağlı farmakokinetik farklılıklar prospektif olarak araştırılmamıştır. Bununla birlikte, sağlıklı beyaz ırk bireyleri ile yapılan çalışmalar, Asyalı (Japonlar) sağlıklı bireyler ile yapılançalışmalarla karşılaştırıldığında farklı bir farmakokinetik profil göstermemiştir. Benzer şekilde,ortopedik cerrahi geçiren hastalar ile yürütülen popülasyon farmakokinetiği analizlerinedayanarak, siyah ve beyaz ırk arasında plazma klerens farklılığı gözlenmemiştir. Vücut ağırlığı:Fondaparinuksun plazma klirensi vücut ağırlığıyla birlikte artar (her 10 kg'de %9 artış). Karaciğer bozukluğu:Orta dereceli karaciğer bozukluğu olan hastalarda (Child-Pugh Kategori B) tek bir subkutan fondaparinuks dozunu takiben, toplam (yani bağlı ve bağlı olmayan) Cmaks ve EAA değerleri,normal karaciğer fonksiyonuna sahip gönüllülerdekine kıyasla sırasıyla %22 ve %39 azalmıştır.Fondaparinuksun düşük plazma konsantrasyonları, karaciğer bozukluğu olan hastalarda plazmaATIII konsantrasyonlarının düşük olmasına bağlı olarak ATIII'ye bağlanmanın azalmasına vedolayısıyla fondaparinuksun renal klirensinin artmasına bağlanmıştır. Sonuç olarak, hafif ila ortadereceli karaciğer bozukluğu olan hastalarda fondaparinuksun bağlı olmayan konsantrasyonlarınındeğişmemesi beklenir ve dolayısıyla farmakokinetiğe dayalı doz ayarlaması gerekmez. Karaciğer bozukluğu olan hastalarda fondaparinuks farmakokinetiği ile ilgili bir çalışma yapılmamıştır (bkz. Bölüm 4.2. ve 4.4.). Klinik çalışmalarAlt ekstremitelerde, major ortopedik cerrahi işlem geçiren hastalarda 9 gün kadar süren tedaviyle venöz tromboembolinin (VTE) önlenmesi:Fondaparinuks klinik programı kalça kırığı, majör diz cerrahisi ya da kalça protezi gibi majör ortopedik cerrahi uygulanan hastalarda proksimal ve distal DVT ve pulmoner emboli (PE) gibivenöz tromboembolinin(VTE) önlenmesinde fondaparinuksun etkililiğini göstermek üzeretasarlanmıştır. Kontrollü Faz II ve Faz III klinik çalışmalarda en az 8000 hasta (kalça kırığı -1,711, kalça protezi - 5,829, majör diz cerrahisi - 1,367) çalışılmıştır. Ameliyattan 6-8 saat sonragünde tek doz 2.5 mg başlanan fondaparinuks, ameliyattan 12 saat önce günde tek doz 40 mg yada ameliyattan 12-24 saat sonra günde iki kez 30 mg başlanan enoksaparin ile karşılaştırılmıştır. 11 Bu çalışmaların havuz analizinde önerilen fondaparinuks rejimi enoksaparine göre, ameliyattan sonra 11 güne dekameliyat tipinden bağımsız olarak VTE oranında anlamlı azalmaya yolaaçmıştır (%54 [%95 GA, %44-63]). Sonlanım noktalarının çoğuna önceden planlanmış venografiile tanı konulmuş ve esas olarak distal DVT saptanmıştır fakat proksimal DVT insidansı daanlamlı derecede azalmıştır. PE dahil olmak üzere semptomatik VTE insidansı tedavi gruplarıarasında anlamlı fark göstermemiştir. Ameliyattan 12 saat önce 40 mg enoksaparin ile karşılaştırma yapılan çalışmada önerilen doz ile tedavi edilen fondaparinuks tedavi grubundaki hastaların %2,8'inde majör kanama saptanırken,bu oran enoksaparin tedavi grubunda %2,6 bulunmuştur. 