Feiba 1000 U Iv İnfüzyon İçin Liyofilize Toz İçeren Flakon Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIFEIBA 1000 U IV infüzyon için liyofilize toz içeren flakon Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir flakon 1000* ünite Faktör VIII İnhibitör By-pass Aktivitesine sahip 400-1200 mg insan plazma proteini içerir. FEIBA bileşiminde aktive Faktör VII yanında büyük bölümü aktif olmayan formda Faktör II, IX ve X bulunmaktadır. Her bir ünite FEIBA, 0,1 ünite kadar Faktör VIII koagülan antijeni (FVIII C:Ag) içerir. Kallikrein-kinin sisteminin faktörleri ancak eser miktarlarda bulunmaktadır. * 1 ünite FEIBA içeren çözelti, yüksek titreli faktör-VIII plazmasının aktive parsiyel tromboplastin zamanını (aPTT) tampon değerin (boş değer) %50'si kadar kısaltır. Yardımcı maddeler:Sodyum klorür 160 mg Sodyum sitrat 80 mg Yardımcı maddeler için 6.1'e bakınız. 3. FARMASOTİK FORMİntravenöz infüzyonluk çözelti için kuru toz ve çözücüsü. Kuru toz beyaz, beyaza yakın veya soluk yeşil renktedir. Sulandırıldıktan sonra oluşan çözeltinin pH'sı 6,8 ile 7,6 arasındadır. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlar- Faktör VIII inhibitörü olan hemofili A hastalarının spontan kanamalarında ve cerrahioperasyonları için - Hemofili hastası olmayıp Faktör VIII'e karşı edinsel inhibitör gelişmiş hastaların spontankanamalarında ve cerrahi operasyonlarında - Yüksek yanıtlı inhibitörü olan (İnhibitör düzeyi>5BU veya inhibitör düzeyi<5BU vefaktör 8'in tekrar verilmesi sonrasında en az bir defa>5BU olan) sık eklem kanaması olanhemofili A hastalarının profilaksisinde 1 Faktör IX eksikliği olan inhibitör geliştirmiş hastaların kanama tedavisi ve cerrahisi için kullanılabilecek alternatiflerin azlığı ve ilacın etki mekanizması dikkate alındığında- Eğer spesifik başka bir tedavisi mevcut değilse, inhibitör gelişmiş hemofili B hastalarındakanamaların tedavisinde ve cerrahisinde endikedir. 4.2. Pozoloji ve uygulama şekliTedavi koagülasyon bozuklukları konusunda uzman bir hekim tarafından başlatılmalı ve bu hekimin gözetiminde devam ettirilmelidir. Pozoloji / uygulama sıklığı ve süresiPozoloji Uygulanacak dozun miktarı, zamanlaması, uygulamanın ne kadar tekrarlanacağı ve tedavinin süresi, kanamanın şiddeti, yeri ve yayılımı yanında hastanın klinik durumuna göre değişir. Dozaj ve uygulama sıklığı her olguda klinik etkililiğe göre ayarlanmalıdır. Genel olarak 50-100 Ünite/kg FEIBA dozu önerilmektedir; kanama şiddetinin daha yüksek doz kullanımını gerektirmediği durumlarda tek uygulamada 100 Ünite/kg ve günlük olarak 200Ünite/kg dozları aşılmamalıdır (Bkz. Bölüm 4.4). Çocuklarda kullanımı:6 yaş altı çocuklardaki kullanımıyla ilgili deneyim yetersizdir. Çocuklarda klinik duruma göre erişkinlerdeki aynı doz şeması adapte edilmelidir. 1) Spontan kanamalarKas, eklem ve yumuşak doku kanamaları:Küçük ve orta dereceli kanamalarda önerilen doz 12 saatlik aralarla 50-75 Ünite/kg'dır. Tedaviye, ağrının kaybolması, eklem şişkinliğinin azalması ya da hareketliliğinin artışı gibiklinik düzelme belirtileri görülene kadar devam edilmelidir. Retroperitonal kanama gibi büyük kas ve yumuşak doku kanamalarında önerilen doz 12 saatlik aralıklarla 100 Ünite/kg'dır. Mukoza kanamaları:Hasta dikkatle izlenerek (kanama bölgesi görülerek, hematokrit ölçümleri tekrarlanarak) 6 saatte bir 50 Ünite/kg önerilir. Kanama durmazsa, günde 200 Ünite/kg'nin üzerine çıkmamayadikkat edilerek doz 100 Ünite/kg'a yükseltilebilir. Diğer ciddi kanamalar:Merkezi sinir sistemi kanamaları gibi ciddi kanamalarda 12 saatlik aralıklarla verilen 100 Ünite/kg dozu önerilir. Bazı hastalara FEIBA, belirgin klinik düzelme görülene kadar 6 saatlik 2 aralarla uygulanabilir. Günlük en yüksek doz olan 200 Ünite/kg'lık doz aşılmamalıdır. 2) Cerrahi girişimlerCerrahi girişimlerde başlangıç dozu olarak ameliyat öncesinde 100 Ünite/kg'lık bir doz ve 612 saat sonra 50-100 Ünite/kg'lık bir doz daha uygulanabilir. Postoperatif idame dozu olarak 6-12 saat aralıklarla 50-100 Ünite/kg'lık dozlar uygulanabilir; doz aralıkları ve ameliyatöncesi ve sonrası tedavinin süresi, uygulanan cerrahi girişime, hastanın genel durumuna vebireysel olarak hastada sağlanan klinik etkinliğe göre belirlenir. (200 Ünite/kg'lık maksimumgünlük doz aşılmamalıdır.) 