Tecentriq 1200 Mg/20 Ml İnfüzyonluk Çözelti Hazırlamak İçin Konsantre Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ"V Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADITECENTRIQ 1200 mg/20 ml infüzyonluk çözelti hazırlamak için konsantre Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMİEtkin madde:20 mL konsantre bir flakon içinde 1200 mg atezolizumab içerir. Dilüsyondan sonra 1 mL solüsyon yaklaşık olarak 4,4 mg atezolizumab içerir (bkz. Bölüm 6.6). Atezolizumab, rekombinant DNA teknolojisiyle Çin hamsteri yumurtalık hücrelerinde üretilen, bir Fc bölgesi değiştirilmiş, hümanize IgG1 anti-programlı ölüm-ligandı 1 (PD-L1) monoklonalantikorudur. Yardımcı madde(ler):Yardımcı maddeler için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMİnfüzyonluk çözelti hazırlamak için steril konsantre Berrak, renksiz ila hafif sarımsı sıvı 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarErken Evre Küçük Hücreli Dışı Akciğer Kanseri (EE-KHDAK) TECENTRIQ, PD-L1 ekspresyonu tümör hücrelerinde (TC) > %50 olan, yetişkin Evre II-IIIA küçük hücreli dışı akciğer kanseri hastalarında, rezeksiyonu ve platin bazlı kemoterapiyitakiben, adjuvan tedavide monoterapi olarak endikedir. Non-skuamöz küçük hücreli dışıakciğer kanserlerinde EGFR ve ALK mutasyonlarının negatif olması gereklidir. Hastalık nüksüveya kabul edilemez toksisite olmadıkça maksimum 1 yıl süreyle kullanımı uygundur. Metastatik Küçük Hücreli Dışı Akciğer Kanseri (KHDAK) TECENTRIQ'in, performans durumu ECOG 0-1 olan, EGFR, ALK, ROS negatif, semptomatik beyin metastazı olmayan, lokal ileri ve/veya metastatik küçük hücreli dışı akciğer kanseri(KHDAK) nedeniyle daha önce 1-2 basamak kemoterapi almış ve progresyon gelişmiş 1hastaların tedavisinde tekrar progresyona kadar kullanımı endikedir. TECENTRIQ, EGFR mutant ya da ALK pozitif olmayan ve PD-L1 ekspresyonu tümör hücrelerinde (TC) > %50 ya da tümör infiltre edici immün hücrelerde > %10 olan, aktif beyinmetastazı olmayan, ECOG 0 veya 1 metastatik küçük hücreli dışı akciğer kanseri yetişkinhastaların birinci basamak tedavisinde monoterapi olarak endikedir. Yassı hücreli KHDAKhastalarında EGFR ve/veya ALK durumunun belirlenmiş olması aranmaz. TECENTRIQ, ECOG performans skoru 0-1 olan, yeterli kardiyak, renal ve hepatik fonksiyonları bulunan, aktif beyin metastazı olmayan, EGFR ya da ALK mutasyonlarıbulunmayan ve eş zamanlı immünsüpresif veya kortikosteroid tedavisi almayan metastatikyassı hücreli olmayan KHDAK hastalarının birinci basamak tedavisinde, nab-paklitaksel vekarboplatinli kemoterapi rejimi ile kombine olarak progresyona kadar kullanımı endikedir.Tedavi sonu progresyonda diğer PD-1 ve PD-L1 inhibitörleri kullanılamaz. Küçük Hücreli Akciğer Kanseri (KHAK) TECENTRIQ'in, karboplatin ve etoposidle kombine olarak yaygın evre küçük hücreli akciğer kanseri olan yetişkin hastaların birinci basamak tedavisinde kullanımı endikedir. Hepatoselüler Kanser TECENTRIQ'in bevacizumab ile kombine olarak, daha önce sistemik tedavi görmemiş, ECOG performans durumu 0 ve 1 olan, Child-Pugh skoru A olan, metastatik veya lokorejyoneltedaviye uygun olmayan rezeke edilemeyen hepatoselüler karsinomlu yetişkin hastalarıntedavisinde kullanımı endikedir. 4.2. Pozoloji ve uygulama şekliTECENTRIQ, kanser tedavisinde deneyimli bir hekimin gözetimi altında uygulanmalıdır. KHDAK hastalarında PD-L1 testi:TECENTRIQ monoterapisiErken evre KHDAK ve birinci basamak metastatik KHDAK hastalarının tedaviye uygunluğu, valide edilmiş bir test ile teyid edilmiş PD-L1 tümör ekspresyonuna göre yapılmalıdır (bkz.Bölüm 5.1). Pozoloji/Uygulama sıklığı ve süresi:TECENTRIQ monoterapisiÖnerilen TECENTRIQ dozu, üç haftada bir intravenöz yoldan uygulanan 1200 mg'dır. TECENTRIQ kombinasyon tedavisiNon-skuamoz KHDAK - TECENTRIQ ile nab-paklitaksel ve karboplatin kombinasyonuİndüksiyon fazında, 1. günde önerilen TECENTRIQ dozu intravenöz yoldan uygulanan 12002mg ve takiben sırasıyla nab-paklitaksel ve karboplatindir. Nab-paklitaksel 8. ve 15. günlerde intravenöz yoldan uygulanır. Bu rejim 4 veya 6 siklus boyunca her 3 haftada bir uygulanır. İndüksiyon fazını, kemoterapi olmadan, yalnızca 3 haftada bir 1200 mg intravenöz yoldan TECENTRIQ uygulanan idame fazı takip eder. KHAK - TECENTRIQ ile karboplatin ve etoposid kombinasyonuİndüksiyon fazında, 1. günde önerilen TECENTRIQ dozu intravenöz yoldan uygulanan 1200 mg ve takiben sırasıyla karboplatin ve etoposiddir. Etoposid 2. ve 3. günlerde intravenöz yoldanuygulanır. Bu rejim 4 siklus boyunca her 3 haftada bir uygulanır. İndüksiyon fazını, kemoterapi olmadan, yalnızca 3 haftada bir 1200 mg intravenöz yoldan TECENTRIQ uygulanan idame fazı takip eder. Hepatoselüler Kanser (HSK)TECENTRIQ ile bevacizumab kombinasyonuÖnerilen TECENTRIQ dozu 1200 mg'dır ve bunu takiben 15 mg/kg bevacizumab ile üç haftada bir intravenöz yoldan uygulanır. Tedavi süresi:2. Basamak Küçük Hücreli Dışı Akciğer Kanseri (KHDAK) ve Hepatoselüler Kanser (HSK)Klinik faydanın kaybedilmesine (bkz. Bölüm 5.1) veya yönetilemeyen toksisiteye kadar hastaların TECENTRIQ ile tedavi edilmeleri önerilmektedir. Erken Evre Küçük Hücreli Dışı Akciğer KanseriHastalık nüksü veya kabul edilemez toksisite olmadıkça hastaların 1 yıl boyunca TECENTRIQ ile tedavi edilmeleri önerilmektedir. 1 yıldan uzun süren tedavi süresi çalışılmamıştır. 1. Basamak Küçük Hücreli Dışı Akciğer Kanseri (KHDAK) (TECENTRIQ monoterapiHastalık progresyonuna veya yönetilemeyen toksisiteye kadar hastaların TECENTRIQ ile tedavi edilmeleri önerilmektedir. 1. Basamak non-skuamoz Küçük Hücreli Dışı Akciğer Kanseri (KHDAK) (TECENTRIQ ile nab-paklitaksel ve karboplatin kombinasyonu)Hastalık progresyonuna veya yönetilemeyen toksisiteye kadar hastaların TECENTRIQ ile tedavi edilmeleri önerilmektedir. Hastalık progresyonundan sonra devam eden TECENTRIQ tedavisiyle atipik yanıtlar (yani ilk olarak hastalık progresyonu ve ardından tümörde küçülme) gözlenmiştir. Hekimin takdirinebağlı olarak hastalık progresyonundan sonra tedavi uygulanması düşünülebilir. Geciken veya atlanan dozlar:Planlanmış bir TECENTRIQ dozu atlanırsa mümkün olan en kısa sürede uygulanmalıdır. Uygulama planı, dozlar arasında 3 haftalık bir aralık korunacak şekilde ayarlanmalıdır. 3Tedavi sırasında doz modifikasyonları:TECENTRIQ için doz azaltımı önerilmez. Doz gecikmesi veya kesilmesi (ayrıca bkz. Bölüm 4.4 ve 4.8):
Not: Toksisite dereceleri, Ulusal Kanser Enstitüsü Advers Olaylar için Ortak Terminoloji Kriterleri, Versiyon 4.0'a (NCI-CTCAE v.4) uygundur. 1 Şiddetten bağımsız olarak 2 Etiyoloji tespiti için detaylı kardiyak değerlendirme gerçekleştirilir ve uygun şekilde yönetilir TECENTRIQ ile tedavi edilen hastalara ilacın riskleri hakkında bilgi veren Hasta Uyarı Kartları verilmelidir. Uygulama şekli:TECENTRIQ intravenöz kullanıma yöneliktir. TECENTRIQ infüzyonları intravenöz puşe veya bolus şeklinde uygulanmamalıdır. İlk TECENTRIQ dozu 60 dakika uygulanmalıdır. İlk infüzyon tolere edilirse, sonraki tüm infüzyonlar 30 dakikada uygulanabilir. Tıbbi ürünün uygulanmadan önceden seyreltilmesi ve kullanımına ilişkin talimatlar için Bölüm 6.6'ya bakınız. Özel popülasyonlara ilişkin ek bilgiler:Karaciğer yetmezliği:Popülasyon farmakokinetik analizine göre hafif veya orta düzeyde karaciğer bozukluğu olan hastalarda doz ayarlaması gerekli değildir. Şiddetli karaciğer bozukluğu olan hastalara ilişkinveri mevcut değildir (bkz. Bölüm 5.2). Böbrek yetmezliği:Popülasyon farmakokinetik analizine göre hafif ve orta derecede böbrek bozukluğu olan hastalarda doz ayarlaması gerekli değildir (bkz. Bölüm 5.2). Şiddetli böbrek bozukluğu olanlaraait bilgi, bu popülasyonda bir sonuca varmak için çok azdır. 8Pediyatrik popülasyon:TECENTRIQ'in çocuklarda ve 18 yaş altındaki adolesanlarda güvenliliği ve etkililiği gösterilmemiştir. Mevcut olan veriler Bölüm 4.8, 5.1 ve 5.2'de anlatılmıştır ancak pozoloji ileilgili herhangi bir tavsiye verilememektedir. Geriyatrik popülasyon:Popülasyon farmakokinetik analizine göre 65 yaş ve üstündeki hastalarda TECENTRIQ doz ayarlaması gerekli değildir (bkz. Bölüm 4.8 ve 5.1). Doğu Kooperatif Onkoloji Grubu (ECOG) performans statüsü >2 ECOG performans statüsü >2 olan hastalar Küçük Hücreli Dışı Akciğer Kanseri (KHDAK), Erken Evre Küçük Hücreli Akciğer Kanseri (EE-KHAK), 2. Basamak Ürotelyal Kanser veHepatoselüler Kanser klinik çalışmalarına dahil edilmemiştir (bkz. Bölüm 4.4 ve 5.1). 