1 haftalık başlangıç profilaksisinden sonra 24 güne kadar tedavi edilen, kalça kırığı operasyonu geçiren hastalarda VTE önlenmesi:Randomize bir çift kör klinik çalışmada, 737 hastaya kalça kırığı ameliyatını takiben 7 +/- 1 gün boyunca günde bir kez 2,5 mg fondaparinuks ile tedavi uygulanmıştır. Bu sürenin sonunda 656hasta ilave 21 +/- 2 gün boyunca günde bir kez 2,5 mg fondaparinuks veya plasebo almak üzererandomize edildi. Fondaparinuks VTE'de plaseboya kıyasla belirgin bir şekilde azalma sağlamıştır (sırasıyla 77 hastaya (%35) karşı 3 hasta (% 1,4). Kaydedilen VTE olaylarının çoğunluğunu (70/80),venografik olarak tespit edilmiş semptomatik olmayan DVT vakaları oluşturmuştur.Fondaparinuks, aynı zamanda plasebo grubunda bildirilen ve ölümle sonuçlanan iki PE'yi deiçeren semptomatik VTE (DVT ve/veya PE) oranında da anlamlı bir azalma sağlamıştır[sırasıyla 1 (%0,3) ve 9 hasta (%2,7)]. Tümü cerrahi uygulanan bölgelerde ortaya çıkan veölümcül olmayan majör kanamalar, 2.5 mg fondaparinuks ile tedavi uygulanan 8 hastada (%2,4)ve plasebo uygulanan 2 (%0,6) hastada gözlenmiştir. Tromboembolik komplikasyon riskinin yüksek olduğu düşünülen abdominal kanser ameliyatı uygulanan hastalar gibi abdominal cerrahi hastalarında, Venöz Tromboembolik Olayların(VTE) önlenmesi:Çift kör bir klinik çalışmada 2.927 hasta, tek bir operasyon öncesi 2.500 IU enjeksiyon ve 2.500 IU'luk bir operasyon sonrası ilk enjeksiyonla 7+2 gün boyunca, günde bir kez 2,5mgfondaparinuks veya günde bir kez 5.000 IU dalteparin almak üzere randomize edilmiştir. Temelcerrahi bölgeleri kolonik/rektal, gastrik, hepatik, kolesistektomi veya diğer biliyer şeklindedir.Hastaların %60'ına kanser için cerrahi uygulanmıştır. Ürolojik (böbrek dışında) veya jinekolojikcerrahi, laparoskopik cerrahi veya vasküler cerrahi uygulanan hastalar çalışmaya dahiledilmemiştir. Bu çalışmada, toplam VTE insidansı fondaparinuks ile %4,6 (47/1.027), dalteparinle ise%6,1 (62/1.021) olmuştur: olasılık oranı düşüşü [%95 CI] = -%25,8 [-%49,7, %9,5]. Tedavi gruplarıarasında toplam VTE oranları açısından söz konusu olan ve istatistiksel açıdan anlamlı olmayanfark, temel olarak asemptomatik distal DVT'deki bir düşüşten kaynaklanmıştır. SemptomatikDVT insidansı tedavi grupları arasında benzerlik sergilemiştir: fondaparinuks grubunda 6hastaya (%0,4) karşılık dalteparin grubunda 5 hasta (%0,3). Kanser ameliyatı geçiren geniş hastaalt grubunda (hasta popülasyonunun %69'u), VTE oranı fondaparinuks grubunda %4,7,dalteparin grubunda ise %7,7 olmuştur. Fondaparinuks