Saxenda 6 Mg/ml Sc Enjeksiyonluk Çözelti İçeren Kullanıma Hazır Kalem Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ'V1. BEŞERİ TIBBİ ÜRÜNÜN ADISAXENDA® 6 mg/mL enjeksiyonluk çözelti içeren kullanıma hazır kalem Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:1 mL çözelti, 6 mg liraglutid* içerir. Bir kullanıma hazır kalem, 3 mL'de 18 mg liraglutidiçerir. Saccharomyces cerevisiae'deYardımcı maddeler:Sodyum hidroksit (pH ayarı için) y.m. Disodyum fosfat dihidrat 1,42 mg/mL Yardımcı maddelerin tam listesi için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti.Berrak ve renksiz veya renksize yakın, izotonik çözelti; pH = 8,15. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarSAXENDA®, başlangıç vücut kitle indeksi (VKİ) aşağıdaki gibi olan erişkin hastaların, vücut ağırlığı yönetiminde düşük kalorili diyet ve artırılmış fiziksel aktiviteye ek tedavi olarakendikedir: > 30 kg/m1 (obez), veya Disglisemi (pre-diyabet veya tip 2 diyabet), hipertansiyon, dislipidemi ya da obstruktifuyku apnesi gibi kilo ile ilişkili en az bir komorbidite varlığında >27 kg/m2 ila < 30kg/m2 (fazla kilolu).Eğer hastalar, 3,0 mg/gün dozunda 12 haftalık tedavinin sonunda başlangıçtaki vücut ağırlıklarının en az %5'ini kaybetmemişlerse, SAXENDA® tedavisi kesilmelidir. 2 4.2. Pozoloji ve uygulama yöntemiPozoloji/uygulama sıklığı ve süresi:Başlangıç dozu günde tek doz 0,6 mg'dır. Doz, gastro-intestinal toleransı artırmak için en az bir hafta aralıklarla her seferinde 0,6 mg artışla günde tek doz 3,0 mg'a yükseltilmelidir(bakınız Tablo1). Eğer bir sonraki doz artışı ardışık 2 hafta için tolere edilemezse, tedaviyikesmek düşünülebilir. Günlük dozun 3,0 mg'dan yüksek olması önerilmez.
Unutulan dozlarEğer doz atlanmışsa ve normalde alınan saati takip eden 12 saatlik zaman dilimi içinde bulunuluyorsa, hasta, mümkün olan en kısa sürede dozu almalıdır. Eğer bir sonraki doza 12saatten daha az bir süre kalmışsa, hasta atlanan dozu almamalıdır ve bir sonraki planlı dozu ilegünde bir kez rejime devam etmelidir. Atlanan bir dozu telafi etmek için ekstra bir doz ya dadaha yüksek bir doz alınmamalıdır. Tip 2 diyabetli hastalarSAXENDA® , başka bir GLP-1 reseptör agonisti ile kombinasyon halinde kullanılmamalıdır. SAXENDA®'ya başlanırken, hipoglisemi riskini azaltmak için, eşzamanlı uygulanan insülin ya da insülin salgılatıcı ilaçların (sülfonilüreler gibi) dozunun azaltılması düşünülmelidir.İnsülin veya insülin sekretagoglarının dozunu ayarlamak için hastanın kendi kendine kanglukoz ölçümleri yapması gerekir (bkz. Bölüm 4.4). Uygulama şekli:SAXENDA® sadece subkutan uygulama içindir. İntravenöz veya intramusküler yolla kesinlikle uygulanmamalıdır. SAXENDA® günde bir kez herhangi bir saatte, yemeklerden bağımsız olarak uygulanır. Karın bölgesine, uyluk bölgesine ya da üst kola enjekte edilmelidir. Doz ayarlaması gerekmeksizinenjeksiyon yer ve uygulama zamanında değişiklik yapılabilir. Ancak, günün en elverişli saatiseçildikten sonra, SAXENDA®'nın her gün yaklaşık olarak aynı saatte uygulanması tercihedilir. Uygulamaya ilişkin daha fazla talimat için bkz. Bölüm 6.6. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği: 11Hafif ila orta dereceli böbrek yetmezliği (kreatinin klirensi >30 mL/dk) olan hastalarda herhangi bir doz ayarlaması gerekmez. SAXENDA®, son dönem böbrek hastalığı dahilşiddetli böbrek yetmezliği (kreatinin klirensi <30 mL/dk) olan hastalarda önerilmez (bkz.bölüm 4.4, 4.8 ve 5.2). Karaciğer yetmezliği:Hafif ila orta dereceli karaciğer yetmezliği olan hastalarda herhangi bir doz ayarlaması gerekmez. SAXENDA®, şiddetli karaciğer yetmezliği olan hastalarda önerilmez ve hafif ilaorta dereceli karaciğer yetmezliği olan hastalarda dikkatli kullanılmalıdır (bkz. Bölüm 4.4 ve5.2). Pediatrik popülasyon:SAXENDA®'nın çocuklar ve 18 yaşın altındaki adölesanlardaki güvenliliği ve etkililiği ortaya konmamıştır (bkz. Bölüm 5.1). Veri bulunmamaktadır. Bu tıbbi ürünün pediatrikhastalarda kullanımı önerilmez. Geriyatrik popülasyon: (>65 yaş)Yaşa dayalı bir doz ayarlaması gerekmemektedir. >75 yaş hastalarda terapötik deneyim sınırlıdır ve bu hastalarda kullanım önerilmez (bkz. Bölüm 4.4 ve 5.2). 4.3. KontrendikasyonlarLiraglutide ya da Bölüm 6.1'de listelenen yardımcı maddelerin herhangi birine karşı aşırı duyarlılık. Kendisinde veya aile öyküsünde medüler tiroid karsinomu olan hastalarda veya çoklu endokrin neoplazma sendromu tip 2 olan hastalarda kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleriKalp yetmezliği bulunan hastalar NYKD (New York Kalp Derneği) sınıf IV olarak değerlendirilen konjestif kalp yetmezliği hastalarında klinik deneyim bulunmamaktadır, bu nedenle bu hastalardaliraglutidin kullanımı önerilmemektedir. Özel popülasyonlar Aşağıdaki hastalarda liraglutidin vücut ağırlığı yönetimindeki güvenliliği ve etkililiği gösterilmemiştir: - 75 yaş ve üzeri hastalar, - Vücut ağırlığı yönetimi için başka ürünlerle tedavi edilen hastalar, - Endokrinolojik veya yeme bozukluklarına veya kilo artışına neden olabilecek tıbbiürünlerle tedaviye sekonder obezitesi olan hastalar, - Şiddetli böbrek yetmezliği olan hastalar, - Şiddetli karaciğer yetmezliği olan hastalar. Bu hastalarda kullanılması önerilmez (bkz. Bölüm 4.2). 