4.3. KontrendikasyonlarTECENTRIQ'in etkin maddesi atezolizumaba veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine aşırı duyarlılığı olan hastalarda kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleriImmünite ile ilişkili advers reaksiyonlar:TECENTRIQ ile tedavi sırasında oluşan immünite ile ilişkili advers reaksiyonların çoğu ilacın kesilmesi ve kortikosteroidlerin veya destekleyici tedavinin başlatılmasıyla geri döndürülebilirolmuştur. Birden fazla vücut sistemini etkileyen immünite ile ilişkili advers reaksiyonlargörülmüştür ve bu reaksiyonlar TECENTRIQ'in son dozundan sonra da oluşabilir. İmmünite ile ilişkili şüpheli advers reaksiyonlar için etyoloj iyi doğrulamak veya diğer nedenleri dışlamak için yeterli değerlendirme yapılmalıdır. Advers etkilerin şiddetine bağlı olarak,TECENTRIQ tedavisine ara verilir ve kortikosteroid uygulanır. Olay < 1. dereceye iyileştiğindekortikosteroid kullanımı >1 ay boyunca azaltılarak kesilmelidir. İmmünite ile ilişkiliistenmeyen reaksiyonların kortikosteroid kullanımı ile kontrol edilemediği hastalarda, klinikçalışmalardan elde edilen sınırlı verilere dayanarak, diğer sistemik immunosupresan ajanlarınkullanımı düşünülebilir. Herhangi bir 3. derece immünite ile ilişkili advers reaksiyon ikinci defa ortaya çıkarsa ve replasman hormonlar ile kontrol edilen endokrinopatiler hariç herhangi bir 4. derece immüniteile ilişkili advers reaksiyon görülürse TECENTRIQ tedavisi tamamen kesilmelidir (bkz. Bölüm4.2 ve 4.8). Immünite ile ilişkili pnömonit:TECENTRIQ ile yürütülen klinik çalışmalarda ölümcül vakalar da dahil olmak üzere pnömonit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar pnömonit belirtileri ve semptomları içinizlenmeli ve immünite ile ilişkili pnömonit dışındaki sebepler dışlanmalıdır. 2. derece pnömonit durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg 9vücut ağırlığı prednizon veya eşdeğeri ile tedavi başlatılmalıdır. Semptomlar <1. dereceye iyileşirse kortikosteroidler >1 ay boyunca azaltılarak kesilmelidir. Olay 12 hafta içinde <1.dereceye iyileşirse ve kortikosteroidler günde <10 mg prednizon veya eşdeğerine düşürülürseTECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece pnömonit durumundaTECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır. Immünite ile ilişkili hepatit:TECENTRIQ ile yürütülen klinik çalışmalarda bazıları ölümcül sonuçlara yol açan hepatit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar hepatit belirtileri ve semptomları içinizlenmelidir. Aspartat aminotransferaz (AST), alanin aminotransferaz (ALT) ve bilirubin, TECENTRIQ ile tedavi öncesinde ve tedavi sırasında periyodik olarak ve klinik çalışmalarda belirtildiği gibiizlenmelidir. Hepatoselüler kanseri olmayan hastalarda 2. derece olay (ALT veya AST >3-5 x NÜS veya kan bilirubin >1,5-3 x NÜS) 5-7 günden uzun süre devam ederse TECENTRIQ ile tedaviye araverilmeli ve günde 1-2 mg/kg vücut ağırlığı prednizon veya eşdeğeri ile tedavi başlatılmalıdır.Olaylar <1. dereceye iyileşirse kortikosteroidler >1 ay boyunca azaltılarak kesilmelidir. Olay 12 hafta içinde <1. dereceye iyileşirse ve kortikosteroidler günde <10 mg prednizon veya eşdeğerine düşürülürse TECENTRIQ ile tedaviye devam edilebilir. 3. veya 4. derece olaylarda(ALT veya AST >5 x NÜS veya kan bilirubin >3 x NÜS) TECENTRIQ ile tedavi kalıcı olarakbırakılmalıdır. Hepatoselüler kanseri olan hastalarda, ALT veya AST başlangıçtaki normal sınırlardan >3 ila <10 x NÜS'e; veya başlangıçtan >1 ila <3 x NÜS'ten >5 ila <10 x NÜS'e; veya başlangıçtan>3 ila <5 x NÜS'ten >8 ila <10 x NÜS'e yükselir ve 5-7 günden uzun süre devam ederseTECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg vücut ağırlığı prednizon veyaeşdeğeri ile tedavi başlatılmalıdır. Olaylar <1. dereceye iyileşirse kortikosteroidler >1 ayboyunca azaltılarak kesilmelidir. 12 hafta içinde olaylar <1. dereceye iyileşirse ve kortikosteroidler günde <10 mg veya eşdeğerine azaltılırsa TECENTRIQ tedavisine devam edilebilir. ALT veya AST >10 x NÜSveya total bilirubin >3 x NÜS'e yükselirse TECENTRIQ tedavisi tamamen kesilmelidir. Immünite ile ilişkili kolit:TECENTRIQ ile yürütülen klinik çalışmalarda diyare veya kolit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar kolit belirtileri ve semptomları için izlenmelidir. 2. veya 3. derece diyare (başlangıca göre >4 dışkı/gün artış) veya kolit (semptomatik) durumunda TECENTRIQ ile tedaviye ara verilmelidir. 2. derece diyare veya kolit durumundasemptomlar >5 gün devam ederse veya nüksederse, günde 1-2 mg/kg vücut ağırlığı prednizonveya eşdeğeri ile tedavi başlatılmalıdır. 3. derece diyare veya kolit durumunda IVkortikosteroidlerle (1-2 mg/kg vücut ağırlığı/gün metilprednizolon veya eşdeğeri) tedavibaşlatılmalı ve semptomlar iyileşmeye başladığında günde 1-2 mg/kg vücut ağırlığı prednizonveya eşdeğerine geçilmelidir. Semptomlar <1. dereceye iyileşirse kortikosteroidler >1 ayboyunca azaltılarak kesilmelidir. Olay 12 hafta içinde <1. dereceye iyileşirse vekortikosteroidler günde <10 mg prednizon veya eşdeğerine düşürülürse TECENTRIQ ile 10tedaviye devam edilebilir. 4. derece (yaşamı tehdit edici; acil müdahale endike) diyare veya kolit durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır. Kolit ile ilişkili potansiyelgastrointestinal perforasyon komplikasyonu dikkate alınmalıdır. Immünite ile ilişkili endokrinopatiler:TECENTRIQ ile yürütülen klinik çalışmalarda hipotiroidizm, hipertiroidizm, adrenal yetmezlik, hipofizit ve diyabetik ketoasidoz dahil olmak üzere tip 1 diabetes mellitus vakalarıgözlenmiştir (bkz. Bölüm 4.8). Hastalar endokrinopatilerin klinik belirtileri ve semptomları için izlenmelidir. Tiroid fonksiyonu TECENTRIQ ile tedavi öncesinde ve tedavi sırasında periyodik olarak izlenmelidir.Başlangıçta anormal tiroid fonksiyon testleri olan hastaların uygun şekilde tedavi edilmesidüşünülmelidir. Tiroid fonksiyonu testleri anormal olan asemptomatik hastalar TECENTRIQ alabilir. Semptomatik hipotiroidizm durumunda TECENTRIQ ile tedaviye ara verilmeli ve tiroidhormonu replasmanı gerektiğinde başlatılmalıdır. İzole hipotiroidizm kortikosteroidlerkullanılmadan replasman tedavisi ile yönetilebilir. Semptomatik hipertiroidizm durumundaTECENTRIQ ile tedaviye ara verilmeli ve bir anti-tiroid tıbbi ürün gerektiği gibi başlatılmalıdır.Semptomlar kontrol altına alındığında ve tiroid fonksiyonu iyileştiğinde TECENTRIQ iletedaviye devam edilebilir. Semptomatik adrenal yetmezlik durumunda TECENTRIQ ile tedaviye ara verilmeli ve intravenöz kortikosteroid (günde 1-2 mg/kg vücut ağırlığı metilprednizolon veya eşdeğeri) iletedavi başlatılmalıdır. Semptomlar iyileştiğinde günde 1-2 mg/kg vücut ağırlığı prednizon veyaeşdeğeri ile tedavi uygulanmalıdır. Semptomlar <1. dereceye iyileşirse kortikosteroidler >1 ayboyunca azaltılarak kesilmelidir. Olay 12 hafta içinde <1. dereceye iyileşirse vekortikosteroidler günde <10 mg prednizon veya eşdeğerine düşürülürse ve hastanın durumureplasman tedavisinde stabilse (gerekli ise) tedaviye devam edilebilir. 2. veya 3. derece hipofizit için TECENTRIQ kesilmeli ve intravenöz kortikosteroidler (1 ila 2 mg/kg vücut ağırlığı/gün metilprednizolon veya eşdeğeri) ile tedavi başlatılmalı ve ihtiyacagöre hormon replasman tedavisi başlatılmalıdır. Belirtiler düzeldiğinde 1 ila 2 mg/kg vücutağırlığı/gün prednizon veya eşdeğeri ile tedavi uygulanmalıdır. Semptomlar < 1. dereceyeiyileşirse, kortikosteroidler > 1 ay boyunca azaltılarak kesilmelidir. Olay, 12 hafta içinde < 1.dereceye iyileşir ve kortikosteroidler günde < 10 mg prednizona veya eşdeğerine düşürülürseve hasta replasman tedavisinde (eğer gerekli ise) stabil kalırsa, tedaviye devam edilebilir. 4.derece hipofizit için TECENTRIQ tedavisi kesilmelidir. Tip 1 diabetes mellitus için insülin tedavisi başlatılmalıdır. >3. derece hiperglisemi (açlık glukozu >250 mg/dL veya 13,9 mmol/L) durumunda TECENTRIQ ile tedaviye ara verilmelidir.