grubundaki hastaların %3,4'ünde ve dalteparin grubundaki hastaların ise %2,4'ünde majör kanama gözlenmiştir. Akut hastalığa bağlı kısıtlı mobilite nedeniyle tromboembolik komplikasyon riski yüksek olan medikal hastalarda, Venöz Tromboembolik Olayların (VTE) önlenmesi:Randomize bir çift kör klinik çalışmada, 839 hastaya 6 ila 14 gün boyunca günde bir kez 2,5 mg fondaparinuks veya plasebo uygulanmıştır. Bu çalışma, > 60 yaşında, en az dört gün boyunca 12 yatak istirahatine gereksinim uyması beklenen ve konjestif kalp yetmezliği NYHA sınıf III/IV ve/veya akut solunum yolu hastalığı ve/veya akut enfeksiyöz ya da enflamatuvar hastalıknedeniyle hastaneye yatırılan akut medikal hastaları içermiştir. Fondaparinuks, genel VTEoranını plaseboya kıyasla anlamlı düzeyde azaltmıştır [sırasıyla 18 hasta (%5,6), 34 hasta(%10,5)]. Olayların çoğunu asemptomatik distal DVT oluşturmuştur. Fondaparinuks aynı zamanda karara bağlanmış ölümcül PE oranını da anlamlı düzeyde azaltmıştır [sırasıyla 0 hasta (%0,0) ve 5 hasta (%1,2)]. Her gruptan 1 hastada (%0,2) majörkanamalar gözlenmiştir. Unstabil Anjina veya STyükselmesi olmayan miyokard infarktüsü (UA/NSTEMI) tedavisi:Çift kör randomize bir non-inferiority (farksızlık) çalışması (OASIS 5), yaklaşık 20.000 UA/NSTEMI hastasına, günde 1 kez, subkutan yolla uygulanan 2.5 mg ARIXTRA® ile günde 2kez subkutan yolla uygulanan 1 mg/kg enoksaparini karşılaştırılmıştır. Tüm hastalaraUA/NSTEMI'ya yönelik standart tıbbi tedavi uygulanmıştır ve hastaların %34'üne PCI, %9'unaise CABG yapılmıştır. Ortalama tedavi süresi fondaparinuks tedavi grubunda 5,5gün,enoksaparin tedavi grubunda 5,2 gündür. PCI yapılmış olduğu takdirde, hastalara son subkutandozun zamanlamasına ve planlanan GP IIb/IIIa inhibitörü kullanımına bağlı olarak ek tedaviolarak ya intravenöz fondaparinuks (fondaparinuks hastaları) ya da ağırlığa göre ayarlanmışintravenöz UFH (enoksaparin hastaları) verilmiştir. Hastaların yaş ortalaması 67'dir ve hastalarınyaklaşık % 60'ı en az 65 yaşındadır. Hastaların yaklaşık % 40'ında ve % 17'sinde sırasıyla hafif(kreatin klerensi >50 ila <80 ml/dakika) ve orta şiddetli (kreatin klerensi >30 ila <50 ml/dakika)böbrek bozukluğu vardır. Randomizasyonu izleyen 9 gün içinde ölüm, miyokard infarktüsü (MI) veya tedaviye dirençli iskemi (RI) primer olarak karara bağlanan birleşik sonlanma noktası olarak değerlendirilmiştir.Dokuzuncu güne kadar fondaparinuks ve enoksaparin ile tedavi edilen hastaların sırasıyla %5,8'inde ve % 5,7'sinde advers olay gelişmiştir (risk oranı 1,01, % 95 GA, 0,90, 1,13, tek taraflınon-inferiority (farksızlık) p değeri = 0,003). 