3 Hafif ve orta dereceli karaciğer yetmezliği olan hastalarda liraglutid, vücut ağırlığı yönetimi açısından araştırılmamış olduğundan bu hastalarda dikkatli kullanılmalıdır (bkz. Bölüm 4.2 ve5.2). Enflamatuvar bağırsak hastalığı ve diyabetik gastroparezisi olan hastalarda sınırlı deneyim bulunmaktadır. Bulantı, kusma ve ishal gibi geçici gastrointestinal advers reaksiyonlarlailişkilendirildiğinden bu hastalarda liraglutid kullanımı önerilmez. PankreatitAkut pankreatit, GLP-1 reseptör agonistleri kullanımıyla gözlenmiştir. Hastalar akut pankreatitin karakteristik semptomları hakkında bilgilendirilmelidirler. Pankreatittenşüpheleniliyorsa, liraglutid tedavisi kesilmelidir; eğer akut pankreatit teyit edilirse, liraglutidetekrar başlanmamalıdır. Kolelitiazis ve kolesistitVücut ağırlığı yönetimine yönelik klinik çalışmalarda, plasebo ile karşılaştırıldığında liraglutid ile tedavi edilen hastalarda daha yüksek bir kolelitiazis ve kolesistit oranıgözlenmiştir. Yüksek miktarda kilo kaybının kolelitiazis riskini ve dolayısıyla kolesistitriskini artırabilmesi, liraglutid ile daha sık görülen bu durumu sadece kısmenaçıklayabilmektedir. Kolelitiazis ve kolesistit, hastaneye yatış ve kolesistektomi gerektirebilir.Hastalar, kolelitiazis ve kolesistitin karakteristik semptomları hakkında bilgilendirilmelidir. Tiroid hastalığıTip 2 diyabete yönelik klinik çalışmalarda, özellikle önceden tiroid hastalığı geçirmiş hastalarda, guatr gibi tiroid ile ilgili advers olaylar rapor edilmiştir. Liraglutid bu nedenletiroid hastalığı olan kişilerde dikkatli kullanılmalıdır. Kalp atım hızıKlinik çalışmalarda liraglutid ile kalp atım hızında bir artış gözlenmiştir (bkz. Bölüm 5.1). Kalp atım hızı, standart klinik uygulama doğrultusunda düzenli aralıklarla izlenmelidir.Hastalar, artmış kalp atım hızının semptomları (çarpıntılar veya istirahat halinde kalbin hızlıattığı hissi) hakkında bilgilendirilmelidir. İstirahat halinde kalp atım hızında klinik olarakanlamlı ve uzamış bir artış deneyimleyen hastalarda liraglutid ile tedavi kesilmelidir. DehidratasyonGLP-1 reseptör agonistleri ile tedavi edilen hastalarda dehidratasyonun renal bozukluk ve akut böbrek yetmezliğini de içeren bulgu ve belirtileri bildirilmiştir. Liraglutid ile tedaviedilen hastalar, gastrointestinal yan etkiler ile ilişkili potansiyel dehidratasyon riskine karşıuyarılmalı ve sıvı kaybına karşı önlem almaları önerilmelidir. Tip 2 diyabet hastalarında hipoglisemiİnsülin ve/veya sülfonilüre grubu bir ilaç ile kombinasyon halinde liraglutid kullanan tip 2 diyabetli hastalar daha yüksek hipoglisemi riski altında olabilir. Hipoglisemi riski insülinve/veya sülfonilüre dozunun düşürülmesiyle azaltılabilir. Tip 2 diyabet hastalarında hiperglisemi 4SAXENDA® diyabet hastalarında insülin yerine kullanılmamalıdır. İnsülin kullanan hastalarda, insülinin birden kesilmesi veya dozunun azaltılmasından sonra diyabetikketoasidoz bildirilmiştir (bkz. Bölüm 4.2). Takip edilebilirlikBiyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. Bu tıbbi ürün her dozunda 1 mmol (23 mg)'dan az sodyum ihtiva eder; yani aslında sodyum içermez. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriIn vitrokoşullarda liraglutidin sitokrom P450 ve plazma proteinlerine bağlanma ile ilişkili farmakokinetik etkileşimlerde yer alma potansiyelinin çok düşük olduğu gösterilmiştir.Liraglutid ile gastrik boşalmadaki küçük gecikme, eşzamanlı olarak oral uygulanan tıbbi ürünlerin emilimini etkileyebilmektedir. Etkileşim çalışmaları emilimde klinik olarakanlamlı bir gecikme göstermemiştir ve dolayısıyla herhangi bir doz ayarlamasıgerekmemektedir. Etkileşim çalışmaları 1,8 mg liraglutid ile yürütülmüştür. Gastrik boşalma hızı üzerindeki etki, liraglutid 1,8 mg ile 3,0 mg (parasetamol EAA0-300 dakika) arasında eşit olmuştur.Liraglutid ile tedavi edilen az sayıda hasta en az bir şiddetli ishal epizodu bildirmiştir. İshal,eşzamanlı alınan oral tıbbi ürünlerin emilimini etkileyebilir. Varfarin ve diğer kumarin türevleriHerhangi bir etkileşim çalışması yapılmamıştır. Varfarin gibi çözünürlüğü zayıf veya terapötik indeksi dar olan etkin maddeler ile klinik olarak anlamlı etkileşimler dışlanamaz.Varfarin veya başka kumarin türevlerini kullanan hastalarda liraglutid tedavisinebaşlandığında daha sık INR (uluslararası normalize edilmiş oran) takibi önerilmektedir. Parasetamol (Asetaminofen)1000 mg tek dozu takiben liraglutid, toplam parasetamol maruziyetini değiştirmemiştir. Parasetamol Cmaks değeri %31 azalmış ve medyan tmaks 15 dakikaya kadar gecikmiştir. Eşzamanlı parasetamol uygulamasında doz ayarlaması gerekmemektedir. Atorvastatin40 mg tek doz atorvastatin uygulamasını takiben liraglutid, toplam atorvastatin maruziyetini değiştirmemiştir. Bu nedenle, liraglutid ile birlikte verildiğinde, atorvastatin için dozayarlaması gerekmemektedir. Liraglutid ile atorvastatin Cmaks %38 azalmış ve medyan tmaks 1ila 3 saate kadar gecikmiştir. GriseofulvinLiraglutid, 500 mg tek doz griseofulvin uygulamasını takiben toplam griseofulvin maruziyetini değiştirmemiştir. Griseofulvin Cmaks değeri %37 artarken, medyan tmaksdeğişmemiştir. Griseofulvin ile düşük çözünürlük ve yüksek geçirgenliğe sahip diğerbileşiklerin dozunun ayarlanması gerekmemektedir. 