İnsülin replasman tedavisinde metabolik kontrol elde edilirse TECENTRIQ ile tedaviye devamedilebilir. Immünite ile ilişkili meningoensefalit:TECENTRIQ ile yürütülen klinik çalışmalarda meningoensefalit gözlenmiştir (bkz. Bölüm 4.8). Hastalar menenjit veya ensefalitin klinik belirtileri ve semptomları için izlenmelidir. Herhangi bir derece menenjit veya ensefalit durumunda TECENTRIQ ile tedavi kalıcı olarak 11bırakılmalıdır. İntravenöz kortikosteroidler (günde 1-2 mg/kg vücut ağırlığı metilprednizolon veya eşdeğeri) ile tedavi başlatılmalı ve hastanın durumu iyileştiğinde 1-2 mg/kg vücut ağırlığıprednizon veya eşdeğerine geçilmelidir. Immünite ile ilişkili nöropatiler:TECENTRIQ alan hastalarda yaşamı tehdit edici olabilen miyastenik sendrom/miyastenia gravis veya Guillain-Barre sendromu gözlenmiştir. Hastalar motor ve duyusal nöropatisemptomları için izlenmelidir. Herhangi bir derece miyastenik sendrom/miyastenia gravis veya Guillain-Barre sendromu durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır. Günde 1-2 mg/kg dozdaprednizon veya eşdeğeri ile sistemik kortikosteroidlerin başlatılması düşünülmelidir. Immünite ile ilişkili pankreatit:TECENTRIQ ile yürütülen klinik çalışmalarda serum amilaz ve lipaz düzeylerinde artışlar dahil olmak üzere pankreatit gözlenmiştir (bkz. Bölüm 4.8). Hastalar akut pankreatiti düşündürenbelirtiler ve semptomlar için yakından izlenmelidir. Serum amilaz veya lipaz düzeylerinde >3. derece artış (> 2 NÜS) veya 2. veya 3. derece pankreatit durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg vücutağırlığı IV metilprednizolon veya eşdeğeri ile tedavi başlatılmalıdır. Semptomlar iyileştiğindegünde 1-2 mg/kg vücut ağırlığı prednizon veya eşdeğeri ile tedavi uygulanmalıdır. Serumamilaz ve lipaz düzeyleri 12 hafta içinde <1. dereceye iyileştiğinde veya pankreatit semptomlarıdüzeldiğinde ve kortikosteroidler günde <10 mg prednizon veya eşdeğerine düşürüldüğündeTECENTRIQ ile tedaviye devam edilebilir. 4. derece veya herhangi bir derecede nüksedenpankreatit durumunda TECENTRIQ ile tedavi kalıcı olarak bırakılmalıdır. Immünite ile ilişkili miyokardit:TECENTRIQ ile yürütülen klinik çalışmalarda ölümcül vakalar da dahil olmak üzere miyokardit gözlemlenmiştir (bkz. Bölüm 4.8). Hastalar miyokarditi düşündüren belirtiler vesemptomlar açısından izlenmelidir. Miyokardit, miyozitin klinik bir belirtisi de olabilir veuygun bir şekilde tedavi edilmelidir. Kardiyak veya kardiyopulmoner semptomları olan hastalar, uygun önlemlerin erken evrede başlatılmasını sağlamak için potansiyel miyokardit açısından değerlendirilmelidir. Miyokarditşüphesi varsa, TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg vücut ağırlığıprednizon veya eşdeğeri ile sistemik kortikosteroidler başlatılmalı ve güncel klinik kılavuzlaragöre tanısal incelemeler ile birlikte hızlı kardiyoloji konsültasyonu yapılmalıdır. Miyokardittanısı konulduğunda, >2. derece miyokardit durumunda TECENTRIQ ile tedavi kalıcı olarakbırakılmalıdır (bkz. Bölüm 4.2). Immünite ile ilişkili nefrit:TECENTRIQ ile yürütülen klinik çalışmalarda nefrit gözlemlenmiştir (bkz. Bölüm 4.8). Hastalar renal fonksiyonda değişiklikler açısından yakından izlenmelidir. 2. derece nefrit durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg vücut ağırlığı prednizon veya eşdeğeri ile sistemik kortikosteroidler başlatılmalıdır. Olay, 12 hafta 12içinde <1. dereceye iyileşir ve kortikosteroidler günde < 10 mg prednizona veya eşdeğerine düşürüldüğünde tedaviye devam edilebilir. 3. veya 4. derece nefrit durumunda TECENTRIQile tedavi kalıcı olarak bırakılmalıdır. Immünite ile ilişkili miyozit:TECENTRIQ ile yürütülen klinik çalışmalarda ölümcül vakalar da dahil olmak üzere miyozit vakaları gözlenmiştir (bkz. Bölüm 4.8). Hastalar miyozit belirtileri ve semptomları açısındanizlenmelidir. Olası miyoziti olan hastalar miyokardit belirtileri açısından izlenmelidir. Bir hastada miyozit belirti ve semptomları gelişirse, yakın takibe alınmalı ve hasta vakit kaybetmeden değerlendirme ve tedavi için bir uzmana yönlendirilmelidir. 2. veya 3. derecemiyozit durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg vücut ağırlığıprednizon veya eşdeğeri ile sistemik kortikosteroidler başlatılmalıdır. Semptomlar < 1.dereceye iyileşirse, klinik olarak belirtildiği gibi kortikosteroidler azaltılarak kesilebilir. Olay,12 hafta içinde <1. dereceye iyileşir ve kortikosteroidler günde < 10 mg prednizona veyaeşdeğerine düşürüldüğünde tedaviye devam edilebilir. 4. derece. veya 3. derece tekrarlayanmiyozit durumunda ya da kortikosteroid dozu başlangıçtan sonraki 12 hafta içinde günde < 10mg prednizon eşdeğerine düşürülemediğinde TECENTRIQ ile tedavi kalıcı olarakbırakılmalıdır. Immünite ile ilişkili şiddetli kutanöz advers reaksiyonlar:TECENTRIQ ile tedavi edilen hastalarda, Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) dahil olmak üzere immünite ile ilişkili şiddetli kutanöz adversreaksiyonlar bildirilmiştir. Hastalar şüpheli şiddetli cilt reaksiyonları açısından izlenmeli vediğer sebepler dışlanmalıdır. Şüpheli şiddetli kutanöz advers reaksiyonları varlığında hastalardaha ileri teşhis ve takip için bir uzmana yönlendirilmelidir. Advers reaksiyonların şiddetine bağlı olarak, 3. derece cilt reaksiyonu durumunda TECENTRIQ ile tedaviye ara verilmeli ve günde 1-2 mg/kg vücut ağırlığı prednizon veyaeşdeğeri ile sistemik kortikosteroidler başlatılmalıdır. Olay, 12 hafta içinde <1. dereceye iyileşirve kortikosteroidler günde < 10 mg prednizona veya eşdeğerine düşürüldüğünde TECENTRIQtedavisine devam edilebilir. 4. derece cilt reaksiyonları durumunda TECENTRIQ ile tedavikalıcı olarak bırakılmalı ve kortikosteroid uygulanmalıdır. Stevens-Johnson sendromu (SJS) ve toksik epidermal nekroliz (TEN) şüphesi olan hastalarda TECENTRIQ ile tedaviye ara verilmelidir. Doğrulanmış Stevens-Johnson sendromu (SJS) vetoksik epidermal nekroliz (TEN) vakalarında TECENTRIQ ile tedavi kalıcı olarakbırakılmalıdır. Daha önce diğer immün sistemi stimüle edici ajanlarla tedavi sırasında şiddetli veya hayatı tehdit eden cilt advers reaksiyonlar yaşayan hastaların TECENTRIQ tedavisine başlanmasıdikkatli olarak değerlendirilmelidir. Immünite ile ilişkili perikardiyal bozukluklarTECENTRIQ ile perikardit, perikardiyal efüzyon ve kardiyak tamponad gibi bazıları ölümcül sonuçlara yol açan perikardiyal bozukluklar gözlenmiştir (bkz. Bölüm 4.8). Hastalar,perikardiyal bozuklukların klinik belirtileri ve semptomları açısından izlenmelidir. 131. derece perikardit şüphesi durumunda TECENTRIQ tedavisi durdurulmalı ve mevcut klinik kılavuzlara göre tanısal çalışmalarla derhal kardiyoloji konsültasyonu başlatılmalıdır. > 2.derece perikardiyal bozukluklardan şüphelenildiğinde, TECENTRIQ tedavisi kesilmeli, 1 ila 2mg/kg vücut ağırlığı/gün prednizon veya eşdeğeri dozunda sistemik kortikosteroidlerle aciltedaviye başlanmalı ve mevcut klinik kılavuzlar doğrultusunda tanısal tetkik ile derhalkardiyoloji konsültasyonu başlatılmalıdır. Bir perikardiyal bozukluk olayı tanısı konulduktansonra, > 2. derece perikardiyal bozukluklar için TECENTRIQ tedavisi kalıcı olarak kesilmelidir(bkz. Bölüm 4.2). Hemofagositik lenfohistiyositozTECENTRIQ alan hastalarda ölümcül vakalar dahil olmak üzere hemofagositik lenfohistiyositoz (HLH) bildirilmiştir (bkz. Bölüm 4.8). Atipik veya uzamış sitokin salınımsendromu varlığı durumunda HLH değerlendirilmelidir. Hastalar HLH'nin klinik belirtileri vesemptomları açısından izlenmelidir. Şüpheli HLH durumunda TECENTRIQ kalıcı olarakkesilmeli ve hastalar ileri tanı ve tedavi için bir uzmana yönlendirilmelidir. immünite ile ilişkili diğer advers reaksiyonlar:TECENTRIQ'in etki mekanizması göz önünde bulundurulduğunda, enfektif olmayan sistitin de dahil olduğu immünite ile ilişkili diğer potansiyel advers reaksiyonlar görülebilir. Diğer sebepleri dışlamak için tüm şüpheli immünite ile ilişkili advers reaksiyonları değerlendiriniz. Hastalar immünite ile ilişkili advers reaksiyonlarına ait işaretler ve semptomlaraçısından gözlenmeli ve reaksiyonun şiddetine göre, klinik gerekliliğe göre tedavimodifikasyonu ve kortikosteroidler ile yönetilmelidir (Bkz. Bölüm 4.2 ve 4.8). Infüzyon ile ilişkili reaksiyonlar:TECENTRIQ ile infüzyon ile ilgili reaksiyonlar gözlenmiştir (bkz. Bölüm 4.8). 1. veya 2. derece infüzyon ile ilişkili reaksiyon görüldüğünde infüzyon hızı düşürülmeli veya tedavi kesilmelidir. 3. veya 4. derece infüzyon ile ilgili reaksiyon görüldüğünde TECENTRIQtedavisi tamamen sonlandırılmalıdır. 