30. güne gelindiğinde, tüm nedenlerden kaynaklanan mortalite insidansı enoksaparin uygulanırken gözlenen %3,5'ten fondaparinuks uygulanırken gözlenen %2,9'a düşmüştür (riskoranı 0,83, %95 CI, 0,71; 0,97, p = 0,02). MI ve RI insidansı üzerindeki etkiler, fondaparinuks veenoksaparin tedavisi grupları arasında istatistiksel açıdan farklılık sergilememiştir. 9. günde fondaparinuks ve enoksaparin uygulanırken gözlenen majör kanama insidansı sırasıyla %2,1 ve %4,1 olmuştur (risk oranı 0,52, %95 CI, 0,44;0,61, p < 0,001). Majör kanama konusundaki etkililik bulguları ve sonuçları, yaşlılar, böbrek bozukluğu olan hastalar, eşzamanlı trombosit agregasyonu inhibitörlerinin tipi (aspirin, tienopiridinler veya GPIIb / IIIa inhibitörleri) gibi önceden belirlenmiş alt gruplarda tutarlılık sergilemiştir. PCI uygulanan ve fondaparinuks veya enoksaparin ile tedavi uygulanan hasta alt grubunda hastaların sırasıyla %8,8'i ve %8,2'si randomizasyonu takip eden 9 gün içinde ölüm/MI/RIyaşamıştır (risk oranı 1,08, %95 CI, 0,92; 1,27). Bu alt grupta 9. Günde fondaparinuks veenoksaparin uygulanırken gözlenen majör kanama insidansı sırasıyla %2,2 ve %4,1 olmuştur(risk oranı 0,43, %95 CI, 0,33;0,57). PCI uygulanan gönüllülerde karara bağlanmış kılavuzkateter trombüsü insidansı, fondaparinuks ile %1,0, enoksaparinle ise %0,3 olmuştur. UFH ile birlikte PCI uygulanan hastalarda kararsız anjina (UA) ya da ST yükselmesi olmayan miyokard infarktüsü (NSTEMI) tedavisi:Açık etiketli fondaparinuks ile tedavi edilen ve PCI uygulanması planlanan yüksek riskli 3235 UA/NSTEMI hastası ile yürütülen çalışmada (OASIS 8/FUTURA), PCI endikasyonu olan 2026hasta UFH doz rejimlerinden birine çift kör olarak randomize edilmiştir. Çalışmaya katılanhastalar 8 gün ya da taburcu olana dek günde bir kez 2.5 mg subkutan fondaparinuks almıştır. 13 Randomize edilen hastalara PCI'nın başlamasından hemen önce düşük doz UFH rejimi (planlanan GP nb/IIIa kullanımından bağımsız olarak 50 U/kg; ACT kılavuzu olmadan) veyastandart doz UFH rejimi (GPIIb/IIIa kullanılmadan: 85 U/kg, ACT kılavuzlu; planlananGPIIb/IIIa kullanımı: 60 U/kg, ACT kılavuzlu) uygulanmıştır.Fondaparinuks tedavisinin süresi ve başlangıç özellikleri her iki UFH grubunda benzerdi. Standart doz UFH ya da düşük doz UFH rejimine randomize edilen gönüllülerde, ortalamaUFH dozu sırasıyla 85 U/kg ve 50 U/kg olmuştur. Primer sonlanım peri-PCI (randomizasyondan PCI sonrası 48 saat) majör ya da minör kanama ya da majör vasküler erişim yeri komplikasyonlarıdır. Standart doz ve düşük doz UFH'ye randomize edilmiş olan hastalarda PCI sırasında karara bağlanmış kılavuz kateter trombüsü insidansları sırasıyla %0,1 (1/1002) ve %0,5 (5/1024)olmuştur. Randomize edilmeyen dört hastada (%0,3) koroner anjiyografi sırasında tanı kataterinde tromboz gelişmiştir. 12 (%0,37) hastada arteriyel tromboz gelişmiş ve bunlardan 7'si anjiyografi ve 5'iPCI sırasında bildirilmiştir.