3) İnhibitör gelişmiş hemofili A hastalarında profilaksi- Yüksek inhibitör titresine sahip ve başarısız olmuş ITI (İmmün tolerans tedavisi) sonrasısık kanamaları olmuş hastalarda veya ITI düşünülmeyen hastalarda kanama profilaksisi:Günaşırı 70-100 Ünite/kg'lık bir doz önerilmektedir. Duruma göre doz günde 100Ünite/kg'a yükseltilebilir ya da giderek azaltılabilir. - Yüksek inhibitör titresine sahip hastalarda ITI (immün tolerans tedavisi) almaktaykengörülen hastaların profilaksisi: FEIBA ,faktör VIII inhibitör titresi <2 BU* (Bethesda Unit) oluncaya kadar günde iki kez 50-100 Ünite/kg dozunda ve faktör VIII uygulamasıyla eş zamanlı olarak kullanılabilir.* 1 Bethesda Ünitesi inkübe edilmiş (37°C'de 2 saat) plazmanın faktör VIII etkinliğinin %50'sini inhibe eden antikor düzeyi olarak tanımlanmıştır.4) FEIBA'nın özel hasta gruplarında kullanımıFaktör IX'a karşı inhibitör gelişmiş hemofili B hastalarındaki kullanımıyla ilgili Bölüm 5.1'e bakınız. Faktör VIII inhibitörlerinin tam ve kalıcı olarak eliminasyonunu sağlamak için FEIBA, Faktör VIII konsantreleri ile kombine olarak uzun süreli tedavilerde de kullanılmıştır. Tedavinin izlenmesiFEIBA'nın etkili olabilmesi için hastada yeterli sayıda ve fonksiyonel olarak sağlam trombosit bulunması gerektiğinden FEIBA ile yürütülen tedaviye yanıt yetersizse birtrombosit sayımı yapılması önerilir. FEIBA'nın etki mekanizmasının kompleks olmasından dolayı etkin maddenin doğrudan izlemi gerçekleştirilemez. Tam kan pıhtılaşma zamanı (WBCT), tromboelastogram (TEG, rdeğeri) ve aPTT değerlerinde genellikle ancak az bir azalma görülür ve klinik etkililik ilekorelasyon göstermeyebilir. Bu nedenle FEIBA tedavisinin izlenmesinde bu testlerin önemidüşüktür (Bkz. Bölüm 4.4). Uygulama şekli:3 Ürünü aşağıda tarif edildiği şekilde sulandırarak kullanıma hazırlayınız ve intravenöz yoldan yavaş infüzyon yoluyla uygulayınız. Uygulama hızı hastanın rahatlığını sağlamalı ve dakikada maksimum 2 Ünite/kg vücut ağırlığını geçmemelidir. FEIBA koruyucu içermediğinden uygulamadan hemen önce sulandırılmalı ve hazırlanan çözelti derhal kullanılmalıdır. Bütün içerik çözünene kadar yavaşça çalkalayınız. FEIBA'nın tamamen çözünmüş olduğundan emin olunmalıdır; aksi takdirde cihazın filtresinden daha az etkin madde geçer. Sulandırıldıktan sonra oluşan çözeltinin herhangi bir partikül içerip içermediği ve renk değişikliği olup olmadığı kontrol edilmelidir. Bulanık olan ya da tortu içeren çözeltilerkullanılmamalıdır. Ambalajı açılmış ürün yeniden kullanılmamalıdır. Steril bariyeri bozulan, ambalajı hasar görmüş ya da bozulma belirtisi gösteren ürünü kullanılmamalıdır. Sulandırmak için yalnızca ambalajında bulunan enjeksiyonluk su ve sulandırma cihazı kullanılmalıdır. FEIBA kutusu dışındaki bir cihaz kullanılması durumunda uygun bir filtre (en az 149 mikrometre çapında deliği olan) kullanılmalıdır. Liyofilize flakonun BAXJECT II Hi-Flow cihaz kullanılarak sulandırılması:1. Gerekliyse çözücü (enjeksiyonluk su) içeren açılmamış flakonu oda sıcaklığına (15°C - 25°C) kadar ısıtınız. Bu işlem için birkaç dakika süreyle ılık su banyosu (maksimum 37°C)kullanılabilir. 2. Kuru toz ve çözücü flakonlarının koruyucu kapaklarını çıkarınız ve her ikisinin de lastiktıpalarını dezenfekte ediniz. Flakonları düz bir yere yerleştiriniz. 3. BAXJECT II Hi-Flow cihazının ambalajını, ambalajdaki kağıt kapağı çekerek cihazıniçine dokunmadan açınız (Şekil a). Bu noktada transfer cihazını, ambalajın içindençıkarmayınız. 4. Ambalajı ters çeviriniz ve şeffaf plastik delici kısmını çözücü flakonun tıpasınauygulayınız (Şekil b). Ambalajı kenarından tutarak BAXJECT II Hi-Flow cihazınınüzerinden çıkarınız (Şekil c). BAXJECT II Hi-Flow cihazının üzerindeki mavi kapağıçıkarmayınız. 5. Çözücü flakon ve BAXJECT II Hi-Flow cihaz kombinasyonunu, çözücü flakon yukarıyagelecek şekilde çeviriniz. Diğer mor renkli plastik delici ucu, toz konsantrenin bulunduğuflakonun tıpasına uygulayınız. Çözücü vakum etkisiyle konsantrenin bulunduğu flakonuniçerisine çekilecektir (Şekil d). 6. Bütün içerik çözünene kadar çalkalamadan hafifçe karıştırınız. FEIBA'nın tamamen 4 çözünmüş olduğundan emin olunmalıdır; aksi takdirde, etkin madde cihazın filtresinden geçmeyecektir.