5 DigoksinTek bir doz 1 mg digoksinin liraglutid ile birlikte uygulanması, digoksinin EAA'sında %16 azalma göstermiştir; Cmaks %31 azalmıştır. Digoksinin medyan tmaks'ı 1 ila 1,5 saatgecikmiştir. Bu sonuçlara göre digoksin dozunda herhangi bir ayarlamaya gerek yoktur. LisinoprilTek bir doz 20 mg lisinoprilin liraglutid ile birlikte uygulanması, lisinoprilin EAA'sında %15 azalma göstermiştir. Cmaks %27 azalmıştır. Lisinopril medyan tmaks'ı liraglutid ile 6 ila 8 saatgecikmiştir. Bu sonuçlara göre lisinopril dozunda herhangi bir ayarlamaya gerek yoktur. Oral kontraseptiflerLiraglutid, oral kontraseptif bir ürünün tek doz uygulanmasını takiben etinilestradiol ve levonorgestrel Cmaks değerlerini sırasıyla %12 ve %13 azaltmıştır. tmaks, liraglutid ile her ikibileşik için 1,5 saat kadar gecikmiştir. Etinilestradiol veya levonorgestrelin her birine toplammaruziyet üzerinde klinik olarak anlamlı etki olmamıştır. Bu nedenle, liraglutid ile beraberuygulandığında, kontraseptif etkide değişiklik olmaması beklenmektedir. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlarda etkileşim çalışması mevcut değildir. Pediyatrik Popülasyon:Pediyatrik hastalarda etkileşim çalışması mevcut değildir. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi X'tir. Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Etinilestradiol veya levonorgestrelin her birine toplam maruziyet üzerinde klinik olarak anlamlı etki olmamıştır. Bu nedenle, liraglutid ile beraber uygulandığında, kontraseptif etkidedeğişiklik olmaması beklenmektedir. Çocuk doğurma potansiyeli olan kadınlara SAXENDA® kullanımı sırasında gebeliği önlemek için etkili kontrasepsiyon kullanmaları tavsiye edilir. Gebelik dönemiLiraglutidin gebe kadınlarda kullanıma ilişkin sınırlı veri mevcuttur. Hayvanlarda yapılan çalışmalarda üreme toksisitesi gösterilmiştir (bkz. Bölüm 5.3). İnsanlara yönelik potansiyelrisk bilinmemektedir. Liraglutid gebelik döneminde kullanılmamalıdır. Eğer hasta gebe kalmak istiyorsa veya tedavi sırasında gebelik oluşursa, liraglutid tedavisi kesilmelidir. Laktasyon dönemiLiraglutidin anne sütüne geçip geçmediği bilinmemektedir. Hayvan çalışmaları liraglutid ve yakın yapısal benzerlikteki metabolitlerinin süte geçişlerinin düşük olduğunu göstermiştir.Klinik dışı çalışmalar, emzirme dönemindeki sıçan yavrularının neonatal gelişiminde 6 tedaviyle ilişkili azalma göstermiştir (bkz. Bölüm 5.3). Deneyim eksikliği nedeniyle; SAXENDA® emzirme döneminde kullanılmamalıdır. Üreme yeteneği/FertiliteCanlı implant sayısında hafif azalma dışında, hayvan çalışmaları fertiliteyle ilgili zararlı etkiler göstermemiştir (bkz. bölüm 5.3). 4.7. Araç ve makine kullanımı üzerindeki etkilerSAXENDA®'nın araç ve makine kullanımı üzerinde etkisi yoktur veya göz ardı edilebilir etkiye sahiptir. Bununla birlikte, baş dönmesi SAXENDA® ile tedavinin daha çok ilk 3 ayındagörülebilir. Baş dönmesi meydana gelirse araç veya makine kullanımında dikkatliolunmalıdır. 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti:SAXENDA® , güvenlilik kapsamında 5.813 obez veya kilo ile ilişkili en az bir komorbiditesi olan fazla kilolu hastanın kaydedildiği 5 çift-kör, plasebo kontrollü çalışmada incelenmiştir.Genel olarak, SAXENDA ile tedavi sırasında gastrointestinal reaksiyonlar en sık bildirilenadvers reaksiyonlar olmuştur (% 67,9) (bkz. Bölüm 'Seçili advers reaksiyonların listesi'). Advers reaksiyonların tablo halinde listesiTablo 2'de yetişkinlerde yapılan uzun süreli faz 2 ve faz 3 kontrollü çalışmalarda ve pazarlama sonrası raporlarda bildirilen advers reaksiyonlar listelenmektedir. Adversreaksiyonlar sistem organ sınıfına ve sıklığa göre sıralanmaktadır. Advers reaksiyonlarınsıklığı faz 2 ve faz 3 klinik çalışma havuzuna dayanmaktadır. Sıklıklar şu şekildetanımlanmaktadır: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldekiverilerden hareketle tahmin edilemiyor). Her bir sıklık gruplamasında, istenmeyen etkiler,azalan ciddiyet sırasına göre sunulmaktadır. 7
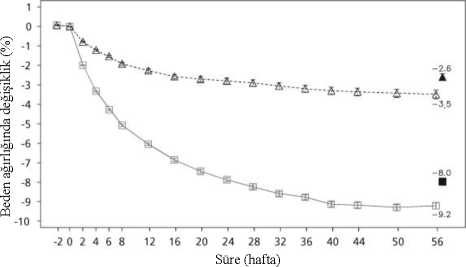
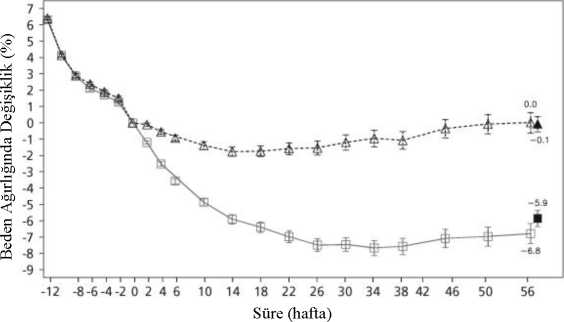