STyükselmesi olan miyokard infarktüsü (STEMI) tedavisi:OASIS 6, STEMI görülen yaklaşık 12.000 hastada günde bir defa uygulanan 2,5 mg fondaparinuksun güvenliliğini ve etkililiğini olağan bakımla [plasebo (%47) veya UFH (%53)]karşılaştırmalı şekilde değerlendirmiş olan çift kör, randomize bir çalışmadır. Tüm hastalararaştırıcının tercihine bağlı olarak primer PCI ile birlikte reperfüzyon (% 31), trombolitikler (%45) ve perfüzyon (% 24) dahil standart STEMI tedavisi almışlardır. Bir trombolitikle tedaviuygulanan hastaların %84'üne fibrine spesifik olmayan bir ajanla (ağırlıklı olarak streptokinaz)tedavi uygulanmıştır. Fondaparinuksla uygulanan tedavinin ortalama süresi 6,2 gün olmuştur.Hastaların yaş ortalaması 61'dir ve bunların yaklaşık % 40'ı en az 65 yaşındadır. Bu hastalarınsırasıyla yaklaşık % 40'ında ve % 14'ünde sırasıyla hafif (kreatin klerensi >50 ila <80ml/dakika) ve orta şiddetli (kreatin klerensi >30 ila <50 ml/dakika) böbrek bozukluğu vardır. 14 Randomizasyonu izleyen 30 gün içinde ölüm ve tekrarlayan miyokard infarktüsü (re-MI) primer olarak karara bağlanan birleşik sonlanma noktası olarak değerlendirilmiştir. Otuzuncu günekadar fondaparinuks ile tedavi edilen ve kontrol grubundaki hastaların sırasıyla %%11,1'inde advers olay gelişmiştir (hazard ratio 0,86, % 95 Cl, 0,77, 0,96, p=0,008).Fondaparinuksla plasebonun karşılaştırıldığı önceden tanımlanmış basamaklandırmada [yanifibrine spesifik olmayan litiklerle tedavi uygulanan hastalar (%77,3), reperfüzyon görülmeyenler(%22), fibrine spesifik litiklerle tedavi uygulananlar (%0,3), primer PCI (%0,4)], 30. Gündeölüm/yeniden Mİ insidansı, plaseboyla gözlenen %14,0'a kıyasla anlamlı bir düşüşle %11,3olmuştur (risk oranı 0,80, %95 CI, 0,69, 0,93, p = 0,003). Fondaparinuks ile UFH'ninkarşılaştırıldığı önceden tanımlanmış basamaklandırmada [primer PCI (%58,5), fibrine spesifiklitikler (%13), fibrine spesifik olmayan litikler (%2,6) ile tedavi uygulanan hastalar vereperfüzyon görülmeyenler (%25,9)], 30. günde fondaparinuks ve UFH'nin ölüm/yeniden Mİinsidansı üzerindeki etkileri istatistiksel açıdan anlamlı farklılık sergilememiştir: sırasıyla %8,3'ekarşılık %8,7 (risk oranı 0,94, %95 CI, 0,79, 1,11, p = 0,460). Bununla birlikte, bubasamaklandırmada yer alan tromboliz yaşayan veya reperfüzyon görülmeyen belirtilmişpopülasyon alt grubunda (yani primer PCI uygulanmayan hastalarda) 30. Günde ölüm/yenidenMİ insidansı, UFH ile gözlenen %14,3'e kıyasla anlamlı bir düşüşle fondaparinuksla %11,5olmuştur (risk oranı 0,79, %95 CI, 0,64, 0,98, p = 0,03).30. günde tüm nedenlerden kaynaklanan mortalite insidansı kontrol grubunda gözlenen %8,9'dan fondaparinuks grubunda gözlenen %7,8'e düşmüştür (risk oranı 0,87, %95 CI, 0,77; 0,98, p =0,02). Mortalitedeki fark, basamak 1'de (plasebo ile karşılaştırma) istatistiksel anlamlılıksergilemiştir fakat basamak 2'de (UFH ile karşılaştırma) sergilememiştir. Mortalite konusundafondaparinuks grubunda gözlenen yararın 180. Gündeki takip periyodu bitimine kadarkorunduğu