Şekil a
Şekil c
Şekil b    Enjeksiyon / İnfüzyon için talimatlar:1. BAXJECT II Hi-Flow cihazındaki mavi koruyucu kapağı çıkarınız. Enjektörü BAXJECT II Hi- Flow cihazına iliştiriniz (ENJEKTÖRE HAVA ÇEKMEYİNİZ) (Şekil e). Ürününenjektör ve BAXJECT II Hi-Flow cihazına iliştiğinden emin olmak için, bir luer kilitlienjektör kullanımı önemle tavsiye edilir. (Birleştirirken dur pozisyonuna kadar enjektörüsaat yönünde çeviriniz.) 2 Bileşkeyi ters çeviriniz (FEIBA flakonu üstte kalacak şekilde). Pistonunu YAVAŞÇA geriye doğru çekerek FEIBA çözeltisini enjektöre çekiniz ve çekme sırasında ürününenjektör ve BAXJECT II Hi-Flow cihazı arasındaki bağlantının sağlandığından eminolunuz (Şekil f). 3. Enjektörü çıkarınız. 4. Enjektörde ürün içinde köpük meydana gelirse, köpük geçene kadar bekleyiniz.Enjektördeki çözeltiyi kelebek infüzyon seti (ya da tek kullanımlık iğne) ile yavaşçaintravenöz enjeksiyon şeklinde uygulayınız. 5
Şekil d
Şekil e
Şekil f  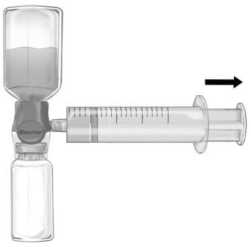  Dakikada maksimum infüzyon hızı her kilogram için 2 ünite'yi aşmamalıdır.FEIBA ile birlikte verilenlerden başka cihazlar kullanılıyorsa, yeterli bir filtrenin kullanıldığından emin olun .Uygulama hızı:Dakikada 2 Ünite/kg infüzyon hızı aşılmamalıdır. Özel popülasyonlara ilişkin ek bilgiler:Böbrek / Karaciğer yetmezliği:Böbrek yetmezliğinde kullanımına ilişkin yeterli veri bulunmamaktadır. Karaciğer fonksiyon testleri bozulmuş olan hastalarda aktive koagülasyon faktörlerinin eliminasyonunun uzamasından dolayı yaygın damar içi pıhtılaşma (DIC) gelişimi riski artar. Pediyatrik popülasyon:6 yaş altı çocuklardaki kullanımıyla ilgili deneyim yetersizdir. Çocuklarda klinik duruma göreerişkinlerdeki aynı doz şeması adapte edilmelidir. Geriyatrik popülasyon:FEIBA'nın geriyatrik popülasyonda kullanımı ile ilgili yalnızca kısıtlı klinik çalışma verileri bulunmaktadır. 4.3. KontrendikasyonlarAşağıdaki durumlarda alternatif terapötik bir tedavi varsa FEIBA kullanılmamalıdır: Ürünün içerdiği etkin maddeye veya yardımcı maddelerden herhangi birine karşı aşırıduyarlılığın bulunması Dissemine İntravasküler Koagülasyon (yaygın damar içi pıhtılaşma; DIC) Akut tromboz veya emboli (miyokart infarktüsü dahil) (Bkz. Bölüm 4.4). 6 4.4. Özel kullanım uyarıları ve önlemleri Virüs güvenliğiFEIBA insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler, ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibi, çeşitlihastalıklara yol açabilen enfeksiyon yapıcı ajanlar içerebilirler. FEIBA'da VaryantCreutzfeldt- Jacob hastalığının bulaşma riski teorik olarak minimumken, klasikCreutzfeldt-Jacob hastalığının bulaşma riski hiçbir kanıtla desteklenmez. Alınanönlemlere rağmen, bu tür ürünler halen potansiyel olarak hastalık bulaştırabilir.Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıp kalmadığının izlenmesi, belirli virüsenfeksiyonlarının halihazırda varlığının test edilmesi ve belirli virüslerin yokedilmesi ve/veya inaktivasyonu ile azaltılmıştır. Bütün bu önlemlere rağmen, buürünler hala potansiyel olarak hastalık bulaştırabilirler. Ayrıca, henüz bilinmeyenenfeksiyon yapıcı ajanların bu ürünlerin içerisinde bulunma ihtimali mevcuttur.HIV, HBV, HCV gibi zarflı virüsler ve HAV gibi zarflı olmayan virüslerin etkisi için önlemlerin alınmasına dikkat edilmelidir. Parvovirus B19 gibi zarflı olmayanvirüslere karşı alınan tedbirler sınırlı sayıda olabilir. Parvovirüs B19 enfeksiyonu,gebelikte (fetal infeksiyon) ve immün yetmezlik ya da kırmızı kan hücre üretimindeartış olan hastalarda tehlikeli olabilir (hemolitik anemi gibi).Doktor, bu ilacı hastaya reçete etmeden veya uygulamadan önce hastası ile risk ve yararlarını tartışmalıdır.İzlenebilirlikBiyolojik tıbbi ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. İnsan plazması kaynaklı faktör VIII inhibitörü ürünlerini düzenli/tekrarlayan şekilde alan hastalarda uygun aşılama (Hepatit A ve B'ye karşı) düşünülmelidir. UYARILAR Aşırı Duyarlılık ReaksiyonlarıFEIBA ürtiker, anjiyoödem, gastrointestinal belirtiler, bronkospazm ve hipotansiyon gibi alerjik tipte aşırı duyarlılık reaksiyonlarının ortaya çıkışını hızlandırabilir. Bu reaksiyonlarciddi ve sistemik olabilir (örn. ürtiker ve anjioödem ile anafilaksi, bronkospazm ve kardiyakşok). Titreme, ateş ve hipertansiyon gibi diğer infüzyon reaksiyonları da raporlanmıştır.Hastalar eritem, deri döküntüsü, yaygın ürtiker, kaşıntı, solunum zorluğu/dispne, göğüstesıkışma hissi, genel bir keyifsizlik durumu, baş dönmesi/sersemlik hali, hafif bir düşmedenalerjik şoka değişen kan basıncı azalmaları gibi aşırı duyarlılık reaksiyonlarının erken 7 belirtileri konusunda bilgilendirilmelidir. Bir infüzyon/aşırı duyarlılık reaksiyonunun ilk belirti ve semptomlarında, FEIBA uygulaması durdurulmalı ve gerekli medikal tedavibaşlatılmalıdır. Ürüne veya bileşenlerinden herhangi birine karşı aşırı duyarlılığı olduğu bilinen ya da kuşkulanılan hastalarda FEIBA'yı yeniden kullanmadan önce, hastadaki bilinen ya dakuşkulanılan aşırı duyarlılığın tipi (alerjik ya da alerjik olmayan) ile potansiyel düzelticive/veya önleyici tedaviler ya da alternatif terapötik ajanlar göz önünde bulundurularak yenidenkullanımdan sağlanacak faydalarla olası risk dikkatle karşılaştırılmalıdır. İnhibitörlerFaktör VIII'e karşı