1. veya 2. derece infüzyon ile ilişkili reaksiyon görülenhastalar yakından izlenerek TECENTRIQ almaya devam edebilir; bu hastalarda antipiretik veantihistaminiklerle premedikasyon değerlendirilebilir. Hastalığa spesifik önlemler TECENTRIQ'in, daha önce tedavi almamış sisplatine uygun olmayan ürotelyal kanseri olan hastalarda kullanılmasıImvigor210 Kohort 1 çalışma popülasyonunun başlangıç ve prognostik hastalık karakterizasyonu, klinikteki sisplatine uygun olmayan ama karboplatin bazlı kombinasyonkemoterapi kullanımına uygun olan hastalar ile genel olarak karşılaştırabilir olmuştur. Herhangibir kemoterapi için uygun olmayan hastalara ait alt grup için veriler yetersizdir, bu nedenleTECENTRIQ bu hastalarda dikkatli kullanılmalı ve bireysel olarak potansiyel risk ve yarardengesi dikkatli olarak değerlendirilmelidir. Hepatoselüler kanseri olan hastalarda TECENTRIQ 'in bevacizumab ile birlikte kullanılmasıChild-Pugh B karaciğer hastalığı olan, TECENTRIQ ile bevacizumab kombinasyon tedavisi14alan HSK hastalarına ait veriler çok sınırlıdır ve mevcut durumda Child-Pugh C karaciğer hastalığı olan HSK hastalarına ait veri bulunmamaktadır. Bevacizumab ile tedavi edilen hastalarda hemoraji riski yüksektir ve TECENTRIQ ile birlikte bevacizumab kullanan hepatoselüler kanserli hastalarda ölümcül olayları da kapsayan şiddetligastrointestinal hemoraji vakaları bildirilmiştir. Hepatoselüler kanseri olan hastalardaTECENTRIQ ile birlikte bevacizumab tedavisine başlamadan önce klinik pratik olaraközofageal varislerin taranması ve tedavisi yapılmalıdır. Kombinasyon tedavisi ile 3. veya 4.derece kanama geçiren hastalarda bevacizumab tedavisi tamamen kesilmelidir. Konu ile ilgilibevacizumab Kısa Ürün Bilgisine bakınız. TECENTRIQ ile birlikte bevacizumab tedavisi sırasında diyabetes mellitus ortaya çıkabilir. Hekimler TECENTRIQ ile birlikte bevacizumab tedavisi öncesinde ve periyodik olarak tedaviboyunca klinik olarak gerekliliklere göre kan glukoz seviyelerini takip etmelidir. 1. Basamak Küçük Hücreli Dışı Akciğer Kanseri tedavisinde monoterapi olarak TECENTRIQ kullanımıHekimler Küçük Hücreli Dışı Akciğer Kanseri (KHDAK) olan hastalarda monoterapi olarak 1. Basamak tedavisine başlamadan önce TECENTRIQ'in gecikmeli etkisini değerlendirmelidir.TECENTRIQ ile kemoterapi karşılaştırmasında randomizasyondan sonraki 2,5 ay boyuncagörülen daha yüksek ölüm vaka sayısını uzun süreli sağkalım faydası takip etmiştir. Erken ölümvakaları ile ilgili spesifik faktör(ler) tespit edilmemiştir (bkz. Bölüm 5.1). Klinik çalışmalardan dışlanan hastalar: Aşağıdaki koşullara sahip hastalar klinik çalışmalara dahil edilmemiştir: Otoimmün hastalık geçmişi, pnömonit geçmişi, aktif beyin metastazı, HIV, hepatit B ya da hepatit C enfeksiyonu(Hepatoselüler kanseri olmayan hastalar için), önemli kardiyovasküler hastalığı ve yetersizhematolojik ve hedef organ fonksiyonu. Kayıttan 28 gün önce canlı, zayıflatılmış bir aşıuygulanan hastalar, çalışmaya başlamadan önceki 4 hafta içerisinde sistemik immün sistemiuyarıcı ajanlar veya 2 hafta içerisinde sistemik immunosupresif ajanlar kullanan hastalar veçalışmaya başlamadan 2 hafta önce terapötik oral veya IV antibiyotik kullanan hastalar klinikçalışmalara alınmamıştır. Biyoteknolojik ürünlerin takip edilebilirliği: Biyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. Hasta Uyarı Kartı: TECENTRIQ reçete eden hekimler, TECENTRIQ tedavisinin risklerini hastalarına açıklamalıdır. TECENTRIQ ile tedavi edilen hastalara ilacın riskleri hakkında bilgi veren HastaUyarı Kartları verilmeli ve kartı her zaman yanlarında taşımaları söylenmelidir. 4.5. Diğer tıbbi ürünlerle etkileşim ve diğer etkileşim biçimleriTECENTRIQ ile herhangi bir resmi farmakokinetik ilaç etkileşimi çalışması yapılmamıştır. TECENTRIQ dolaşımdan katabolizma ile temizlendiğinden metabolik ilaç-ilaç etkileşimleribeklenmemektedir. 15TECENTRIQ ile tedaviye başlamadan önce, TECENTRIQ'in farmakodinamik aktivitesine ve etkililiğine yapabilecekleri potansiyel etkiler nedeniyle sistemik kortikosteroidlerin veyaimmunosupresanların kullanılmasından kaçınılmalıdır. Bununla birlikte, sistemikkortikosteroidler veya diğer immunosupresif maddeler, TECENTRIQ tedavisine başladıktansonra immünite ile ilişkili advers reaksiyonların tedavisinde kullanılabilir (bkz. Bölüm 4.4). Özel popülasyonlara ilişkin ek bilgiler:TECENTRIQ ile herhangi bir farmakokinetik ilaç etkileşimi çalışması yapılmamıştır.Pediyatrik popülasyon:TECENTRIQ ile pediyatrik popülasyonda herhangi bir farmakokinetik ilaç etkileşimi çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiye:Gebelik kategorisi: D Çocuk doğurma potansiyeli bulunan kadınlar/doğıım kontrolü (kontrasepsiyon):Çocuk doğurma potansiyeline sahip kadınlar TECENTRIQ ile tedavi sırasında ve tedaviden 5 ay sonraya kadar etkili bir doğum kontrol yöntemi kullanmalıdır. Gebelik dönemi:TECENTRIQ'in hamile kadınlar üzerinde etkisine dair herhangi bir veri bulunmamaktadır. TECENTRIQ ile gelişimsel çalışmalar ve üreme çalışmaları yapılmamıştır. Hayvançalışmalarıyla, PD-L1/PD-1 yolak inhibisyonunun fare gebelik modellerinde immünite ileilişkili, fetüs ölümüyle sonuçlanan fetüs gelişiminin reddine sebep olduğu gösterilmiştir (bkz.Bölüm 5.3). Bu sonuçlar, etki mekanizmasına bağlı olarak potansiyel bir risk oluşturmakta olup,gebelik döneminde TECENTRIQ uygulamasının artmış düşük veya ölü doğum oranları dahilolmak üzere fetal zarara sebep olabileceğini göstermektedir. TECENTRIQ bir insan G1 immunoglobulinidir (IgG1) ve IgG1'in plasenta bariyerini aştığı bilinmektedir. Bu nedenle, TECENTRIQ'in anneden gelişmekte olan fetüse geçme potansiyelibulunmaktadır. Gebe kadınların klinik durumu TECENTRIQ ile tedavi gerektirmedikçe gebelik sırasında TECENTRIQ kullanılmamalıdır. Laktasyon dönemi:TECENTRIQ'in anne sütüne geçip geçmediği bilinmemektedir. TECENTRIQ bir monoklonal antikordur ve ilk gelen sütte bulunması ve daha sonra da az miktarda sütte bulunmasıbeklenmektedir. Yeni doğanlar ve infantlar üzerindeki risk dışlanamaz. Emzirmenin çocuk içinfaydaları ve tedavinin anne için faydaları dikkate alınarak emzirmenin durdurulması veyaTECENTRIQ tedavisinin durdurulması kararlaştırılmalıdır. 16Üreme yeteneği/fertilite:TECENTRIQ'in fertilite üzerindeki olası etkilerine ilişkin veri bulunmamaktadır. TECENTRIQ'in doğurganlık üzerindeki etkisini değerlendirme amaçlı reprodüktif vegelişimsel toksisite çalışmaları yapılmamıştır. Bununla birlikte, 26 haftalık yinelenen doztoksisitesi çalışmasında, önerilen dozda TECENTRIQ kullanan hastalarda menstrüel siklustaeğri altında kalan alanı yaklaşık 6 katına çıkardığı ve geri dönüşümlü olduğu görülmüştür (bkz.Bölüm 5.3). Erkek reprodüktif organları üzerinde etki görülmemiştir. 4.7. Araç ve makine kullanımı üzerindeki etkilerTECENTRIQ'in araç ve makine kullanma yeteneği üzerinde küçük bir etki göstermesi söz konusudur. Yorgunluk hisseden hastalara semptomlar hafifleyene kadar araç ve makinekullanmamaları tavsiyesinde bulunulmalıdır (bkz. Bölüm 4.8). 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti:TECENTRIQ monoterapisinin güvenliliği, çoklu tümör tiplerinde 4349 hastadaki havuzlanmış verilere dayandırılmıştır. En yaygın advers reaksiyonlar (>%10); yorgunluk (%30,1), iştahkaybı (%21,3), bulantı (%20), döküntü (%19,3), pireksi (%19), öksürük (%18,6), diyare (%18),dispne (%17,2), artralji (%16,7), asteni (%13,2), pirürit (%13,2), sırt ağrısı (%12,8), kusma(%12,5), idrar yolu enfeksiyonu (%11,5) ve baş ağrısı (%10,3) olmuştur. TECENTRIQ'in diğer tıbbi ürünlerle kombinasyonunda kullanımının güvenliliği, çoklu tümör tiplerinde 4535 hastada değerlendirilmiştir. En yaygın advers reaksiyonlar (>%20); anemi(%36,8), nötropeni (36,6), bulantı (%35,5), yorgunluk (%33,1), trombositopeni (%27,1), diyare(%27,6), döküntü (%27,8), alopesi (%28,1), konstipasyon (%25,8), iştah azalması (%24,7),periferal nöropati (%24,4) olmuştur. Adjuvan Küçük Hücreli Dışı Akciğer Kanseri koşulunda TECENTRIQ kullanımıKüçük hücreli dışı akciğer kanseri (KHDAK) hasta popülasyonunda adjuvan ortamda TECENTRIQ'in güvenlilik profili (IMpower010), genel olarak ileri evre ortamında genelhavuzlanmış monoterapi güvenlilik profili ile tutarlı olmuştur. Bununla birlikte, ileri evrehastalığı olan havuzlanmış monoterapi popülasyonunda immün sistemle ilişkili adversreaksiyonlarının insidansı %38,4 iken, IMpower010 çalışmasında atezolizumabın immünsistemle ilişkili advers reaksiyonlarının insidansı %51,7 olmuştur. Adjuvan ortamda immünsistemle ilgili yeni advers reaksiyonlar saptanmamıştır. Ciddi advers reaksiyonlara dair detaylı bilgiler Bölüm 4.4 Özel Kullanım Uyarıları ve Önlemleri bölümünde verilmektedir. Advers reaksiyonların tablo halinde listesiAdvers İlaç Reaksiyonları (ADR), MedDRA sistem organ sınıfına (SOC) ve sıklık kategorilerine göre Tablo 2'de TECENTRIQ monoterapi veya kombinasyon tedavisi içinlistelenmiştir. Kombinasyon tedavisi kullanılan klinik çalışmalarda raporlanmasa bile, tekbaşına TECENTRIQ ile veya kemoterapiler ile ortaya çıkabileceği bilinen advers reaksiyonlar,kombinasyon tedavisi sırasında ortaya çıkabilir. 17 Aşağıdaki sıklık kategorileri kullanılmıştır: Çok yaygın (>1/10), yaygın (>1/100 ila <1/10), yaygın olmayan (>1/1000 ila <1/100), seyrek (>1/10.000 ila <1/1000), çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahminedilemiyor). Her bir sıklık grubunda advers reaksiyonlar azalan ciddiyet sırasına göreyazılmıştır.