olmayan hastalarda bildirilen hipoglisemi (hastaların kendi bildirdiği ve kan glukoz ölçümleri ile doğrulanmamış semptomlara dayalı). Daha fazla bilgi için bkz. bölüm 'Seçili adversreaksiyonların listesi'. **İnsomnia ağırlıklı olarak tedavinin ilk 3 ayında görülmüştür. ***Bkz. Bölüm 4.4. **** Kontrollü faz 2, 3a ve 3b klinik çalışma sonuçlarına göre Seçili advers reaksiyonların listesi:Tip 2 diyabeti olmayan hastalarda hipoglisemi Diyet ve egzersiz ile kombinasyon halinde SAXENDA® ile tedavi edilen tip 2 diyabeti olmayan aşırı kilolu veya obez hastalarla yürütülen klinik çalışmalarda hiç şiddetlihipoglisemik olay (üçüncü şahıstan yardım gerektiren) bildirilmemiştir. Hipoglisemik olaysemptomları, SAXENDA® ile tedavi edilen hastaların %1,6'sı ve plasebo uygulananhastaların %1,1'i tarafından bildirilmiştir; ancak bu olaylar kan glukoz ölçümleri iledoğrulanmamıştır. Olayların büyük çoğunluğu hafif olmuştur. Tip 2 diyabeti olan hastalarda hipoglisemi Diyet ve egzersiz ile kombinasyon halinde SAXENDA® ile tedavi edilen tip 2 diyabetli aşırı kilolu veya obez hastalarla yürütülen klinik çalışmalarda şiddetli hipoglisemi(üçüncü şahıstanyardım gerektiren), SAXENDA® ile tedavi edilen hastaların %0,7'si tarafından ve sadecesülfonilüre ile eşzamanlı tedavi edilen hastalarda bildirilmiştir. Ayrıca, bu hastalardakaydedilmiş semptomatik hipoglisemi, SAXENDA® ile tedavi edilen hastaların %43,6'sı ve 8 plasebo ile tedavi edilen hastaların %27,3'ü tarafından bildirilmiştir. Sülfonilüre ile eşzamanlı tedavi görmeyen hastalardan, SAXENDA® ile tedavi edilen hastaların %15,7'si ve plasebo iletedavi edilen hastaların %7,6'sı tarafından kaydedilmiş semptomatik hipoglisemi(semptomların eşlik ettiği <3,9 mmol/L plazma glukozu olarak tanımlı) bildirilmiştir. İnsülin ile tedavi edilen tip 2 diyabeti olan hastalarda hipoglisemi Diyet ve egzersiz ile birlikte insülin ve liraglutid 3,0 mg/gün ve 2 OAD'ye kadar tedavi edilen tip 2 diyabetli aşırı kilolu veya obez hastalarda yürütülen bir klinik çalışmada, ciddihipoglisemi (üçüncü taraf yardımı gerektiren) vakaları liraglutid 3,0 mg/gün ile tedavi edilenhastaların %1,5'inde bildirilmiştir. Bu çalışmada, belgelenmiş semptomatik hipoglisemivakaları (semptomlar eşliğinde plazma glukozu <3,9 mmol/L olarak tanımlanan) liraglutid 3,0gün/gün ile tedavi edilen hastaların % 47,2'sinde ve plasebo ile tedavi edilen hastaların %51,8'inde rapor edilmiştir. Eşzamanlı olarak sülfonilüre ile tedavi edilen hastalar arasında,liraglutid 3,0 mg/gün ile tedavi edilen hastaların % 60,9'unda ve plasebo ile tedavi edilenhastaların % 60,0'ında belgelenmiş semptomatik hipoglisemik olaylar olduğu rapor edilmiştir.Gastrointestinal advers reaksiyonlar Gastrointestinal olayların çoğu epizodu hafif ila orta şiddette, geçici olmuş ve büyük çoğunluğunda tedavinin kesilmesi gerekmemiştir. Reaksiyonlar genellikle tedavinin ilkhaftasında ortaya çıkmış ve tedavi devam ettikçe birkaç gün veya hafta içinde kaybolmuştur. >65 yaşın üzerindeki hastalar SAXENDA® ile tedavi edildiklerinde daha fazla gastrointestinal etki yaşayabilir. Hafif ila orta dereceli böbrek yetmezliği (kreatinin klirensi >30 mL/dk) olan hastalar SAXENDA® ile tedavi edildiklerinde daha fazla gastrointestinal etki deneyimleyebilir. Akut böbrek yetmezliği GLP-1 reseptör agonistleri ile tedavi edilen hastalarda akut böbrek yetmezliği bildirimleri olmuştur. Bildirilen olayların büyük çoğunluğu önceden, hacim kaybına neden olan bulantı,kusma ve ishal yaşamış hastalarda görülmüştür (bkz. Bölüm 4.4). Alerjik reaksiyonlar Liraglutidin ticari kullanımı ile hipotansiyon, taşikardi, dispne ve ödem gibi semptomların eşlik ettiği az sayıda anafilaktik reaksiyon olgusu bildirilmiştir. Anafilaktik reaksiyonlarpotansiyel olarak yaşamı tehdit edici olabilir. Eğer anafilaktik reaksiyondan şüpheleniyorsa,liraglutid kesilmelidir ve tedaviye yeniden başlanmamalıdır (bkz. Bölüm 4.3). Enjeksiyon yeri reaksiyonları SAXENDA® ile tedavi edilen hastalarda enjeksiyon yeri reaksiyonları bildirilmiştir. Bu reaksiyonlar genellikle hafif ve geçicidir ve büyük çoğunluğu devam eden tedavi sırasındakaybolmuştur. Taşikardi Klinik çalışmalarda taşikardi, SAXENDA® ile tedavi edilen hastaların %0,6'sında ve plasebo uygulanan hastaların %0,1'inde bildirilmiştir. Olayların büyük kısmı hafif ila orta dereceliolmuştur. Olaylar izoledir ve büyük çoğunluğu SAXENDA® ile devam eden tedavi sırasındadüzelmiştir. 9 Pediatrik Popülasyon SAXENDA® pediyatrik hastaların kullanımında önerilmemektedir. Gastrointestinal rahatsızlıklar bugüne kadar tamamlanan iki doz yükseltme çalışmasında en sık bildirilenadvers olaylardır. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99) 4.9. Doz aşımı ve tedavisiLiraglutidin klinik çalışmalar ve ticari kullanımında 72 mg'a (vücut ağırlığı yönetiminde önerilen dozun 24 katı) ulaşan doz aşımları bildirilmiştir. Bildirilen olaylar şiddetli bulantı,şiddetli kusma ve şiddetli hipoglisemiyi içermektedir. Doz aşımı durumunda, hastanın klinik belirti ve semptomlarına göre gerekli destekleyici tedavi başlatılmalıdır. Hasta, dehidratasyonun klinik belirtileri açısından gözlemlenmelidir vekan glukozu izlenmelidir. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Diyabette kullanılan ilaçlar, Glukagon benzeri peptid-1 (GLP-1) analogları ATC kodu: A10BJ02 Etki mekanizmasıLiraglutid, endojen insan GLP-1'inin aminoasit dizilişine %97 homoloji gösteren, açillenmiş bir glukagon benzeri peptid-1 (GLP-1) analoğudur. Liraglutid, GLP-1 reseptörüne (GLP-1R)bağlanır ve aktive eder. GLP-1, iştah ve besin alımının bir fizyolojik regülatörüdür; fakat etki mekanizması bütünüyle net değildir. Hayvan çalışmalarında, liraglutidin periferik uygulaması, iştah regülasyonundarolü olan spesifik beyin bölgelerinde, liraglutidin spesifik GLP-1R aktivasyonu yoluylaspesifik tokluk sinyallerini artırdığı ve spesifik açlık sinyallerini azalttığı, böylelikle dahadüşük vücut ağırlığına yol açtığı gözlenmiştir. GLP-1 reseptörleri, kalp, damarlar, bağışıklık sistemi ve böbreklerdeki spesifik lokasyonlarda konumlanmaktadır. Aterosklerotik fare modellerinde liraglutid, aortik plak progresyonunuönlemiştir ve plaktaki enflamasyonu azaltmıştır. Ayrıca liraglutidin, plazma lipidleri üzerinede faydalı etkisi olmuştur. Liraglutid, halihazırda yerleşmiş plakların boyutunu azaltmamıştır. Farmakodinamik etkiler10 Liraglutid insanda vücut ağırlığını; temelde viseral yağdaki kayıpların subkutan yağ kaybından daha büyük olduğu yağ kütlesi kaybı yoluyla düşürür. Liraglutid, doygunluk vetokluk duygularını artırarak, aynı zamanda da açlık ve ileriye dönük gıda tüketim beklentisihissini azaltarak ve bu şekilde besin alımında azalmaya yol açarak iştahı düzenler. Liraglutid,plasebo ile karşılaştırıldığında enerji tüketimini artırmaz. Liraglutid glukoza bağımlı bir şekilde insülin salgısını uyarır ve glukagon salgısını azaltır, bunun neticesinde açlık ve tokluk glukoz düzeyi düşer. Glukoz düşürücü etki, pre-diyabet vediyabet hastalarında, normoglisemili hastalara kıyasla daha belirgindir. Klinik çalışmalar,liraglutidin HOMA-B'ye göre beta hücresi fonksiyonunu ve pro-insülin/insülin oranınıiyileştirdiğini ve koruduğunu öne sürmektedir. Klinik etkililik ve güvenlilikVücut ağırlığı yönetiminde azalmış kalori alımı ve artmış fiziksel aktivite ile bir arada liraglutidin etkililiği ve güvenliliği, toplam 5.358 hastayı içeren dört adet faz 3 randomize, çiftkör, plasebo kontrollü çalışmada incelenmiştir. Çalışma 1 (SCALE Obezite & Pre-Diyabet - 1839):Obeziteli (BMI >30 kg/m2), ya da dislipidemi ve/veya hipertansiyon ile birlikte aşırı kilolu (BMI >27 kg/m2) 3.731 hasta, başlangıçtaki BMI (>30 kg/m2 or <30 kg/m2) vetaramadaki pre-diyabet durumuna göre belirlenmiştir. 3.731 hastanın tümü tedavinin 56.haftasında randomize edilmiştir ve taramadaki pre-diyabetik 2.254 hasta tedavinin 160.haftasında randomize edilmiştir. Her iki tedavi periyodu, 12 haftalık ilaçsız/plasebogözlemsel takip süreci ile takip edilmiştir. Enerji kısıtlı diyet ve egzersizdanışmanlığının yaşam tarzına etkisi tüm hastalar için tedavinin arka planınıoluşturmuştur. Çalışma 1'in 56 haftalık bölümü, 3.731 randomize hastanın tümünde(tamamlayan 2.590 ) vücut ağırlığı kaybını değerlendirmiştir. Çalışma 1'in 160 haftalıkbölümü, pre-diyabetli 2.254 randomize hastada (tamamlayan 1.128) tip 2 diyabetbaşlama zamanını değerlendirmiştir. Çalışma 2 (SCALE Diyabet - 1922):Yeterli düzeyde kontrol edilmemiş tip 2 diyabetli(HbA1c aralık %7-10) 846 randomize (628'i tamamlamıştır) obez ve aşırı kilolu hastadakilo kaybının değerlendirildiği 56 haftalık çalışma. Çalışma başlangıcındaki mevcuttedavi ya yalnızca diyet ve egzersiz, tek ajanlar olarak metformin, bir sülfonilüre, birglitazon ya da bunların herhangi bir kombinasyonudur.Çalışma 3 (SCALE Uyku Apnesi- 3970):Orta veya şiddetli obstrüktif uyku apnesiolan 359 randomize (276'sı tamamlamıştır) obez hastada uyku apnesi şiddeti ve kilokaybının değerlendirildiği 32 haftalık bir çalışma.Çalışma 4 (SCALE idame - 1923):422 randomize (305'i tamamlamıştır) obez vehipertansiyonu veya dislipidemisi olan aşırı kilolu hastada düşük kalori diyeti ilesağlanan önceki >%5 kilo kaybından sonra, kilo idamesinin ve kilo kaybınındeğerlendirildiği 56 haftalık çalışma.Vücut ağırlığıÇalışılan tüm gruplarda obez/aşırı kilolu hastalarda plasebo ile karşılaştırıldığında liraglutid ile daha üstün kilo kaybı elde edilmiştir. Tüm çalışma popülasyonlarında, plaseboya kıyaslaliraglutid ile daha yüksek bir oranda hasta > %5 ve >%10 kilo kaybı elde etmiştir (Tablo 3-5).Çalışma 1'in 160. haftalık kısmında kilo kaybı esasen ilk yılda meydana gelmiştir ve 160 11 hafta boyunca sürdürülmüştür. Çalışma 4'te, plasebo ile karşılaştırıldığında liraglutid ile daha fazla hasta tedavi başlangıcı öncesindeki kilo kaybını korumuştur (sırasıyla %48,9 ve %81,4).