nötralize edici antikor (inhibitörler) oluşumu, hemofili A hastalarının tedavisinde bilinen bir komplikasyondur. Bu inhibitörler genellikle faktör VIII prokoagülanaktiviteye yönelik olan IgG immünoglobülinleridir ve modifiye tetkik kullanılarak her mlplazmada Bethesda Ünitesi (BU) olarak ölçülür. İnhibitör gelişme riski, faktör VIII'emaruziyetin yanı sıra hastalığın şiddeti ile ilişkilidir ve bu risk ilk 50 maruziyet gününde enyüksek seviyededir; ancak risk yaygın görülmemesine rağmen yaşam boyu devam eder. İnhibitör gelişiminin klinik önemi inhibitör titresine bağlı olacaktır; düşük titrenin teşkil ettiği yetersiz klinik yanıt riski, yüksek titreli inhibitörlere kıyasla daha az olacaktır. Genel olarak, koagülasyon faktörü VIII ürünleri ile tedavi edilen tüm hastalar, uygun klinik gözlem ve laboratuvar testleri ile inhibitörlerin gelişimi açısından dikkatle izlenmelidir. Eğerbeklenen faktör VIII aktivitesinin plazma düzeylerine ulaşılamazsa veya yeterli doz ilekanama kontrol altına alınamazsa faktör VIII inhibitörü varlığı açısından test yapılmalıdır.İnhibitör düzeyleri yüksek olan hastalarda faktör VIII tedavisi etkili olmayabilir ve diğertedavi seçenekleri dikkate alınmalıdır. Böyle hastaların tedavisi hemofili ve faktör VIIIinhibitörleri tedavisi konusunda deneyimli hekimler tarafından yönlendirilmelidir. Trombotik ve Tromboembolik OlaylarFEIBA tedavisi sırasında yaygın damar içi pıhtılaşma (DIC), venöz tromboz, pulmoner embolizm, miyokard enfarktüsü ve inme dahil olmak üzere trombotik ve tromboembolikolaylar oluşmuştur. Bu olayların bazıları 200 Ünite/kg/gün'den yüksek dozlarla ya da tromboembolik olayların olduğu diğer risk faktörlerinin (DIC, ileri aterosklerotik hastalık, ezilme yaralanması veyasepsis gibi) bulunduğu hastalarda görülmüştür. Eş zamanlı rekombinant Faktör VIIa kullanılanhastalarda tromboembolik olay gelişim riski artabilir. FEIBA'nın yüksek dozlarındatrombotik ve tromboembolik olayların riski artabilir. Böyle risk faktörlerinin olası varlığı her zaman doğumsal ve edinsel hemofili hastalarında düşünülmelidir. FEIBA artmış tromboembolik komplikasyon riski olan hastalarda, sadece hiçbir terapötik 8 alternatif olmaması durumunda ve dikkatle kullanılmalıdır. Tromboembolik komplikasyon riskinin arttığı durumlar; bunlar ile sınırlı olmamak üzere: hastanın özgeçmişinde koronerkalp hastalığı, karaciğer hastalığı, DIC, arteriyel veya venöz tromboz bulunması, hastanınpost-operatif immobilizasyon döneminde bulunması ile hastanın geriyatrik hasta veya yenidoğan olmasıdır. FEIBA ile yapılan klinik çalışmalarda trombotik mikroanjiopati (TMA) bildirilmemiştir. TMA vakaları deneklerin ani gelişen kanamalara yönelik aldıkları tedavi rejiminin bir parçası olarakFEIBA kullanılan emisizumab çalışmasında bildirilmiştir (bkz. emisizumabın Avrupa KamuDeğerlendirme Raporu'nda (EPAR) bulunan klinik tartışma; ayrıca bkz. Oldenburg ve ark.,Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med 2017:377:809-818).FEIBA'nın emisizumab alan hastalarda ani kanamalar için güvenliliği ve etkililiği henüzkanıtlanmamıştır. Bu nedenle, emisizumaba maruz kalan hastalarda FEIBA için yarar-risk değerlendirmesi yapılması gerekmektedir ve hastalar hekimleri tarafından yakından izlenmelidir (ayrıca bkz.bölüm 4.5). Trombotik ve tromboembolik olayların işaret ve semptomları görülür görülmez infüzyon hemen durdurulmalı ve uygun tanısal ve terapötik önlemler uygulanmaya başlanmalıdır. Kanamanın ciddiyeti daha yüksek doz kullanımını gerektirmedikçe, tek uygulamada 100 Ünite/kg ve günlük 200 Ünite/kg dozları aşılmamalıdır. Ürün kanamayı durdurmak içinkullanıldığında, terapötik başarıyı sağlamak için sadece kesin olarak gerektiği süre boyuncaverilmelidir. Tedavinin İzlenmesiTek uygulamada 100 Ünite/kg'lık ve günlük olarak 200 Ünite/kg'lık dozlar aşılmamalıdır. 100 Ünite/kg ve üzeri dozların uygulandığı hastalar dikkatle, özellikle de DIC ve/veya akutkoroner iskemi semptomlarının gelişimi ve diğer trombotik veya tromboembolik olaylarınsemptomları açısından izlenmelidir. Yüksek dozlarda FEIBA ancak kesin olarak gerektiğisüre boyunca - kanama durana kadar - kullanılmalıdır. Kan basıncı veya kalp atım hızında klinik olarak anlamlı değişiklikler, solunum zorluğu, öksürük ya da göğüs ağrısı ortaya çıkarsa infüzyon derhal durdurulmalı, uygun tanı ve tedaviönlemler uygulanmaya başlanmalıdır. Fibrinojen düzeylerinde bir azalma olması, trombositsayısında bir azalma olması ve/veya fibrin/fibrinojen yıkım ürünlerinin (FDP) ortaya çıkmasıDIC açısından anlamlı laboratuvar parametreleridir. Diğer DIC göstergeleri arasında trombinzamanı, protrombin zamanı ve aPTT'nin açık bir şekilde uzaması bulunur. İnhibitörlühemofili hastalarında veya faktör VIII, faktör IX ve/veya faktör XI'e karşı kazanılmışinhibitörü olan hastalarda aPTT, altta yatan hastalığa bağlı olarak uzayabilir. FEIBA tedavisi uygulanan inhibitörlü hemofili hastalarında veya koagülasyon faktörlerine karşı kazanılmış inhibitörleri olan hastalarda aynı zamanda kanamaya eğilim ve tromboz riski 9 artabilir. Laboratuvar testleri ve klinik etkililikEtkililiğin kanıtı olabilecek aPTT, tam kan pıhtılaşma zamanı (WBCT) ve tromboelastogram (TEG) gibi in vitrotestlerle klinik tablonun korelasyon göstermesi şart