gösterilmiştir. Bir trombolitikle revaskülarizasyon uygulanan hastalarda fondaparinuks, 30. günde kontrol grubunda %13,6 olan ölüm/yeniden Mİ insidansını anlamlı düzeyde azaltarak %10,9'adüşürmüştür (risk oranı 0,79, %95 CI, 0,68;0,93, p = 0,003). Başlangıçta reperfüzyongörülmeyen hastalarda 30. Günde ölüm/yeniden Mİ insidansı kontrol grubunda %15'kenfondaparinuks grubunda %12,1 olmuştur (risk oranı 0,79, %95 CI, 0,65; 0,97, p = 0,023). PrimerPCI uygulanan hastalarda 30. Günde ölüm/yeniden Mİ insidansı iki grup arasında istastistikselfarklılık sergilememiştir (fondaparinuks grubunda %6,0, kontrol grubunda %4,8; risk oranı 1,26,%95 CI, 0,96; 1,66). Fondaparinuks ile tedavi uygulanan hastaların %1,1'i ve kontrol hastalarının %1,4'ü 9. Günde şiddetli bir kanama yaşamıştır. Bir trombolitik uygulanan hastalar arasında şiddetli kanama,fondaparinuks hastalarının %1,3'ünde ve kontrollerin %2,0'ında ortaya çıkmıştır. Başlangıçtareperfüzyon gözlenmeyen hastalarda şiddetli kanama insidansı fondaparinuks için %1,2,kontroller içinse %1,5 olmuştur. Primer PCI uygulanan hastalarda şiddetli kanama insidansıfondaparinuks için %1,0, kontroller içinse %0,4 olmuştur. Primer PCI uygulanan gönüllülerde karara bağlanmış kılavuz kateter trombüsü insidansı, fondaparinuks alanlarda %1,2, kontrol gönüllülerinde ise %0 olmuştur. Şiddetli kanama konusundaki etkililik bulguları ve sonuçları, yaşlılar, böbrek bozukluğu olan hastalar, eşzamanlı trombosit agregasyonu inhibitörlerinin tipi (aspirin, tienopiridinler) gibiönceden belirlenmiş alt gruplarda tutarlılık sergilemiştir. Derin Ven Trombozunun (DVT) eşlik etmediği akut semptomatik spontan yüzeyel ven trombozu görülen hastaların tedavisi:Randomize, çift kör bir klinik çalışmaya (CALISTO), kompresyonlu ultrason incelemesiyle doğrulandığı üzere, alt ekstremitelerde en az 5 cm uzunluğunda akut, semptomatik, izole,spontan yüzeyel ven trombozu bulunan 3002 hasta dahil edilmiştir. Hastalar eşzamanlı DVT 15 veya safeno-femoral bileşkenin 3 cm yakınında yüzeyel ven trombozu sergiledikleri takdirde çalışmaya dahil edilmemiştir. Hastalar şiddetli karaciğer bozukluğu (kreatinin klirensi<30ml/dak), düşük vücut ağırlığı (<50kg), aktif kanser, semptomatik PE veya yakın zamanlıDVT/PE (<6 ay) ya da yüzeyel ven trombozu (<90 gün) öyküsü veya skleroterapi ya da IV yolkomplikasyonu ile ilişkili yüzeyel ven trombozu görüldüğü veya yüksek kanama riski sözkonusu olduğu takdirde çalışmaya dahil edilmemiştir. Hastalar 45 gün süresince elastik çoraplar, analjezik ve/veya topikal NSAİİ anti-enflamatuvar ilaçlara ek olarak günde bir defa 2,5 mg fondaparinuks veya plasebo almak üzere randomizeedilmiştir. Takip 77. Güne kadar devam etmiştir. Medyan yaşı 58 olan çalışma popülasyonunun%64'ü kadındır ve %4,4'ü <50 ml/dakikalık bir kreatinin klirensine sahiptir. 