a İdrar yolları enfeksiyonu, sistit, piyelonefrit, escherichia idrar yolları enfeksiyonu, bakteriyel idrar yolları enfeksiyonu, böbrek enfeksiyonu, akut piyelonefrit, kronik piyelonefrit, piyelit,böbrek apsesi, streptokokal idrar yolu enfeksiyonu, üretrit, fungal idrar yolları enfeksiyonu,psödomonal idrar yolları enfeksiyonu raporlarını içerir. b Pnömoni, bronşit, alt solunum yolu enfeksiyonu, enfeksiyöz plevral efüzyon, trakeobronşit, atipik pnömoni, akciğer apsesi, kronik obstrüktif solunum yolu hastalığının enfektifalevlenmesi, parakanseröz pnömoni, piyopnömotoraks, plevral enfeksiyon, prosedür sonrasıpnömoni raporlarını içerir. c Kanda kreatinin yükselmesi, hiperkreatinemi raporlarını içerir d Trombositopeni ve platelet sayısında azalma raporlarını içerir. e Nötropeni, nötrofil sayısında azalma, febril nötropeni, nötropenik sepsis, granülositopeni raporlarını içerir. f Beyaz kan hücresi sayısında azalma ve lökopeni raporlarını içerir. g Lenfopeni, lenfosit sayısında azalma raporlarını içerir. h İnfüzyonla ilişkili reaksiyon, sitokin salınım sendromu, aşırı duyarlılık ve anafilaksi raporlarını içerir. 191 Anti-tiroid antikor pozitifliği, otoimmün hipotiroidizm, otoimmün tiroidit, kanda tiroid stimüle edici hormon anomalisi, kanda tiroid stimüle edici hormon azalması, kanda tiroid stimüle edicihormon artışı, ötiroid hasta sendromu, guatr, hipotiroidizm, immünite ile ilişkili hipotiroidizm,miksödem, miksödem koması, primer hipotiroidizm, tiroid bozukluğu, tiroid hormonlarındadüşüş, tiroid fonksiyon testi anomalisi, tiroidit, akut tiroidit, tiroksin azalması, serbest tiroksinazalması, serbest tiroksin artışı, tiroksin artışı, triiyodotironin azalması, anormal serbesttriiyodotironin, serbest triiyodotironin azalması, serbest triiyodotironin artışı, sessiz tiroidit,kronik tiroidit raporlarını içerir. o Periferal nöropati, otoimmün nöropati, periferal duyusal nöropati, polinöropati, herpes zoster, periferal motor nöropati, nevraljik amyotrofi, periferal sensorimotor nöropati, toksik nöropati,aksonal nöropati, lumbosakral pleksopati, nöropatik artropati, periferal sinir enfeksiyonu, nevrit,immünite ile ilişkili nöropati raporlarını içerir. p Guillain-Barre sendromu ve demiyalizan polinöropati raporlarını içerir. q Ensefalit, otoimmün ensefalit, menenjit, fotofobi raporlarını içerir.r Myastenia gravis raporlarını içerir. s Miyokardit, otoimmün miyokardit, immünite ile ilişkili miyokardit raporlarını içerir. * Pnömonit, akciğer infiltrasyonu, bronşiolit, immünite ile ilişkili pnömonit, interstisyel akciğer hastalığı, alveolit, akciğer opasitesi, pulmoner toksisite ve radyasyon pnömoniti raporlarınıiçerir. u İshal, acil dışkılama, sık bağırsak hareketleri, hemorajik diyare, gastrointestinal hipermotilite raporlarını içerir. v Kolit, otoimmün kolit, iskemik kolit, mikroskobik kolit, ülseratif kolit, diversiyon koliti, immünite ile ilişkili enterokolit raporlarını içerir. ab Miyozit, rabdomiyoliz, polimiyalji romatika, dermartomiyozit, kas apsesi, idrarda miyoglobin varlığı raporlarını içerir. ac Proteinüri, idrarda protein varlığı, hemoglobinüri, idrar anomaliliği, nefrotik sendrom, albuminüri raporlarını içerir. ad Nefrit, otoimmün nefrit, Henoch-Schonlein Purpura nefriti, paraneoplastik glomerulonefrit, tubülointerstisyel nefrit raporlarını içerir. ae Hipokalemi, kanda potasyum azalması raporlarını içerir. af Hiponatremi, kanda sodyum azalması raporlarını içerir. ag Hipoksi, oksijen satürasyonu azalması, pO2 azalması raporlarını içerir. 20ah Alopesi, madarozis, alopesi areata, alopesi totalis, hipotrikoz raporlarını içerir. aı Hipertansiyon, kan basıncı artışı, hipertansif krizler, sistolik kan basıncı artışı, diyastolikhipertansiyon, uygun şekilde kontrol edilemeyen kan basıncı, hipertansif retinopati, hipertansifnefropati, esansiyel hipertansiyon, ortostatik hipertansiyon raporlarını içerir.aj Sepsis, septik şok, ürosepsis, nötropenik sepsis, pulmoner sepsis, bakteriyel sepsis, klebsiellasepsisi, abdominal sepsis, kandida sepsis, echerichia sepsis, psödomonal sepsis, stafilokokalsepsis raporlarını içerir.ak Bulöz dermatit, eksfoliyatif döküntü, eritema multiforme, genel eksfoliyatif dermatit, toksik cilt erüpsiyonu, Steven-Johnson sendromu, eosinofili ve sistemik semptomları olan ilaçreaksiyonu, toksik epidermal nekrolizis, kutanöz vaskülit raporlarını içerir.al Enfektif olmayan ve immün aracılı sistit raporlarını içerir.am Nazofarenjit, nazal konjesyon ve rinorea raporlarını içerir. an Psoriyazis, psoriyaziform dermatit, guttate psoriyazis (sedef hastalığı) raporlarını içerir.ao Perikardit, perikardiyal efüzyon, kardiyak tamponad ve konstriktif perikardit raporlarını içerir Seçilen advers reaksiyonların açıklaması:Aşağıdaki veriler, klinik çalışmalarda TECENTRIQ monoterapi ile oluşan anlamlı advers reaksiyonlarla ilgili bilgileri yansıtır (bkz. Bölüm 5.1). TECENTRIQ kombinasyonu ile ortayaçıkan advers reaksiyonlara ait detaylı bilgiler, eğer advers reaksiyon TECENTRIQmonoterapiye göre klinik olarak anlamlı bir farklılık gösteriyorsa karşılaştırmalı olarakverilmiştir. Bu advers reaksiyonlar için yönetim kılavuzları Bölüm 4.2 ve 4.4'te açıklanmaktadır. İmmünite ile ilişkili pnömonit: Pnömonit, TECENTRIQ monoterapi alan hastaların %3 (130/4349) kadarında meydana gelmiştir. 130 hasta içinde iki olay ölümcül olmuştur. Başlangıca kadar geçen medyan süre 4ay (aralık: 3 gün - 29,8 ay) olmuştur. Medyan süre 1,6 ay (aralık: 1 (bir) gün - 27,8+ ay; "+"sansürlenmiş bir değeri gösterir) olmuştur. Pnömonit, 29 hastada (%0,7) TECENTRIQ'inbırakılmasına yol açmıştır. Kortikosteroid kullanımı gerektiren pnömonit, TECENTRIQmonoterapi alan hastaların %1,7'sinde (76/4349) meydana gelmiştir. İmmünite ile ilişkili hepatit: Hepatit, TECENTRIQ monoterapi alan hastaların %1,7'sinde (75/4349) meydana gelmiştir. 75 hasta içinde iki ölümcül olay olmuştur. Başlangıca kadar geçen medyan süre 1,6 ay (aralık: 7gün - 18,8 ay) olmuştur. Medyan süre 2,1 ay (aralık: 1 (bir) gün - 22+ ay; "+" sansürlenmiş birdeğeri gösterir) olmuştur. Hepatit, 13 hastada (<%0,3) TECENTRIQ'in bırakılmasına yolaçmıştır. Kortikosteroid kullanımı gerektiren hepatit, TECENTRIQ monoterapi alanhastaların %0,5'inde (22/4349) meydana gelmiştir. İmmünite ile ilişkili kolit: Kolit, TECENTRIQ monoterapi alan hastaların %1,1'inde (50/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5,1 ay (aralık: 15 gün - 17,2 ay) olmuştur. Medyan süre1,2 ay (aralık: 1 (bir) gün - 35,9+ ay; "+" sansürlenmiş bir değeri gösterir) olmuştur. Kolit, 17hastada (%0,4) TECENTRIQ'in bırakılmasına yol açmıştır. Kortikosteroid kullanımıgerektiren kolit TECENTRIQ monoterapi alan hastaların %0,6'sında (24/4349) meydanagelmiştir. 21İmmünite ile ilişkili endokrinopatiler: Tiroid bozuklukları Hipotiroidizm, TECENTRIQ monoterapi alan hastaların %7,6'sında (331/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 4,3 ay (aralık: 1 (bir) gün - 34,5 ay) olmuştur.Adjuvan küçük hücreli dışı akciğer kanseri ortamında TECENTRIQ monoterapisi alanhastaların %17,4'ünde (86/495) hipotiroidizm meydana gelmiştir. Başlangıca kadar geçenmedyan süre 4 ay (aralık: 22 gün - 11,8 ay) olmuştur. Hipertiroidizm, TECENTRIQ monoterapi alan hastaların %2,1'inde (93/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 2,6 ay (aralık: 1 (bir) gün - 24,3 ay) olmuştur.Adjuvan küçük hücreli dışı akciğer kanseri ortamında TECENTRIQ monoterapi alanhastaların %6,5'inde (32/495) hipertiroidizm meydana gelmiştir. Başlangıca kadar geçenmedyan süre 2,8 ay (aralık: 1 (bir) gün - 9,9 ay) olmuştur. Adrenal yetmezlik Adrenal yetmezlik, TECENTRIQ monoterapi alan hastaların %0,5'inde (21/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 6,1 ay (aralık: 2 (iki) gün - 21,4 ay) olmuştur.Adrenal yetmezlik 5 (beş) hastada (%0,1) TECENTRIQ'in bırakılmasına neden olmuştur.Kortikosteroid kullanımı gerektiren adrenal yetmezlik TECENTRIQ monoterapi alanhastaların %0,4'ünde (17/4349) meydana gelmiştir. Hipofizit Hipofizit, TECENTRIQ monoterapi alan hastaların %0,1'inden azında (4/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 56,1 aydır (aralık: 23 gün - 13,7 ay). Üç hastada(< %0,1) kortikosteroid kullanımı gerekmiştir ve 1 hastada (< %0,1) TECENTRIQ tedavisidurdurulmuştur. Hipofizit, nab-paklitaksel ve karboplatinle kombinasyon halinde atezolizumab almış olan hastaların %0,4'ünde (2/473) meydana gelmiştir. Başlangıca kadar geçen medyan süre, 5,2 ay(aralık: 5,1 - 5,3 ay) olmuştur. Her iki hastada kortikosteroid kullanılması gerekmiştir. Diabetes mellitus Diabetes mellitus, TECENTRIQ monoterapi alan hastaların % 0,5'inde (20/4349) meydana gelmiştir. Medyan süre 5,5 ay (aralık: 4 (dört) gün - 29 ay) olmuştur. Diabetes mellitus,hastaların %0,1'inden azında (3/4349) TECENTRIQ'in bırakılmasına yol açmıştır. Diabetes mellitus, TECENTRIQ ile birlikte bevacizumab alan hepatoselüler kanserli hastaların % 2,0'ında (10/493) meydana gelmiştir. Başlangıca kadar geçen medyan süre 4,4 ay(aralık: 1,2 ay - 8,3 ay) olmuştur. TECENTRIQ tedavisinin bırakılmasına neden olacak birdiabetes mellitus olayı görülmemiştir. İmmünite ile ilişkili meningoensefalit: Meningoensefalit, TECENTRIQ monoterapi alan hastaların %0,4'ünde (18/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 16 gün (aralık: 1 (bir) gün - 12,5 ay) olmuştur.Medyan süre 22 gün (aralık: 6 gün 14,5+ ay; "+" sansürlenmiş bir değeri gösterir) olmuştur. 