değildir. Bu nedenlebu değerleri normale döndürmek için FEIBA dozu arttırılmamalıdır; aksine olası bir aşırı dozabağlı DIC olayını tetikleyebilme riski nedeniyle kesinlikle dozun arttırılmaması gerekir.Trombosit sayısının önemiFEIBA'nın etkili olabilmesi için hastada yeterli sayıda ve fonksiyonel olarak sağlam trombosit bulunması gerektiğinden FEIBA ile yürütülen tedaviye yanıt yetersizse bir trombosit sayımıyapılması önerilir. ÖNLEMLER Trombotik ve Tromboembolik KomplikasyonlarAşağıdaki durumlarda FEIBA yalnızca uygun koagülasyon faktörü konsantreleri kullanılarak yapılan tedaviye yanıt beklenmediğinde, örneğin yüksek bir inhibitör titresi varlığı ve yaşamıtehdit edebilen boyutta (örn. travma veya ameliyat sonrası) bir kanama veya kanama riskivarsa kullanılabilir: - Yaygın damar içi pıhtılaşma (DIC): Laboratuvar bulgularının ve/veya klinik semptomlarınbulunması. - Karaciğer hasarı: Aktive koagülasyon faktörlerinin klerensinin uzamasından dolayıkaraciğer işlevleri bozulmuş hastalarda DIC gelişmesi riski artmıştır. - Koroner kalp hastalığı, akut tromboz ve/veya embolizm. FEIBA kullanan hastalar DIC, akut koroner iskemi ve diğer trombotik ve tromboembolik olayların işaret ve semptomlarının gelişimi açısından izlenmelidir. Trombotik vetromboembolik olayların ilk işaret ve semptomları görülür görülmez infüzyon hemendurdurulmalı ve uygun tanısal ve terapötik önlemler uygulanmaya başlanmalıdır. By-pass Ajanlarına Karşı Yetersiz YanıtHastaya özel faktörler nedeniyle by-pass yapan bir ajana yanıt değişik olabilmekte ve belirli bir kanama durumunda bir ajanla yetersiz yanıt alınan hastalarda, başka bir ajanla yanıtalınabilmektedir. By-pass yapan bir ajanla yetersiz yanıt alınması durumunda, diğer bir ajanınkullanılması düşünülmelidir. Anamnestik Yanıtİnhibitörlü hastalarda FEIBA uygulanması başlangıçta inhibitör düzeylerinde anamnestik bir yükselmeye yol açabilmektedir. FEIBA uygulamaya devam edildiğinde, inhibitörler zamanlaazalabilir. Klinik ve yayınlanmış veriler FEIBA'nın etkililiğinin azalmadığını öngörmektedir. 10 Laboratuvar Testleri ile EtkileşimYüksek dozda FEIBA uygulaması, pasif olarak geçen Hepatit B yüzey antikorlarının geçici yükselişi, serolojik testlerde pozitif sonuçların yanlış yorumlanmasına neden olabilir. FEIBA, kan grubu izohemaglütininleri (anti-A ve anti-B) içermektedir. Antikorların eritrosit antijenlerine (örn. A, B, D) pasif transmisyonu, antiglobülin testi (Coombs testi) gibi alyuvarantikorlarına yönelik bazı serolojik testlerle etkileşime girebilir. Pediyatrik popülasyonVaka raporları ve kısıtlı klinik çalışma verileri FEIBA'nın 6 yaş altı çocuklarda kullanılabileceğini göstermektedir. Çocuklarda klinik duruma göre erişkinlerdeki aynı dozşeması adapte edilmelidir. Geriyatrik popülasyonFEIBA'nın geriyatrik popülasyonda kullanımı ile ilgili yalnızca kısıtlı klinik çalışma verileri bulunmaktadır. İnhibitörlü hemofili B hastalarında profilaktik kullanımHastalığın nadir görülmesi nedeniyle hemofili B hastalarındaki kanamanın profilaksisinde FEIBA uygulamasıyla ilişkili olarak ancak kısıtlı klinik veri (vaka raporu olarak 4 hasta ve090701 profilaksi çalışmasında 1 hasta) bulunmaktadır. Enfeksiyöz ajanların bulaşmasıİnsan kanı ya da plazmasından hazırlanan tıbbi ürünlerin kullanımından kaynaklanan enfeksiyonların önlenmesi için alınan standart önlemler arasında, donörlerin seçimi, bireyselbağışların ve plazma havuzlarının belirli enfeksiyon göstergeleri için takibi ve virüslerininaktivasyonu/uzaklaştırılması için etkili üretim aşamalarının kullanılması yer almaktadır.Buna rağmen insan kanı ya da plazmasından hazırlanan tıbbi ürünler uygulandığında,enfeksiyon ajanlarının bulaşma olasılığı tam olarak ortadan kaldırılamayabilir. Bu durumhenüz bilinmeyen ya da yeni ortaya çıkan virüsler ve diğer hastalık etkenleri için de geçerlidir. Alınan önlemlerin HIV, HBV, HCV gibi zarflı virüslerle HAV gibi zarfsız virüsler için etkili olduğu düşünülmektedir. Alınan önlemlerin Parvovirüs B19 gibi bazı zarfsız virüslere karşıetkisi ise kısıtlıdır. Parvovirus B19 virüsü en ciddi olarak gebe kadınları (fetusta enfeksiyonaneden olabilmektedir), immün yetmezlikli hastaları veya artmış eritrosit döngüsü olanhastaları (örn. hemolitik anemi durumu) etkilemektedir. Yardımcı maddeler ile ilişkili durumlarFEIBA her flakonunda yaklaşık 80 mg sodyum içerir. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır. 11 Bu miktar, bir yetişkin için Dünya Sağlık Örgütü (WHO) tarafından önerilen maksimum günlük 2 g sodyum alımının %4'üne eşdeğerdir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriFEIBA ve rekombinant Faktör VIIa, antifibrinolitiklerin veya emisizumabın kombine ve ardışık kullanımı ile ilgili yeterli ve kontrollü çalışmalar yürütülmemiştir. Traneksamik asit veaminokaproik asit gibi sistemik antifibrinolitikler FEIBA tedavisinde birlikte kullanıldığındatromboembolik olayların olasılığı dikkate alınmalıdır. Bu nedenle, FEIBA uygulamasındansonra yaklaşık 6 ila 12 saatlik sürede antifibrinolitikler kullanılmamalıdır. Rekombinant Faktör VIIa ile eş zamanlı kullanımı durumunda, mevcut in vitroverilere ve klinik gözlemlere göre olası bir ilaç etkileşimi (potansiyel olarak tromboembolik olay gibiadvers olaylarla sonuçlanan) göz ardı edilemez.Emisizumabla yapılan bir klinik çalışmada elde edilen klinik deneyim, ani kanamaya yönelik uygulanan bir tedavi rejimi kapsamında FEIBA ile birlikte kullanıldığında emisizumabla birilaç etkileşimi potansiyeli olduğuna ve bunun sonucunda tromboembolik olaylar ve trombotikmikroanjiopati olabileceğine işaret etmektedir (bkz. Bölüm 4.4). Özel popülasyonlara ilişkin ek bilgilerFEIBA'nın rekombinant faktör VIIa ya da antifibrinolitik ile birlikte veya ardışık kullanımına dair yeterli ve iyi kontrollü çalışmalar yoktur (pediyatik ve özel popülasyonlar dahil). Pediyatrik popülasyonFEIBA'nın rekombinant faktör VIIa ya da antifibrinolitik ile birlikte veya ardışık kullanımına dair yeterli ve iyi kontrollü çalışmalar yoktur (pediyatik ve özel popülasyonlar dahil). 