47. Güne kadar meydana gelen birleşik semptomatik PE, semptomatik DVT, semptomatik yüzeyel ven trombozu uzaması, semptomatik yüzeyel ven trombozu rekürrensi veya ölümşeklindeki primer etkililik sonucu, plasebo hastalarında %5,9'ken 2,5 mg fondaparinuksalanlarda anlamlı düşüş göstermiş ve %0,9 olmuştur (bağıl risk düşüşü: %85,2; %95 CI, %73,7 -%91,7 [p<0,001]). Aynı zamanda primer sonucun her bir tromboembolik bileşeninin insidansıda fondaparinuks alan hastalarda şu şekilde anlamlı azalma sergilemiştir: semptomatik PE [0'a(%0) karşılık 5 (%0,3) (p=0,031)], semptomatik DVT [3'e (%0,2) karşılık 18 (%1,2); bağıl riskdüşüşü %83,4 (p<0,001)], semptomatik yüzeyel ven trombozu uzaması [4'e (%0,3) karşılık 51(%3,4); bağıl risk düşüşü %92,2 (p<0,001)], semptomatik yüzeyel ven trombozu rekürrensi [5'e(%0,3) karşılık 24 (%1,6); bağıl risk düşüşü %79,2 (p<0,001)]. Tedavi gruplarında mortalite oranlarının düşük ve benzer olduğu gözlenmiştir ve fondaparinuks grubunda 2 (%0,1) ölüm, plasebo grubunda ise 1 (%0,1) ölüm meydana gelmiştir. Etkililik 77. Güne kadar korunmuştur ve varikoz venlere sahip hastaları ve diz altında yüzeyel ven trombozuna sahip hastaları da içeren önceden tanımlanmış tüm alt gruplarda tutarlılıksergilemiştir. 1 (%0,1) fondaparinuks hastasında ve 1 (%0,1) plasebo hastasında tedavi sırasında majör kanama ortaya çıkmıştır. 5 (%0,3) fondaparinuks hastasında ve 8 (%0,5) plasebo hastasındaklinik açıdan anlamlı majör olmayan kanama ortaya çıkmıştır. 5.3. Klinik öncesi güvenlilik verileriKlinik dışı veriler güvenlilik farmakolojisi, tekrarlı doz toksisitesi ve genotoksisitenin incelendiği klasik çalışmalar temelinde insanlar açısından özel bir risk ortaya koymamaktadır. Hayvançalışmaları, sınırlı maruziyet nedeniyle, üreme toksisitesi üzerindeki etkiler bakımından yeterlideğildir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiSodyum klorür Enjeksiyonluk su Hidroklorik asit Sodyum hidroksit 6.2. GeçimsizliklerGeçimsizlik ile ilgili çalışma yapılmadığından, ARİXTRA® diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3. Raf ömrü36 ay 16 6.4. Saklamaya yönelik özel tedbirlerARİXTRA® %0.9'luk serum fizyolojik serum setine ilave edilirse hemen infüze edilmelidir, ancak 24 saat kadar oda sıcaklığında saklanabilir. 25°C altındaki oda sıcaklığında saklayınız. Dondurmayınız. 6.5. Ambalajın niteliği ve içeriğiARİXTRA® tek kullanımlık enjektör Tip I, 1 ml'lik cam hazne, buna bağlı 27 gauge x 12,7 mm'lik iğne ve brombutil veya klorbütil elastomer pistondan oluşur. 0,5 ml'de 2,5 mg fondaparinuks sodyum içeren, mavi renkli, otomatik güvenlik sistemi olan, 10 adet kullanıma hazır enjektör. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerSubkutan enjeksiyon, standart bir enjektör ile uygulandığı şekilde yapılır. İntravenöz uygulama var olan bir damar yolu aracılığıyla doğrudan veya küçük hacimli serum fizyolojik seti (25 veya50 ml'lik) kullanılarak yapılmalıdır. Parenteral çözeltiler uygulama öncesi herhangi bir partikül varlığı veya renk bozukluğu yönünden incelenmelidir. Subkutan yolla yapılan kendi kendine uygulama ile ilgili talimatlar Kullanma Talimatında yer almaktadır. ARİXTRA® kullanıma hazır enjektörün iğne koruma sistemi, enjeksiyon sonrası olabilecek iğne yaralanmalarını önlemek için geliştirilmiştir. 