22Kortikosteroid kullanımını gerektiren meningoensefalit TECENTRIQ tedavisi alan hastaların %0,2'sinde (10/4349) meydana gelmiştir ve sekiz hastada (%0,2) TECENTRIQ'inbırakılmasına yol açmıştır. İmmünite ile ilişkili nöropatiler: Guillain-Barre sendromu ve demiyalizan polinöropati, TECENTRIQ monoterapi alan hastaların %0,1'inde (6/4349) meydana gelmiştir: Bu olay için başlangıca kadar geçen medyansüre 4,1 ay (aralık: 17 gün - 8,1 ay) olmuştur. Medyan süre 8 ay (aralık: 19 gün - 24,5 ay+, "+"sansürlenmiş bir değeri gösterir). 1 (bir) hasta (< %0,1), Guillain-Barre sendromu nedeniyleTECENTRIQ kullanımını bırakmıştır. Kortikosteroid kullanımı gerektiren Guillain-Barresendromu TECENTRIQ monoterapi alan hastaların %0,1'inden azında (3/4349) meydanagelmiştir. Miyastenik sendrom: Miyestenia gravis, TECENTRIQ monoterapi alan hastaların %0,1'inden azında (1 (bir)/4349) meydana gelmiştir. Başlangıca kadar geçen süre 1,2 aydır. İmmünite ile ilişkili pankreatit: Yüksek amilaz ve yüksek lipaz dahil olmak üzere pankreatit, TECENTRIQ monoterapi alan hastaların %0,7'sinde (32/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5,5ay (aralık: 1 (bir) gün - 24,8 ay) olmuştur. Medyan süre 24 gün (aralık: 3 gün - 22,4+ ay; "+"sansürlenmiş bir değeri gösterir) olmuştur. Pankreatit, 3 hastada (<%0,1) TECENTRIQ'inbırakılmasına yol açmıştır. Kortikosteroid kullanımı gerektiren pankreatit vakalarıTECENTRIQ monoterapi alan hastaların %0,1'inde (5/4349) meydana gelmiştir. İmmünite ile ilişkili miyokardit: Miyokardit, TECENTRIQ monoterapi alan hastaların %0,1'inden azında (3/4349) meydana gelmiştir.Adjuvan küçük hücreli dışı akciğer kanseri ortamında 3 vakadan biri ölümcülolmuştur. Başlangıca kadar geçen medyan süre 2,1 ay (aralık: 1,5 - 4,9 ay) olmuştur. Geçenmedyan süre 14 gün (aralık: 14 gün - 2,8 ay) olmuştur. Miyokartidiki hastada (<%0,1)TECENTRIQ'in bırakılmasına neden olmuştur. İki hastada (< %0,1) kortikosteroidkullanımına gerek duyulmuştur. İmmünite ile ilişkili nefrit: Nefrit, TECENTRIQ alan hastaların %0,2'sinden az hastada (10/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 5 ay (aralık: 2 gün - 17,5 ay) olmuştur. Nefrit 5 (beş)hastada (< %0,1) TECENTRIQ'in bırakılmasına neden olmuştur. Dört hastada (< %0,1)kortikosteroid kullanımına gerek duyulmuştur. İmmünite ile ilişkili miyozit: Miyozit, TECENTRIQ monoterapi alan hastaların %0,5'inde (20/4349) meydana gelmiştir. Başlangıca kadar geçen medyan süre 3,3 aydır (aralık: 12 gün ile 11 ay). Medyan süre 5,7 ay(aralık: 2 (iki) gün ile 36,9 +ay; + sansürlenmiş bir değeri gösterir). Miyozit 2 (iki) hastada(< %0,1) TECENTRIQ'in bırakılmasına neden olmuştur. 7 (yedi) hastada (%0,2) 23kortikosteroid kullanımına gerek duyulmuştur. İmmünite ile ilişkili şiddetli kutanöz advers reaksiyonlar: TECENTRIQ monoterapi alan hastaların %0,6'sında (28/4349) şiddetli kutanöz advers reaksiyonlar meydana gelmiştir. 28 hastadan birinde ölümcül olay meydana gelmiştir.Başlangıca kadar geçen medyan süre 5,2 aydır (aralık: 4 (dört) gün ile 15,5 ay). Medyan süre 2.4 ay (aralık: 1 (bir) gün ile 37,5 +ay; + sansürlenmiş bir değeri gösterir). Şiddetli kutanözadvers reaksiyonlar 3 (üç) hastada (< %0,1) TECENTRIQ tedavisinin bırakılmasına nedenolmuştur.TECENTRIQ monoterapi alan hastaların %0,2'sinde (9/4349) kortikosteroidkullanımına gerek duyulmuştur. İmmünite ile ilişkili perikardiyal bozukluklar: TECENTRIQ monoterapisi alan hastaların %1,1'inde (47/4349) perikardiyal bozukluklar meydana gelmiştir. Medyan başlangıç süresi 1,4 aydır (aralık: 6 gün ila 17,5 ay). Medyan süre 1.4 aydır (aralık: 0 ila 19,3+ ay; "+" sansürlenmiş bir değeri gösterir). Perikardiyal bozukluklar3 (< %0,1) hastada TECENTRIQ tedavisinin kesilmesine neden olmuştur. Kortikosteroidkullanımını gerektiren perikardiyal bozukluklar hastaların %0,2'sinde (7/4349) meydanagelmiştir. İmmünojenisite: Çoklu faz III çalışmaları karşılaştırılmasında, hastaların %13,1 ile %54,1'i tedaviye yeni başlayan anti-ilaç antikorları (ADA) geliştirmiştir. Tedavi sonucu oluşmuş anti-ilaç antikoru(ADA) gelişen hastaların başlangıçta genel olarak sağlık durumu ve hastalık karakteristikleriaçısından daha zayıf olduğu görülmüştür. Başlangıçtaki bu sağlık ve hastalıkkarakteristiklerindeki dengesizlikler, farmakokinetik, etkililik ve güvenlilik analizlerininyorumlanmasında karışıklık yaratabilmektedir. Anti-ilaç antikorlarının (ADA) etkililiğeetkisini araştırmak için başlangıçtaki sağlık ve hastalık karakteristiklerindeki dengesizlikleriayarlayan keşif analizleri yapılmıştır. Bu analizlerde ADA geliştiren hastaların, ADAgeliştirmeyen hastalara kıyasla etkililik faydasında azalma ihtimali gözardı edilmemiştir. Anti-ilaç antikorlarının başlangıca kadar geçen medyan süresi 3 (üç) ila 5 (beş) hafta olmuştur. TECENTRIQ monoterapisi (N=3460) ve kombinasyon tedavileri (N=2285) ile tedavi edilen hastalardan elde edilen hasta havuzu verilerinden, ADA-pozitif popülasyonunda ADA-negatifpopülasyonuna kıyasla gözlenen advers olayların sıklığı sırasıyla: Monoterapi için; 3. ve 4.derece advers olaylar %46,2'ye karşı %39,4, ciddi advers olaylar %39,6'ya karşı %33,3,tedavinin kesilmesine neden olan advers olaylar %8,5'e karşı %7,8 iken; Kombinasyon tedavisiiçin 3. ve 4. derece advers olaylar %63,9'a karşı %60,9, ciddi advers olaylar %43,9'a karşı 35,6,tedavinin kesilmesine neden olan advers olaylar % 22,8'e karşı %18,4 olmuştur (kombinasyontedavisi için). Ancak mevcut verilerden yola çıkarak olası ilaç advers reaksiyonlarının yolağıhakkında kesin sonuçlara varılamamaktadır. Pediyatrik popülasyon TECENTRIQ'in çocuklar ve adölesanlardaki güvenliliği bilinmemektedir. 69 pediyatrik hastada (<18 yaş) yapılan bir klinik çalışmada yeni bir güvenlilik sinyali oluşmamıştır vegüvenlilik profili erişkinlerinki ile karşılaştırılabilirdir. 24Geriyatrik popülasyon TECENTRIQ monoterapi alan 65 yaş ve üzerindeki hastalar ile daha genç hastalar arasında genel olarak bir güvenlilik farklılığı gözlemlenmemiştir. Impower150, IMpower133 ve IMpower110 çalışmalarından elde edilen veriler, 75 yaş ve üzeri hasta grubu hakkında değerlendirme yapılması için çok sınırlıdır. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr[email protected];4.9. Doz aşımı ve tedavisiTECENTRIQ doz aşımına ilişkin bilgi mevcut değildir. Doz aşımı durumunda, hastalar advers reaksiyon belirtileri veya semptomları bakımından yakından izlenmeli ve uygun semptomatik tedavi başlatılmalıdır. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antineoplastikler ve immünomodülatör ajanlar, monoklonal antikorlar ve antikor ilaç konjugatları, PD-1/PDL-1 (Programlanmış Hücre Ölüm Proteini 1/ ÖlümLigandı 1) İnhibitörleri ATC kodu: L01FF05 Etki mekanizması:PD-L1, tümör hücreleri ve/veya tümör infiltre eden immün hücrelerinde eksprese olabilir ve tümör mikroortamında anti-tümör immün yanıtının inhibisyonuna katkıda bulunabilir. PD-L1'in T hücrelerinde ve antijen sunan hücrelerde bulunan PD-1 ve B7.1 reseptörlerinebağlanması sitotoksik T hücresi aktivitesini, T hücresi çoğalmasını ve sitokin üretimini baskılar. Atezolizumab Fc bölgesi değiştirilmiş, hümanize bir immünoglobülin G1 (IgG1) monoklonal bir antikorudur; doğrudan PD-L1'e bağlanır ve PD-1 ve B7.1 reseptörlerinin ikili blokajınısağlayarak, antikor bağımlı sellüler sitotoksisiteyi indüklemeden antitümör immün yanıtınyeniden aktive edilmesi de dahil, immün yanıtın PD-L1/PD-1 aracılı inhibisyonunu serbestbırakır. Atezolizumab, PD-L2/PD-1 etkileşimini koruyarak PD-L2/PD-1 aracılı inhibitörsinyallerin devam etmesine izin verir. Klinik etkililik ve güvenlilik:Küçük hücreli dışı akciğer kanseri: 25Erken evre küçük hücreli dışı akciğer kanserinin adjuvan tedavisi IMpower010 (GO29527): Rezeke edilmiş KHDAK'lı hastalarla sisplatin bazlı kemoterapiden sonra randomize faz III çalışmaEvre IB (tümörler > 4cm) - IIIA KHDAK'lı hastaların adjuvan tedavisi için atezolizumabın etkililiğini ve güvenliliğini değerlendirmek üzere bir faz III, açık etiketli, çok merkezli,randomize çalışma olan GO29527 (IMpower010) yürütülmüştür (Kanser evrelendirmesistemine ilişkin Uluslararası Kanser Kontrolü Birliği/Amerikan Ortak Kanser Komitesi, 7.Baskısına göre). Aşağıdaki seçim kriterleri, terapötik endikasyona dahil olan ve 7. baskı evreleme sistemine göre evre II - IIIA hasta popülasyonunu yansıtan yüksek nüks riski olan hastaları tanımlar: Tümör boyutu > 5 cm veya N1 veya N2 durumunun eşlik ettiği herhangi bir boyuttaki tümörler veya torasik yapılara invaze olan tümörler (doğrudan parietal plevra, göğüs duvarı, diyafram,frenik sinir, mediastinal plevra, parietal perikard, mediasten, kalp, büyük damarlar, trakea,rekürren laringeal sinir, özofagus, vertebral gövde, karinaya invaze) veya ana bronşu karinanın< 2 cm distalinde tutan ancak karina tutulumu olmayan tümörler veya tüm akciğerin atelektazisiveya obstrüktif pnömonisi ile bağlantılı tümörler veya primer olarak aynı lobda veya farklıipsilateral lobda ayrı nodül(ler)e sahip tümörler. Çalışma, mediasten, kalp, büyük damarlar, trakea, rekürren laringeal sinir, özofagus, vertebral cisim ve karinaya invaze tümörleri olan N2 durumundaki hastaları veya farklı bir ipsilaterallobda ayrı tümör nodülleri olan hastaları içermemiştir. Toplamda 1280 kaydedilmiş hasta tam tümör rezeksiyonu yaptırmış ve 4 sikluse kadar sisplatin bazlı kemoterapi almaya uygun olmuştur. Sisplatin bazlı kemoterapi rejimleri Tablo 3'te tarifedilmektedir. Tablo 3: Adjuvan tedavi rejimleri (IMpower010)1. ve 8. gün intravenöz olarak 30 mg/m2 vinorelbin 1. gün intravenöz olarak 75 mg/m2 dosetaksel_1. ve 8. gün intravenöz olarak 1250 mg/m2 gemsitabin 1. gün intravenöz olarak 500 mg/m2pemetreksed (non-skuamöz)Sisplatin bazlı adjuvan kemoterapi: Belirtilen tedavi rejimlerinden biri ile her 21 günlük siklusun 1.gününde IV olarak75 mg/m2 sisplatin Sisplatin bazlı kemoterapinin (dört siklusa kadar) tamamlanmasından sonra toplamda 1005 hasta 1:1 oranında atezolizumab (Kol A) veya en iyi destekleyici bakımı (BSC) (Kol B) almayarandomize edilmiştir. Atezolizumab, hastalık nüksü veya kabul edilemez toksisite görülmediğisürece 16 döngüye kadar her 3 haftada bir IV infüzyonla 1200 mg'lık sabit dozda uygulanmıştır.Randomizasyon cinsiyet, hastalık evresi ve PD-L1 ekspresyonuna göre sınıflandırılmıştır. Hastalar, otoimmün hastalık öyküsüne sahiplerse; randomizasyondan önceki 28 gün içinde canlı, attenüe aşı uygulaması yapılmışsa; randomizasyondan önceki 4 hafta içinde sistemikimmunostimulatör ajanlar veya 2 hafta içinde immunosupresif ilaçlar uygulanmışsa hariç 26tutulmuştur. Tümör değerlendirmeleri randomizasyon fazının başlangıcında ve 1. siklus, 1. gününün ardından ilk yıl boyunca her 4 ayda bir ve ardından beş yıla kadar her 6 ayda bir vesonrasında yılda bir yürütülmüştür. ITT popülasyonunun demografik bilgileri ve başlangıçtaki hastalık özellikleri tedavi kolları arasında iyi dengelenmiştir. Medyan yaş 62 olup (aralık: 26 ila 84) hastaların %67'si erkektir.Hastaların çoğu Beyaz (%73) ve %24'ü Asyalıdır. Çoğu hasta sigara içicisidir veya geçmişteiçmiştir (%78) ve hastalarda başlangıç ECOG performans durumu 0 (%55) veya 1'dir (%44).Genelde hastaların %12'sinde evre IB, %47'sinde evre II ve %41'inde evre IIIA hastalıkmevcuttur. Tümörleri VENTANA PD-L1 (SP263) Analizi ile ölçüldüğü üzere TC'de PD-L1ekspresyonu > %1 ve > %50 olan tümörleri olan hastaların yüzdesi sırasıyla %55 ve %26olmuştur. Birincil etkililik sonucu ölçümü araştırmacı tarafından değerlendirilen hastalıksız sağkalımdır (HS). Hastalıksız sağkalım, randomizasyon tarihinden aşağıdakilerden birinin meydana geldiğitarihe kadar geçen süre olarak tanımlanmıştır: ilk belgelenmiş hastalık nüksü, yeni primerKHDAK veya herhangi bir nedenle ölüm (hangisi daha önce olursa). Birincil etkililik hedefi,PD-L1 > %1 TC evre II IIIA hasta popülasyonunda HS'yi değerlendirmektir. Kritik ikinciletkililik hedefleri, PD-L1 > %50 TC evre II-IIIA hasta popülasyonunda HS'yi ve ITTpopülasyonunda genel sağkalımı (GS) değerlendirmektir. Ara hastalıksız sağkalım analizi zamanında, çalışma birincil sonlanım noktasını sağlamıştır. Medyan takip süresi yaklaşık 32 aydır. EGFR mutasyonları veya ALK yeniden düzenlemeleriolmayan PD-L1 > %50 TC evre II- IIIA hastalarının analizinde (n = 209), BSC koluna kıyaslaatezolizumab kolunda HS'de klinik olarak anlamlı bir iyileşme gözlenmiştir (Tablo 4). Genelsağkalım (GS) verileri EGFR mutasyonları ve ALK yeniden düzenlemeleri olmaksızın, PD-L1> %50 TC evre II- IIIA hasta popülasyonunda genel olarak bildirilen yaklaşık %16,3 oranındaölüm ile hastalıksız sağkalımda ara analizi sırasında olgunlaşmamıştır. Genel sağkalımınkeşifsel analizi bu hasta popülasyonunda 0,39'luk sınıflandırılmış bir HR (%95 GA:0,18, 0,82)ile en iyi destekleyici bakıma karşı atezolizumab lehine bir eğilimi düşündürmüştür. EGFR mutasyonları ve ALK yeniden düzenlemeleri olmayan PD-L1 > %50 TC evre II- IIIA hasta popülasyonu için ana etkililik bulguları Tablo 4'te özetlenmektedir. Hastalıksız sağkalımiçin Kaplan-Meier eğrisi Şekil 1'de sunulmaktadır. Tablo 4: EGFR mutasyonları veya ALK yeniden düzenlemeleri olmayan PD-L1 ekspresyonu > %50 TC evre II- IIIA hasta popülasyonunda etkililik özeti (IMpower010)
sağkalım oranı (%)HS=Hastalıksız sağkalım, GA=Güven Aralığı, NE=öngörülemiyor Şekil 1: EGFR mutasyonları veya ALK yeniden düzenlemeleri olmayan PD-L1 ekspresyonu > %50 TC evre II- IIIA hasta popülasyonunda (IMpower010) hastalıksızsağkalım için Kaplan-Meier eğrisi
1.0 -0.9 -0.8-0.7 -0.6-0.5 -0.4 -0.3 -0.2-0.1 -0.0- A Kolu: Tecentriq B Kolu: En iyi destek tedavisiI I-1 I-24 27 30 33Süre (ay)36 39 42 45 48 51 549 12 15 18 21Risk altındaki Hasta SayısıA Kolu: Tecentriq 106 101 98B Kolu: En iyi destek 103 98 84tedavisiNE=öngörülemiyor 67521513261743325642897278578464876588689678En iyi destekleyici tedavi koluna kıyasla atezolizumab kolunda gözlenen hastalıksız sağkalım iyileşmesi, hem non-skuamoz KHDAK hastaları (sınıflandırılmamış risk oranı: 0,35 , %95 GA:0,18, 0,69; medyan hastalıksız sağkalım NE'ye (NE= öngörülemiyor) karşı 35,7 ay) hem deskuamoz KHDAK hastaları (sınıflandırılmamış risk oranı: 0,60 ,%95 GA: 0,29 , 1,26; medyanhastalıksız sağkalım 36,7'ye karşı NE (NE=öngörülemiyor) ay) içeren EGFR mutasyonlarıveya ALK yeniden düzenlemeleri olmayan PD-L1 >%50 TC evre II-IIIA hasta popülasyonundaönceden belirtilmiş alt grupların çoğu arasında tutarlı şekilde gösterilmiştir. Birinci basamak küçük hücreli dışı akciğer kanseri IMpower130 (GO29537): Daha önce kemoterapi uygulanmamış metastatik non-skuamoz KHDAK hastalarında nab-paklitaksel ve karboplatinle kombinasyona yönelik randomize faz III çalışma.Daha önce kemoterapi uygulanmamış metastatik non-skuamoz KHDAK hastalarında nab-paklitaksel ve karboplatinle kombinasyon halinde atezolizumabın etkililik ve güvenliliğini değerlendirmek üzere bir faz III, açık etiketli, randomize çalışma (GO29537 (IMpower130))gerçekleştirilmiştir. EGFR mutasyonları veya ALK rearanjmanları olan hastalarda daha öncetirozin kinaz inhibitörleriyle tedavi almış olma koşulu aranmıştır. Hastalar, Amerikan Ortak Kanser Komitesi (AJCC) 7. baskıya göre evrelendirilmiştir. Otoimmün hastalık öyküsü olan, randomizasyondan önceki 28 gün içinde canlı ve zayıflatılmış 28aşı uygulanmış, randomizasyondan önceki 4 hafta içinde immünostimülatör ajan veya randomizasyondan önceki 2 hafta içinde sistemik immünosüpresif ilaç uygulanmış ve aktifveya tedavi edilmemiş MSS metastazları olan hastalar çalışmaya alınmamıştır. Daha önceCD137 agonistleri veya immün kontrol noktası blokaj tedavileri (anti-PD-1 ve anti-PD-L1terapötik antikorları) almış hastalar çalışmaya uygun değildir. Bununla birlikte, daha önce anti-CTLA-4 tedavisi almış olan hastalar, en son dozun randomizasyondan en az 6 hafta öncealınmış olması ve anti-CTLA-4'ten kaynaklanan şiddetli immün ilişkili advers etki öyküsü(NCI 3 ve 4. Derece) olmaması koşuluyla çalışmaya alınmıştır. Tümör değerlendirmeleri, 1.Siklustan sonra ilk 48 hafta boyunca 6 haftada bir ve daha sonra 9 haftada bir yapılmıştır.Tümör numuneleri, tümör hücreleri (TC) ve tümörü infiltre eden immün hücreler (IC)üzerindeki PD-L1 ekspresyonu bakımından değerlendirilmiştir ve bulgular, aşağıda açıklanananalizler için PD-L1 ekspresyonu alt gruplarının tanımlanmasında kullanılmıştır. EGFR mutasyonları veya ALK rearanjmanları olanlar dahil olmak üzere hastalar, Tablo 5'te açıklanan tedavi rejimlerinden biri uygulanmak üzere çalışmaya alınmış ve 2:1 oranındarandomize edilmiştir. Randomizasyon grupları; cinsiyet, karaciğer metastazları varlığı ve TCve IC üzerindeki PD-L1 ekspresyonuna göre oluşturulmuştur. Tedavi rejimi B alan hastalarınhastalık progresyonundan sonra çapraz geçiş yaparak atezolizumab monoterapisi almasına izinverilmiştir.