4.6. Gebelik ve laktasyonGenel tavsiye:Gebelik Kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon):Hasta hamile kaldığında veya hamilelik kararı aldığında doktorunu bilgilendirmesi gerektiği hususunda uyarılmalıdır. Doğum kontrol yöntemleri ile herhangi bir etkileşimbildirilmemiştir. Hekim FEIBA'nın ve bazı hormonal kontraseptiflerin protrombotikrisklerini göz önünde bulundurarak uygun bir kontrasepsiyon yöntemine karar vermelidir. Gebelik dönemiFEIBA'nın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan araştırmalar, gebelik / ve-veya / embriyonal / fetal gelişim / ve-veya / doğum / ve-veya / doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir. FEIBA gerekli olmadıkça gebelik döneminde kullanılmamalıdır (bkz. bölüm 5.3). İnsanlarayönelik potansiyel risk bilinmemektedir. 12 Hekimler potansiyel riskleri değerlendirmeli ve gebelik ve doğum sonrası dönemde artan tromboembolik olay riski ve artan DIC riski ile ilişkili hamilelik komplikasyon riskini gözönünde bulundurarak sadece açıkça gerekliyse FEIBA reçete etmelidir. Dikkatli tıbbi izlemgereklidir. Parvovirüs B19 enfeksiyonu ile ilgili bilgi için Bölüm 4.4'e bakınız. Laktasyon dönemiFEIBA'nın emziren kadınlarda kullanımı ile ilgili yeterli veri bulunmamaktadır. FEIBA emziren annelerde kullanılmamalıdır. Üreme yeteneği / FertiliteFEIBA ile hayvan üreme çalışmaları gerçekleştirilmemiştir ve kontrollü klinik çalışmalarda FEIBA'nın fertilite üzerindeki etkileri belirlenmemiştir. 4.7. Araç ve makine kullanımı üzerindeki etkilerFEIBA'nın araç ve makine kullanımı yeteneği üzerinde etkisi yoktur ya da ihmal edilebilir düzeydedir. 4.8. İstenmeyen etkilerGüvenlilik profili özetiAşırı duyarlılık ya da alerjik reaksiyonlar (bunlar arasında anjiyoödem, infüzyon bölgesinde yanma ve batma, titreme, sıcak basması, yaygın ürtiker, baş ağrısı, kurdeşen, kan basıncındadüşme, letarji, bulantı, huzursuzluk, taşikardi, göğüste sıkışma hissi, karıncalanma, kusma,hırıltılı solunum yer alabilir) nadiren gözlenmiştir ve bazı olgularda şiddetli anafilaksiye kadarilerleyebilir (şok dahil). İlgili aşırı duyarlılık reaksiyonları ile birlikte, fare, sığır ve/veya hamster proteinine karşı antikor gelişini çok nadiren gözlenmiştir. FEIBA da dahil olmak üzere faktör VIII ile tedavi edilmiş hemofili A hastalarında nötralize edici antikorlar (inhibitörler) gelişebilir (bkz. bölüm 5.1). Bu tür inhibitörler oluşursa, durum,yetersiz klinik yanıt şeklinde kendini gösterebilir. Bu gibi durumlarda uzman hemofilimerkezleriyle bağlantı kurulması önerilmektedir. Bu bölümde sunulan advers reaksiyonlar, kanama öyküsü olan pediyatrik ve yetişkin hastaların tedavisi için Hemofili A veya B hastalarında ve Faktör VIII veya IX'a inhibitörlühastalarda FEIBA ile yürütülen 2 çalışmanın yanı sıra pazarlama sonrası deneyimlerdenraporlanmıştır. Bir çalışma ayrıca Faktör VIII'e karşı inhibitör gelişmiş hemofili hastalarında(49 hastadan 2'si) yürütülmüştür. Ek olarak advers reaksiyonlar arasında kanadıkça tedavi ileprofilaktik tedaviyi karşılaştıran üçüncü bir çalışmada görülen advers reaksiyonlar da yeralmaktadır. Advers ilaç reaksiyonlarının sıklık sınıflandırması şu şekildedir: Çok yaygın (>1/10); yaygın 13 (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000), çok seyrek, izole raporlar dahil (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahminedilemiyor). Kan ve lenf sistemi hastalıklarıÇok yaygın: *Faktör VIII inhibisyonu (HTGH) Yaygın olmayan: *Faktör VIII inhibisyonu (TGH) Bilinmiyor: Yaygın damar içi pıhtılaşma (DIC) İnhibitör düzeyinde yükselme (anamnestik yanıt)(a) *Sıklık, şiddetli hemofili A hastalarının yer aldığı, tüm FVIII ürünleriyle ilgili çalışmalara dayanmaktadır. HTGH (Daha önce hiç tedavi görmemiş hastalar) TGH (Daha önce tedavi görmüş hastalar) Bağışıklık sistemi hastalıklarıYaygın: Aşırı duyarlılıkc Bilinmiyor: Ürtiker Anafilaktik reaksiyon Sinir sistemi hastalıklarıYaygın: Başağrısıc Baş dönmesi / sersemlik halib Bilinmiyor: Parestezi Hipoestezi Trombotik inmeEmbolik inmeUykuya meyil (Somnolans) Tat alma bozukluğu (Disguzi) Kardiyak hastalıklarBilinmiyor: Miyokard infarktüsü Taşikardi Vasküler hastalıklarYaygın: Hipotansiyonc Bilinmiyor: Tromboz Venöz tromboz Arteriyel tromboz Embolizm (tromboembolik komplikasyonlar) Hipertansiyon Yüz ve boyunda kızarma (flushing) 14 Solunum, göğüs bozuklukları ve mediastinal hastalıklarBilinmiyor: Pulmoner embolizm Bronkospazm Hırıltılı soluk alıp vermeÖksürük Zorlu soluk alıp verme (dispne) Gastrointestinal hastalıklarBilinmiyor: Kusma Diyare Karında rahatsızlık hissi Bulantı Deri ve deri altı doku hastalıklarıYaygın: Döküntüc Bilinmiyor: Yüzde hissizlik Anjiyoödem Ürtiker Kaşıntı (Pirürit) Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarBilinmiyor: Enjeksiyon yerinde ağrı Halsizlik Sıcaklık hissiTitreme Ateş yükselmesi Göğüste ağrıGöğüste rahatsızlık hissi AraştırmalarYaygın: Hepatit B yüzey antikorunun pozitifleşmesi Bilinmiyor: Kan basıncında düşme (a)(b)(c)MedDRA sınıflandırma sisteminde yer almayan inhibitör düzeyinde yükselme (anamnestik yanıt) FEIBA uygulanmasından sonra mevcut inhibitör düzeylerinde olanyükselmedir (Bkz Bölüm 4.4). Orijinal ve profilaksi çalışmalarında raporlanan advers ilaç reaksiyonudur. Yalnızca profilaksi çalışmasında bulunan sıklıktır. Profilaksi çalışmalarında raporlanan advers ilaç reaksiyonudur. Yalnızca profilaksi çalışmasında bulunan sıklıktır. Sınıf reaksiyonları Plazma kaynaklı ürünlere karşı gelişen aşırı duyarlılık reaksiyonlarının diğer semptomlarına 15 letarji ve huzursuzluk dahildir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99) 4.9. Doz aşımı ve tedavisiFEIBA'nın yüksek dozları trombotik ve tromboembolik olayların riskini ((DIC), miyokard infarktüsü, venöz tromboz ve pulmoner embolizm dahil) arttırabilir. Raporlanantromboembolik advers olayların bazıları 200 Ünite/kg'dan yüksek dozlarda veyatromboembolik olaylar için diğer risk faktörleri olan hastalarda meydana gelmiştir. Trombotikve tromboembolik olayların işaret ve semptomları görülürse, infüzyon hemen durdurulmalı veuygun tanısal ve terapötik önlemler uygulanmaya başlanmalıdır (Bkz. Bölüm 4.4). 5. FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özelliklerFarmakoterapötik Grubu: Kan koagülasyon faktörleri ATC kodu: B02BD03 FEIBA yetmişli yılların başlarında geliştirilmiş ve hem in vitro,in vivoolarak Faktör VIII inhibitörü by-pass edici etkinliği gösterilmiş olmasına rağmen, etki mekanizması halabilimsel tartışma konusudur. Aktivite ölçümlerinde bulunduğu üzere FEIBA, potensindenbağımsız olarak hemen hemen eşit miktarlarda prokoagülan (protrombin FVII, FIX, FX) veantikoagülan (protein C) protrombin kompleksi zimojenlerinden oluşur; prokoagülan enzimiçeriği nispeten düşüktür. Dolayısıyla FEIBA, protrombin kompleksi faktörlerininproenzimlerini içerirken, en fazlası FVIIa olacak şekilde bunların aktivasyon ürünlerininküçük miktarlarını içermektedir. [Turecek PL and Schwarz HP. Chapter 4: Factor EightInhibitor Bypassing Activity, in Production of Plasma Proteins for Therapeutic Use, eds.Joseph Bertolini, Neil Goss, John Curling, Wiley 2013, ISBN: 978-0-470-92431-0].Günümüzdeki bilimsel çalışmalar, FEIBA'nın etki mekanizmasında aktive protrombin kompleksinin spesifik bileşenleri, protrombin (Faktör II) ve aktive Faktör X'un (Faktör Xa)rolü bulunduğuna işaret etmektedirler. [Turecek PL, Varadi K, Gritsch H, et al. Factor Xa andProthrombin: Mechanism of Action of FEIBA. Vox Sang. 77: 72-79, 1999] FEIBA, kanamaları trombin üretimini uyararak ve kolaylaştırarak kontrol altına alır; bu sürecin kritik aşaması protrombinaz kompleksinin oluşumudur. Bir dizi in vitroin vivobiyokimyasal çalışmada, FEIBA'nın aktivitesinde FXa ve protrombinin kritik bir rolü olduğugösterilmiştir. FEIBA'nın en önemli hedefinin protombinaz kompleksi olduğu bulunmuştur.FEIBA, protrombin ve FXa dışında, inhibitör gelişmiş hemofili hastalarında hemostazıkolaylaştırabilen diğer protrombin kompleksi proteinlerini de içerir.16 İnhibitör gelişmiş hemofili B hastalarının tedavisiHastalığın nadir görülüyor olması nedeniyle, faktör IX'a karşı inhibitör gelişmiş hemofili B hastalarında deneyim kısıtlıdır. FEIBA, gereksinime göre, profilaktik olarak ya da cerrahigirişimlerde kullanılan klinik çalışmalarda toplam beş inhibitör gelişmiş hastadakullanılmıştır: İnatçı yüksek titreli inhibitörü olan hemofili A ya da B hastalarında gerçekleştirilen prospektif açık etiketli ve paralel tasarımlı randomize bir klinik çalışmada (090701, PROOF), 36 hasta12 ay ± 14 gün profilaktik tedavi ya da gereksinim halinde tedavi alacak şekilde randomizeedilmiştir. Profilaksi kolundaki 17 hastaya günaşırı 85 ± 15 Ünite/kg FEIBA uygulanmış; gerikalan 19 hastaya ise gereksinim halinde hekimin belirlediği miktarlarda FEIBAuygulanmıştır. Gereksinim halinde tedavi alan gruptaki iki inhibitörlü hemofili B hastası veprofilaksi grubundaki bir hemofili B hastası tedavi edilebilmiştir. Tüm kanama tipleri için ortanca ABR (yıllık kanama sıklığı) profilaksi grubunda (ortanca ABR = 7,9), gereksinime göre uygulamanın yapıldığı gruptakinde bulunandan (ortanca ABR=28,7) daha düşüktü; buna göre ortanca ABR açısından iki tedavi kolu arasındaki rakam%72,5'luk bir azalmaya karşılık geliyordu. FEIBA'nın perioperatif dönemde kullanıldığı bir başka prospektif girişimsel olmayan gözetim çalışmasında (PASS-INT-003, SURF) 23 hastada toplam 34 cerrahi işlem uygulandı.Hastaların büyük bölümü (18 hasta) inhibitör gelişmiş hemofili A hastasıydı. Kalan hastalarınikisi inhibitör gelişmiş hemofili B hastası ve üçü inhibitör gelişmiş kalıtsal olmayan(kazanılmış) hemofili A hastasıydı. FEIBA'ya maruziyet süresi 1 ile 28 gün idi (ortalama 9gün, ortanca 8 gün). Ortalama kümülatif doz olarak 88,347 Ünite (ortanca kümülatif doz =59,000 Ünite) kullanıldı. İnhibitör gelişmiş hemofili B hastaları için en uzun FEIBAmaruziyet süresi 21 gün ve uygulanan maksimum doz 7324 Ünite idi. Bu çalışmalara ek olarak faktör IX inhibitörlü hemofili B hastalarındaki kanamaların tedavi ve önlenmesi için FEIBA kullanılan 36 vaka bildirimi bulunmaktadır. Bunlardan 24'ü inhibitörlühemofili B hastalarında gereksinime göre, dördü profilaktik olarak ve sekizi ise cerrahigirişim sırasında FEIBA kullanılan vakadır. FEIBA'nın faktör X, XI ve XIII'e karşı kazanılmış inhibitörleri olan hastalarda kullanımıyla ilgili izole raporlar da bulunmaktadır. 