Çalışmalar 1-4 için kilo kaybı, yanıt veren hastalar, süre ve kilo değişikliğinin kümülatifdağılımı (%) ile ilgili spesifik veriler Tablo 3-7 ve Şekil 1, 2, ve 3'te gösterilmektedir. Liraglutid (3,0 mg) tedavisi ile 12 hafta sonra kilo kaybı yanıtıErken dönemde yanıt veren hastalar, liraglutidin tedavi dozunda kalınan 12 hafta sonunda (doz yükseltme fazında 4 hafta ve tedavi dozunda 12 hafta) >%5 kilo kaybına ulaşan hastalarolarak tanımlanmıştır. Çalışma 1'in 56 haftalık bölümünde, hastaların %67,5'i 12 haftasonrasında >%5 kilo kaybına ulaşmıştır. Çalışma 2'de, hastaların %50,4'ü 12 hafta sonra >%5kilo kaybı elde etmiştir. Liraglutid ile tedaviye devam edilen, 1 yıllık tedavi sonrası erkendönemde yanıt veren bu hastaların %86,2'sinin >%5 kilo kaybına ve %51'inin >%10 kilokaybına ulaşacağı öngörülmektedir. 1 yıllık tedaviyi tamamlayan erken dönemde yanıt verenhastalarda öngörülen ortalama kilo kaybı, başlangıçtaki vücut ağırlıklarının %11,2'sidir(erkekler için 9,7 ve kadınlar için %11,6). Liraglutidin tedavi dozunda kalınan 12 haftasonunda <%5 kilo kaybına ulaşan hastalar arasında, 1 yıl sonra >%10 kilo kaybı elde etmeyenhastaların oranı %93,4'tür. Glisemik kontrolLiraglutid ile tedavi, normoglisemi, pre-diyabet ve tip 2 diyabetli alt popülasyonlarda glisemik parametrelerde anlamlı ölçüde düzelme sağlamıştır. Çalışma 1'in 56 haftalıkbölümünde, plasebo ile karşılaştırıldığında liraglutid ile tedavi edilen daha az sayıda hastadatip 2 diyabet gelişmiştir (%1,1 karşısında %0,2). Plasebo ile karşılaştırıldığında, başlangıçtapre-diyabeti olan daha fazla hastada pre-diyabet tersine döndürülmüştür (%69,2 karşısında%32,7). Çalışma 1'in 160 haftalık bölümünde primer etkinlik sonlanım noktası, tip 2diyabetes mellitus gelişen hastaların oranıdır ki burada tip 2 diyabetes mellitus başlangıcınakadar geçen süre değerlendirilmiştir. 160. haftada, tedavi süresince, SAXENDA® tedavisialan hastaların %3'ü ve plasebo tedavisi alanların %11'i tip 2 diyabetes mellitus tanısıalmıştır. 3,0 mg liraglutid ile tedavi edilen hastalar için tip 2 diyabetes mellitusun tahminibaşlama zamanı 2,7 kat daha uzun olmuştur (% 95 güven aralığı [1,9- 3,9] ve pleaseboya karşıliraglutid için tip 2 diyabetes mellitus gelişme riski oranı 0,2'dir. Kardiyometabolik risk faktörleriLiraglutid ile tedavi sistolik kan basıncında ve bel çevresinde, plaseboya kıyasla anlamlı derecede düzelme sağlamıştır (Tablo 3, 4 ve 5). Apne-Hipopne İndeksi (AHI)Plasebo ile karşılaştırıldığında liraglutid ile tedavi, obstrüktif uyku apnesinin AHI'de başlangıçtaki değerden değişme ile değerlendirilen şiddetini anlamlı ölçüde azaltmıştır (Tablo6). Tablo 3 Çalışma 1: 56 haftada vücut ağırlığı, glisemi ve kardiyometabolik parametrelerde başlangıca göre değişiklik12
13 160. haftada >5% vücut ağırlığı kaybeden hasta oranı, % (95% CI)'
160. haftada >10% vücut ağırlığı kaybeden hasta oranı, % (95% CI)24,4
Tam Analiz Seti: Vücut ağırlığı için, HbA1c, FPG, kan basıncı ve bel çevresi başlangıç değerleri ortalamalardır, hafta 160'da başlangıca göre değişiklikler, öngörülen ortalamalardır(en düşük kareler) ve hafta 160'daki tedavi kontrastları öngörülen tedavi farklarıdır. Vücutağırlığının >%5/>%10'unu kaybeden hastaların oranları için öngörülen olasılık oranlarıverilmektedir. Kayıp başlangıç sonrası değerleri, ileri taşınan en son gözlem kullanılarakgirilmiştir. ** p<0,0001. GA= güven aralığı. FPG=açlık plazma glukozu. SS= standart sapma. S Saxenda A Plasebo ¦ * İleri taşınan son gözlem (LOCF)Tüm planlı vizitleri tamamlayan hastalar için gözlenen değerlerŞekil 1 Çalışma 1 (0-56 hafta)'de vücut ağırlığında zamana göre başlangıç değerinden değişiklik
1 40 ^ 3020100Şekil 2 Çalışma 1'de 56 haftalık tedaviden sonra kilo değişikliğinin yüzde dağılımı (%)14
Tam Analiz Seti. Vücut ağırlığı için, HbAic, FPG, kan basıncı ve bel çevresi, başlangıç değerleri ortalamalardır, hafta 56'da başlangıca göre değişiklikler, öngörülen ortalamalardır(en düşük kareler) ve hafta 56'daki tedavi kontrastları öngörülen tedavi farklarıdır. Vücutağırlığının >%5/>%10'unu veren hastaların oranları için öngörülen olasılık oranlarıverilmektedir. Kayıp başlangıç sonrası değerleri, ileri taşınan en son gözlem kullanılarakgirilmiştir. * p<0,05. ** p<0,0001. GA= güven aralığı. FPG=açlık plazma glukozu. SS=standart sapma. 15 Tablo 6 Çalışma 3: 32 haftada vücut ağırlığı ve Apne- Hipopne İndeksinde başlangıca göre değişikliklerSAXENDA Plasebo (N=179) SAXENDA vs.(N=180) plasebo
Tam Analiz Seti. Başlangıç değerleri ortalamalardır, hafta 32'de başlangıca göre değişiklikler, öngörülen ortalamalardır (en düşük kareler) ve hafta 32'deki tedavi kontrastları öngörülentedavi farklarıdır. (%95 GA). Vücut ağırlığının >%5/>%10'unu veren hastaların oranları içinöngörülen olasılık oranları verilmektedir. Kayıp başlangıç sonrası değerleri, ileri taşınan enson gözlem kullanılarak girilmiştir. * p<0,05. ** p<0,0001. GA= güven aralığı. SS= standartsapma.