17 Kendi kendine uygulama için talimatlar: ARİXTRA® güvenlik enjektörünün bölümleri:1. İğne kapağı 2. Piston 3. Tutma yeri 4. Emniyet kılıfı Şekil 1. Otomatik iğne koruma sistemli enjektör  BASAMAK BASAMAK ARİXTRA® KULLANIMI1. Ellerinizi sabun ve su ile yıkayınız. Havlu ile kurulayınız. 2. Enjektörü karton kutudan çıkarınız ve şunları kontrol ediniz: Son kullanma tarihinin geçmediğini, Çözeltinin berrak ve renksiz olduğunu ve partikül içermediğini, Enjektörün açılmamış ve zarar görememiş olduğunu 3. Rahat bir pozisyonda oturunuz veya uzanınız. Alt karın bölgesinde göbek deliğinin en az 5 cm aşağısında bir nokta belirleyiniz.Enjeksiyon işlemini her seferinde alt karınbölgesinin sağ ve sol taraflarına dönüşümlüolarak uygulayınız. Bu, enjeksiyon yerindeki rahatsızlığın azalmasına yardımcı olacaktır. Eğer altkarın bölgesine enjeksiyon mümkün değilise, doktorunuz veya hemşirenize danışınız. 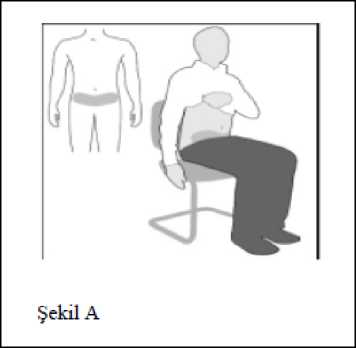
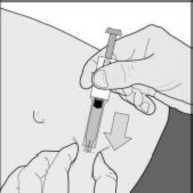
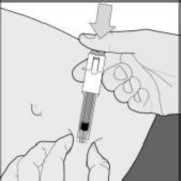
18 4. Enjeksiyon bölgesini alkollü pamukla temizleyiniz. 5. İğne kapağını önce döndürerek (şekil B1), sonra da düzdoğrultuda enjektör gövdesinden uzağa doğru çekerekçıkarınız (şekil B2). İğne koruyucusunu atınız. Önemli Not- Enjeksiyondan önce iğneye dokunmayınız veya herhangi biryüzeye temas etmesini önleyiniz. - Enjektör içinde küçük bir hava kabarcığının olmasınormaldir. Herhangi bir ürün kaybını önlemek için havakabarcığını enjeksiyondan önce çıkarmaya çalışmayınız, budurum ilaç kaybına neden olabilir. 6. Önceden temizlenmiş olan bölgedeki deriyi, boğumoluşturacak şekilde, enjeksiyon sona erene kadar başparmağınız ve işaret parmağınız arasında hafifçe tutunuz (şekil C). 7. Enjektörü tutma yerinden sağlamca tutunuz.İğnenin tamamını deri boğumuna dik açı ilederiiçine sokunuz (şekil D). 8. Pistonu sonuna kadar iterek içeriğinin tümünü enjekteediniz. (şekil E). 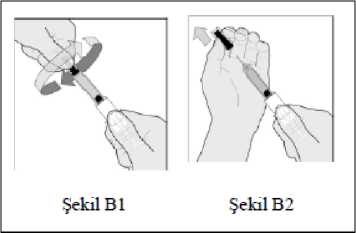 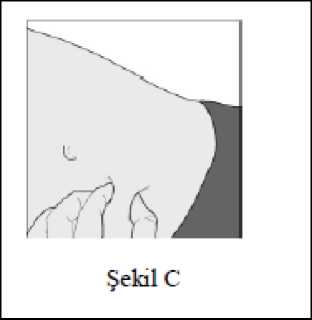
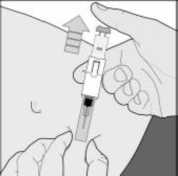
19 9. Piston serbest bırakıldığında iğne otomatikolarak deriden çıkacak ve bundan sonra,içinde kalacağı emniyet kılıfının içineçekilecektir (şekil F).
Kullanılmış enjektörü evsel atık olarak atmayınız, hemşireniz ya da doktorunuzun tavsiye ettiği şekilde imha ediniz. Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kotrolü Yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİVLD Danışmanlık, Tıbbi Ürünler ve Tanıtım Hizmetleri A.Ş. Büyükdere Cad. No:127 Astoria İş Merkezi A Blok Kat:8 Esentepe, Şişli- İstanbulTelefon: (212) 340 76 84 8. RUHSAT NUMARASI13.05.2015 - 2015/363 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEMETARİHİİlk ruhsat tarihi: 13.05.2015 Ruhsat yenileme tarihi: 10. KUB'UN YENİLENME TARİHİ20
|
İlaç BilgileriArixtra 2,5 Mg /0,5 Ml Enjeksiyonluk SolüsyonEtken Maddesi: Fondaparinuks Sodyum Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.