5.2. Farmakokinetik özelliklerGenel özelliklerEmilim:FEIBA'nın etki mekanizması halen kesin olarak bilinmediğinden, farmakokinetik özellikleri hakkında kesin yargılarda bulunmak mümkün değildir. Dağılım:FEIBA'nın etki mekanizması halen kesin olarak bilinmediğinden, farmakokinetik özellikleri 17 hakkında kesin yargılarda bulunmak mümkün değildir. Biyotransformasyon:FEIBA'nın etki mekanizması halen kesin olarak bilinmediğinden, farmakokinetik özellikleri hakkında kesin yargılarda bulunmak mümkün değildir. Eliminasyon:FEIBA'nın etki mekanizması halen kesin olarak bilinmediğinden, farmakokinetik özellikleri hakkında kesin yargılarda bulunmak mümkün değildir. Hastalardaki karakteristik özelliklerFEIBA'nın etki mekanizması halen kesin olarak bilinmediğinden, hastalardaki karakteristik özellikleri hakkında kesin yargılarda bulunmak mümkün değildir. Farmakokinetik/farmakodinamik ilişki(ler):Bilgi bulunmamaktadır. 5.3. Klinik öncesi güvenlilik verileriFEIBA'nın normal farelere, FVIII knockout farelere ve sıçanlara, insanlarda önerilen günlük maksimum dozun üzerinde uygulandığı (>200 Ünite/kg) akut toksisite çalışmalarına göre,FEIBA ile ilişkili yan etkilerin büyük oranda ürünün farmakolojik özelliklerindenkaynaklanan hiperkoagülasyonun sonucunda ortaya çıktığı sonucuna varılabilir. Hayvanlarda, heterolog proteinlere karşı gelişen antikorların sonucu etkilemesi nedeniyle, tekrarlayan doz toksisite çalışmaları mümkün değildir. İnsan kanındaki pıhtılaşma faktörlerinin karsinojen ya da mutajen olarak görülmemelerinden dolayı, özellikle heterolog türlerde deneysel çalışmaların yapılmasına gerek duyulmamıştır. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiToz: Sodyum klorür Sodyum sitratÇözücü: Enjeksiyonluk su 6.2. GeçimsizliklerFEIBA, bölüm 6.6'da bahsedilen çözücüsü dışında başka ilaçlarla karıştırılmamalıdır. Tüm kan pıhtılaşma faktör preparatlarında geçerli olduğu gibi, başka ilaçlarla karıştırılması etkililik ve toleransta bozulmalara neden olabilir. Uygulama diğer ilaçların da kullanıldığı birvenöz uygulama setinden yapılıyorsa, FEIBA infüzyonu öncesi ve sonrası venöz uygulamasetinin, izotonik sodyum klorür gibi uygun çözeltilerle yıkanması önerilmektedir. 18 İnsan plazmasından elde edilen koagülasyon faktörleri belli enjeksiyon/infüzyon cihazlarının iç yüzeyleri tarafından adsorbe edilebilirler. Bu durum tedavinin başarısız olması ilesonuçlanabilir. Bu nedenle FEIBA ile sadece onaylanmış plastik infüzyon cihazlarıkullanılmalıdır. 6.3. Raf ömrü24 ay. Rekonstitüye edildikten sonra oda sıcaklığında (25°C'ye kadar) 3 saat boyunca kimyasal ve fiziksel kullanımda stabilitesi kanıtlanmıştır. Mikrobiyolojik açıdan, rekonstitüsyon metodu mikrobiyal kontaminasyon riskini (kontrollü ve valide edilmiş aseptik koşullar) engellemediği sürece, ürün hemen kullanılmalıdır. Hemenkullanılmadığı takdirde, kullanımdaki saklama süreleri ve koşulları kullanıcınınsorumluluğundadır. Rekonstitüye edilmiş çözelti buzdolabında saklanmamalıdır. 6.4. Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklığında saklanmalıdır. Dondurulmamalıdır. Işıktan korumak amacıyla orijinal ambalajı içerisinde saklanmalıdır. Rekonstitüye edilerek kullanıma hazır hale getirilmiş ilacın saklanması için bkz. Bölüm 6.3. 6.5. Ambalajın niteliği ve içeriğiFEIBA kuru toz; yüzeyi muamele edilmiş Tip II hidrolitik renksiz cam flakon içinde sunulmaktadır Çözücü; yüzeyi muamele edilmiş Tip I hidrolitik renksiz cam flakon içinde sunulmaktadır. Flakonlar, bütil lastik tıpa ve koruyucu kapakla kapalıdır. Ambalaj içeriği: - 1 adet liyofilize toz içeren flakon - 1 adet 20 mL enjeksiyonluk su içeren çözücü flakon - 1 adet Baxject II Hi-Flow cihazı (iki flakondaki ürünün birbiriyle karıştırılarak birenjektöre alınmasını sağlayan iğnesiz transfer cihazı) - 1 adet tek kullanımlık enjektör - 1 adet tek kullanımlık iğne - 1 adet klempli kelebek iğne (enjeksiyon için kanatlı set). 19 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerAmbalajı açılmış ürün yeniden kullanılmamalıdır. Steril bariyeri bozulan, ambalajı hasar görmüş ya da bozulma belirtisi gösteren ürün kullanılmamalıdır. Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİTakeda İlaç Sağlık Sanayi Ticaret Limited Şirketi Levent-Şişli/İSTANBUL 8. RUHSAT NUMARASI2016/359 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 28.04.2016 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ20 |
İlaç BilgileriFeiba 1000 U Iv İnfüzyon İçin Liyofilize Toz İçeren FlakonEtken Maddesi: Faktör Viii İnhibitör By-pass Aktivitesine Sahip İnsan Plazma Proteini Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.