16
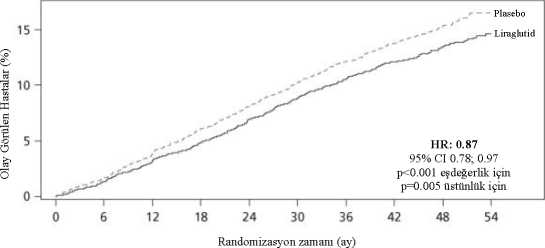
Şekil 3 Çalışma 4'te vücut ağırlığında zamana göre randomizasyondaki değere kıyasla değişiklikHafta 0'dan önce hastalar sadece düşük kalori diyeti ve egzersiz ile tedavi edilmiştir. Hafta 0'da hastalar SAXENDA® veya plasebo almak üzere randomize edilmiştir. İmmünojenisiteProtein veya peptid içeren tıbbi ürünlerin potansiyel immünojenik özellikleriyle uyumlu olarak, liraglutid ile tedaviyi takiben hastalar anti-liraglutid antikorları geliştirebilirler. Klinikçalışmalarda liraglutid ile tedavi edilen hastaların %2,5'i anti-liraglutid antikorlarıgeliştirmiştir. Antikor oluşumu liraglutidin etkililiğinin azalmasıyla ilişkilendirilmemiştir. Kardiyovasküler değerlendirmeMajör advers kardiyovasküler olaylara (MACE), harici bağımsız bir uzmanlar grubu tarafından karar verilmiş olup, ölümcül olmayan miyokard enfarktüsü, ölümcül olmayan inmeve kardiyovasküler ölüm şeklinde tanımlanmıştır. SAXENDA® ila uzun süreli klinikçalışmalarda liraglutid ile tedavi edilen hastalarda 6 MACE ve plasebo verilen hastalarda 10MACE gözlenmiştir. Olasılık oranı ve %95 GA liraglutid için plasebo karşısında 0,33 [0,12;0,90] bulunmuştur. Faz 3 klinik çalışmalarda liraglutid ile kalp atım hızında başlangıca göredakikada ortalama 2,5 atımlık bir artış gözlenmiştir (çalışmalarda 1,6 ila 3,6 atım/dakikaaralığında). Kalp atım hızı yaklaşık 6 hafta sonra pik değere ulaşmıştır. Kalp atım hızındakibu ortalama artışın uzun vadedeki klinik etkisi tespit edilmemiştir. Kalp atım hızındakideğişiklik, liraglutid kesildikten sonra geri dönüşlü olmuştur (bkz. Bölüm 4.4). Liraglutid'in Diyabetteki Etkisi ve Etkinliği - Kardiyovasküler Sonuçların Değerlendirilmesi: Liraglutidin uzun dönemli kardiyovasküler sonuç çalışması olan LEADER çalışmasına, tip 2diyabeti yeterince kontrol edilemeyen 9.340 hasta dahil edilmiştir. Bunların büyük çoğunluğukardiyovasküler hastalığa sahiptir. Hastalar, standart tedaviye ek olarak ya günlük 1,8 mgdoza kadar (4.668) liraglutid ya da plasebo (4.672) koluna randomize olarak dağıtılmıştır.Gözlem süresi 3,5 yıl- 5 yıl arasıdır. Yaş ortalaması 64 olup, ortalama BKİ 32,5 kg/m2'dir.Başlangıçta ortalama HbA1c 8,7'dir ve 3 yıl sonra liraglutid verilen hastalarda % 1,2, plaseboverilen hastalarda % 0,8 oranında iyileşmiştir. Primer sonlanım noktası, randomizasyondanitibaren herhangi bir majör kardiyovasküler advers olayın ((MACE): kardiyovasküler ölüm,ölümcül olmayan miyokard enfarktüsü veya ölümcül olmayan inme) ilk meydana geldiğizamana dek geçen süredir. 17 Liraglutid % 13'lük bir risk azalması ile, HR 0,87, [0,78, 0,97] [% 95 CI] (p = 0,005) plaseboya kıyasla (sırasıyla liraglutid ve plasebo gruplarında gözlenen 100 hasta yılı başına3,90 a karşın 3,41) majör kardiyovasküler olayların (primer sonlanım noktası, MACE) oranınıanlamlı ölçüde azaltmıştır (Bakınız şekil 4).
Şekil 4: İlk MACE'ye dek geçen sürenin Kaplan-Meier Analizi-Tam Analiz Seti Popülasyonu5.2. Farmakokinetik özelliklerGenel özelliklerEmilim:Subkutan uygulama sonrasında liraglutidin emilimi yavaş olmuş, dozdan sonraki yaklaşık 11 saatte maksimum konsantrasyona ulaşmıştır. Obez (VKİ 30-40 kg/m2) hastalarda 3 mgliraglutid uygulamasının ardından erişilen ortalama liraglutid kararlı durum konsantrasyonu(EAAt/24) yaklaşık 31 nmol/L'dir. Liraglutid maruziyeti doz ile orantılı şekilde artmıştır.Subkutan uygulama sonrasında liraglutidin mutlak biyoyararlanımı yaklaşık %55'tir. Dağılım:Subkutan uygulamayı takiben ortalama görünür dağılım hacmi 20-25 L'dir (yaklaşık 100 kg ağırlığında bir kişi için). Liraglutid büyük oranda plazma proteinine bağlanır (>%98). Biyotransformasyon:Sağlıklı gönüllülere tek bir [3H]-liraglutid dozunun uygulanmasından 24 saat sonra plazmadaki başlıca bileşen değişmemiş liraglutid olmuştur. İki minör metabolit tespitedilmiştir (toplam plazma radyoaktivite maruziyetinin <%9 ve <%5'i). Eliminasyon:Liraglutid, ana atılım yolu olarak spesifik bir organ olmaksızın büyük proteinlere benzer biçimde endojen olarak metabolize olur. Bir [3H]-liraglutid dozundan sonra idrarda ya dafeçeste değişmemiş liraglutid bulunmamıştır. Uygulanan radyoaktivitenin sadece minör birkısmı idrarda veya feçeste liraglutid ile ilişkili metabolitler şekilde atılmıştır (sırasıyla %6 ve 18 %5). İdrar ve feçes radyoaktivitesi büyük oranda ilk 6-8 gün süresince atılmıştır ve sırasıyla üç minör metabolite karşılık gelmiştir. Liraglutidin subkutan uygulanmasının ardından ortalama klirens, yaklaşık 13 saatlik bir eliminasyon yarı ömrü ile yaklaşık 0,9-1,4 L/saattir. Hastalardaki karakteristik özelliklerGeriyatrik popülasyon:Aşırı kilolu ve obez hastalara (18 ila 82 yaş) ait verilerin popülasyon farmakokinetiği analizine dayalı olarak yaşın liraglutidin farmakokinetiği üzerinde klinik olarak anlamlı biretkisi olmamıştır. Yaşa dayalı herhangi bir doz ayarlaması gerekmemektedir. Cinsiyet:Popülasyon farmakokinetiği analizlerinin sonuçlarına dayalı olarak kadınlarda ağırlığa göre düzeltilmiş liraglutid klirensi erkeklere kıyasla %24 daha düşüktür. Maruziyet-yanıt verilerinegöre, cinsiyete dayalı herhangi bir doz ayarlaması gerekmemektedir. Etnik köken:Beyaz, Siyah, Asyalı ve Hispanik/Hispanik harici gruplardan aşırı kilolu ve obez hastaları içeren popülasyon farmakokinetik analizinin sonuçlarına dayalı olarak etnik kökenin,liraglutidin farmakokinetiği üzerinde klinik olarak anlamlı herhangi bir etkisi olmamıştır. Vücut ağırlığı:Liraglutide maruziyet, başlangıç vücut ağırlığındaki artış ile azalmaktadır. Liraglutidin 3,0 mg günlük dozu, klinik çalışmalarda maruziyet yanıtı açısından değerlendirilen 60-234 kg vücutağırlığı aralığında yeterli sistemik maruziyetler sağlamıştır. Liraglutid maruziyeti >234 kgvücut ağırlığına sahip hastalarda çalışılmamıştır. Karaciğer yetmezliği:Liraglutidin farmakokinetiği, bir tek doz (0,75 mg) çalışmasında çeşitli derecelerde karaciğer yetmezliği olan hastalarda değerlendirilmiştir. Sağlıklı olgular ile karşılaştırıldığında hafif ilaorta dereceli karaciğer bozukluğu olan hastalarda liraglutid maruziyeti %13-23 azalmıştır.Şiddetli karaciğer bozukluğu (Child Pugh skoru >9) olan hastalarda ise maruziyet önemlioranda daha düşük olmuştur (%44). Böbrek yetmezliği:Bir tek doz (0,75 mg) çalışmasında liraglutide maruziyet, böbrek fonksiyonu normal olan kişiler ile karşılaştırıldığında böbrek yetmezliği olan hastalarda daha düşük bulunmuştur.Liraglutid maruziyeti, hafif (kreatinin klirensi, CrCl 50-80 mL/dk), orta (CrCl 30-50 mL/dk)ve şiddetli (CrCl <30 mL/dk) böbrek yetmezliği olan hastalarda ve diyaliz gerektiren sondönem böbrek hastalığı olan olgularda sırasıyla %33, %14, %27 ve %26 azalmıştır. Pediatrik popülasyon:Farmakokinetik özellikler sırasıyla 12-17 yaş (14 hasta, vücut ağırlığı 80-122 kg) ve 7-11 yaş (16 hasta, vücut ağırlığı 45-87 kg) olan obeziteli pediyatrik popülasyondaki klinik farmakolojiçalışmalarında değerlendirilmiştir. 19 Adölesanlardaki (12-17 yaş) liraglutid maruziyeti obeziteli yetişkinlerdeki ile benzer olmuştur. 3,0 mg liraglutid ile ilişkili maruziyetin, 7 ila 11 yaş arası çocuklar, adölesanlar veyetişkin gönüllüler arasında, vücut ağırlığı doğrulanmasından sonra karşılaştırılabilir olduğubulunmuştur. 5.3. Klinik öncesi güvenlilik verileriGüvenlilik farmakolojisi, tekrarlı doz toksisitesi veya genotoksisite için yapılan konvansiyonel çalışmalardan elde edilen klinik dışı veriler, insana özel bir tehlike ortayakoymamıştır. Sıçan ve faredeki iki yıllık karsinojenite çalışmalarında ölümcül olmayan tiroid C hücresi tümörleri görülmüştür. Sıçanda, advers etkinin görülmediği düzey (NOAEL) gözlenmemiştir.Bu tümörler 20 ay süreyle tedavi edilen maymunlarda görülmemiştir. Kemirgenlerdeki bubulgulara kemirgenlerin özellikle hassas oldukları genotoksik olmayan, spesifik GLP-1reseptörü aracılı bir mekanizma neden olmaktadır. İnsanlar için anlamlılığı olasılıkla düşüktürfakat tamamen olasılık dışı bırakılamamaktadır. Tedavi ile ilişkili başka tümör bulunmamıştır. Hayvan çalışmaları, fertilite bakımından doğrudan zararlı etkiler göstermemektedir fakat en yüksek dozda erken embriyonik ölümlerde hafif bir artış olduğuna işaret etmektedir.Gestasyonun ortasına denk gelen dönemde liraglutid uygulaması maternal ağırlıkta ve fetüsbüyümesinde azalmaya neden olmuş, sıçanda kaburga kemikleri ve tavşanda iskeletvaryasyonları üzerinde şüpheli etkiler söz konusu olmuştur. Sıçanda liraglutid maruziyetisırasında neonatal büyüme azalmış ve yüksek doz grubunda sütten kesme sonrasında bu etkidevam etmiştir. Yavruların büyümesindeki yavaşlamaya, doğrudan bir GLP-1 etkisine bağlıyavru süt alımında azalmanın mı yoksa azalmış kalori alımına bağlı olarak maternal sütüretiminde azalmanın neden olup olmadığı bilinmemektedir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiDisodyum fosfat dihidrat Propilen glikol Fenol Hidroklorik asit (pH ayarlaması için) Sodyum hidroksit (pH ayarlaması için) Enjeksiyonluk su 6.2. GeçimsizliklerSAXENDA®'ya eklenen maddeler liraglutidin bozunmasına neden olabilir. Geçimlilik çalışmaları bulunmadığından, bu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3. Raf ömrü30 ay. 20 İlk kullanımdan sonra:6.4. Saklamaya yönelik özel tedbirlerBuzdolabında (2-8°C'de) saklanmalıdır. Dondurulmamalıdır. Dondurucu kısmın uzağında tutulmalıdır. İlk kullanımdan sonra:6.5. Ambalajın niteliği ve içeriğiPolipropilen, poliasetal, polikarbonat ve akrilonitril bütadien stirenden oluşan çok dozlu, tek kullanımlık, kullanıma hazır kalem içinde bir piston (bromobütil) ve bir lamine kauçuk kapak(bromobütil/poliizopren) ile kapatılmış bir kartuş (tip 1 cam). Her bir kalem, 3 mL çözelti içerir ve 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg ve 3,0 mg dozlarını verebilir. 1, 3 ya da 5 adet kullanıma hazır kalemden oluşan ambalaj boyutları. Tüm ambalaj boyutları pazarda olmayabilir. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliği gereklerine uygun olarak imha edilmelidir. Eğer berrak ve renksiz veya renksize yakın görünümde değilse çözelti kullanılmamalıdır. Eğer donmuş ise SAXENDA® kullanılmamalıdır. Enjeksiyon kalemi, 8 mm boya ve 32G inceliğe kadar olan NovoFine® veya NovoTwist® tek kullanımlık iğne uçlarıyla kullanılmak üzere tasarlanmıştır. İğne uçları kutunun içinde bulunmamaktadır. Hastaya her enjeksiyon sonrasında kullanılan enjeksiyon iğnesini Tıbbi Atıkların Kontrolü Yönetmeliği gerekliliklere uygun olarak atması gerektiği ve enjeksiyon kalemini ucundaiğne takılı olmaksızın saklaması gerektiği bildirilmelidir. Bu, kontaminasyon, enfeksiyon veilacın kalemden sızıntı yapmasını önleyecektir. Ayrıca, dozlamanın doğru olmasını dasağlayacaktır. 7. RUHSAT SAHİBİNovo Nordisk Sağlık Ürünleri Tic. Ltd. Şti. Nispetiye Cad. Akmerkez E3 Blok Kat:7 21 34335 Etiler - İstanbul Türkiye Tel: 0 212 385 40 40Faks: 0 212 282 21 20 8. RUHSAT NUMARASI2017/452 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 21.06.2017 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ
22 1
|
İlaç BilgileriSaxenda 6 Mg/ml Sc Enjeksiyonluk Çözelti İçeren Kullanıma Hazır KalemEtken Maddesi: Liraglutid Kullanma talimatı ve kısa ürün bilgileri |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.