a Atezolizumab, araştırmacı değerlendirmesine göre belirlenen klinik fayda kaybına kadar uygulanır b Nab-paklitaksel, her siklusun 1, 8 ve 15. günlerinde uygulanır c Nab-paklitaksel ve karboplatin, 4-6. sikluslar tamamlanana kadar veya progresif hastalık veya kabul edilemez toksisiteye kadar (hangisi daha erken gerçekleşirse) uygulanır ITT-WT olarak tanımlanan çalışma popülasyonunun (n=679) demografik özellikleri ve başlangıçtaki hastalık özelliklerinin tedavi kolları arasında dengeli olduğu görülmüştür.Medyan yaş 64'tür (aralık: 18-86 yaş). Hastaların çoğu erkek (%59) ve beyazdır (%90).Hastaların %14,7'sinde başlangıçta karaciğer metastazı olduğu ve çoğu hastanın (%90) sigarakullanmakta olduğu veya geçmişte kullandığı görülmüştür. Hastaların çoğunun başlangıçECOG performans durumunun 1 olduğu (%59) ve <%1 oranında PD-L1 ekspresyonusergilediği (yaklaşık %52) görülmüştür. İndüksiyon tedavisinin ardından yanıt durumu stabilhastalık, kısmi yanıt veya tam yanıt olan 107 Kol B hastası arasında 40 hasta pemetrexed geçişidame tedavisi almıştır. Primer analiz, EGFR mutasyonları veya ALK rearanjmanları olanlar hariç olmak üzere, ITT-WT olarak tanımlanan bütün hastalarda (n=679) gerçekleştirilmiştir. Hastalarda medyan sağkalım takip süresi 18,6 ay olup, hastalar kontrole kıyasla atezolizumab, nab-paklitaksel ve 29karboplatinle OS ve PFS'de iyileşme göstermiştir. Temel bulgular, Tablo 6'da özetlenmiştir ve OS ve PFS'ye ait Kaplan-Meier eğrileri sırasıyla Şekil 2 ve 4'te sunulmuştur. PD-L1ekspresyonuna göre OS ve PFS için elde edilen keşifsel bulgular, sırasıyla Şekil 3 ve 5'teözetlenmiştir. Karaciğer metastazları olan hastalar, nab-paklitaksel ve karboplatine kıyaslaatezolizumab, nab-paklitaksel ve karboplatinle PFS veya OS'de iyileşme göstermemiştir(sırasıyla PFS için 0,93 HR, %95 CI: 0,59, 1,47 ve OS için 1,04 HR, %95 CI: 0,63, 1,72). Atezolizumab, nab-paklitaksel ve karboplatin kolundaki hastaların %7,3'üne karşılık nab-paklitaksel ve karboplatin kolundaki hastaların %59'u hastalık progresyonunun ardından çapraz geçiş tedavisi olarak atezolizumab (bütün hastaların %41'i) dahil olmak üzere herhangi birkanser immünoterapisi almıştır. Daha uzun takip süresine (medyan: 24,1 ay) sahip bir keşifsel analizde, her iki koldaki medyan OS birincil analize göre değişmemiş olup, HR=0,82'dir (%95 CI: 0,67, 1,01). Tablo 6: Primer analizde IMpower130'da kanıtlanan etkililiğin özeti (ITT-WT popülasyon)
30
t Cinsiyet ve TC ve IC üzerindeki PD-L1 ekspresyonuna göre gruplandınlmıştır A Doğrulanmış ORR ve DoR, keşifsel sonlanım noktalarıdırPFS=progresyonsuz sağkalım; RECIST=Solid Tümörlerde Yanıt Değerlendirme Kriterleri v1.1.; GA=güven aralığı; ORR=objektif yanıt oranı; DOR=yanıt süresi; OS=Genel sağkalımŞekil 2: Genel sağkalıma ait Kaplan-Meier eğrileri (IMpower130)
Nab-paklitaksel + 228 21820e 180 178 187 181 1M 147 138 132 124 110 100 88 80 75 85 58 48 38 312417 138831karboplatin31Şekil 3: PD-L1 ekspresyonuna göre genel sağkalııııı gösteren meta analiz diyagramı (IMpowerl30)
PD-L1 Ekspresyon Düzeyi TC > %50 veya IC > %10TC < %50 ve > %1 veya IC <%10 ve > %1TC ve IC < %1ni%}130(19)193 (28)356(521  rrT-WT3 ITT-WT için gruplandırılmış risk oram; diğer bütün alt gruplar içingruplandırılmamış risk oranı
679(100) 10Risk Oranı3 0.220Tecentriq + nab-paklitaksel + karboplatin lehineŞekil 4: Progresyonsuz sağkalıma ait Kaplan-Meier eğrileri (IMpowerl30)
228 214 174 150 136 110 00 75 61 48 40 35 20 23 18 15 76553322132Şekil 5: PD-L1 ekspresyonuna göre progresyonsuz sağkalııııı gösteren meta analiz diyagramı (IMpowerl30)
a ITT-WT için gruplandırılmış risk oranı;diğer bütün alt gruplar için gruplandırılmamış risk oranıIMpower110 (GO29431): Kemoterapi tedavisi almamış metastatik KHDAK hastalarında yapılan randomize Faz III çalışmaKemoterapi kullanmamış, metastatik KHDAK hastalarında atezolizumabın etkililiğini ve güvenliliğini değerlendirmek için Faz III, açık etiketli, çok merkezli, randomize bir çalışmaolan rMpower110 yürütülmüştür. Hastalardaki PD-L1 ekspresyonu >%1 TC (PD-L1 tümörhücrelerinin >%1'i boyanmıştır) veya >%1 IC (tümör bölgesinin >%1 'ini kapsayan tümör-infiltre edici immün hücreleri PD-L1 boyanmıştır) VENTANA PD-L1 (SP142) Testinedayanmaktadır. Toplamda 572 hasta randomize edilmiş ve 1:1 oranında atezolizumab (A kolu) veya kemoterapi (B kolu) verilmiştir. Atezolizumab, araştırmacı tarafından değerlendirildiği şekilde klinik faydakaybına veya kabul edilmeyen toksisiteye kadar üç haftada bir intravenöz yolla 1200 mg sabitdozla verilmiştir. Kemoterapi rejimleri Tablo 7'de gösterilmektedir. Randomizasyon cinsiyet,ECOG performans statüsü, histoloji ve TC ile IC'de PD-L1 tümör ekspresyonu ilekatmanlaştırılmıştır.
gemsitabina,c (1000 mg/m2) aSisplatin, karboplatin, pemetreksed ve gemsitabin 4 veya 6 siklusun tamamlanmasına veya progresif hastalığa veya kabul edilemez toksisiteye kadar uygulanmaktadırbPemetreksed, progresif hatalığa veya kabul edilemez toksisiteye kadar her 21 gün idame rejimiolarak uygulanmaktadır dKontrol kolundan (platin bazlı kemoterapi) atezolizumab koluna (A kolu) geçişe izin verilmemiştir Otoimmün hastalık, randomizasyondan önceki 28 gün içinde canlı, zayıflatılmış aşı uygulaması, 4 hafta içinde sistemik immün sistemi uyarıcı ajanların uygulanması veya randomizasyondanönceki 2 hafta içinde sistemik immün sistemi baskılayıcı ilaçlar öyküsü, aktif veya tedaviedilmemiş merkezi sinir sistemi metastazları olan hastalar çalışma dışı bırakılmıştır. Tümördeğerlendirmeleri, 1. siklus, 1.gün ve bunu takip eden ilk 48 hafta boyunca 6 haftada bir vedaha sonra her 9 haftada bir yürütülmüştür. EGFR mutasyonları veya ALK yeniden düzenlemeleri olmayan (n=554) PD-L1 ekspresyonu > %1 TC veya > %1 IC olan hastalarda demografik ve çalışma başlangıcı hastalık özellikleritedavi kolları arasında iyi dengelenmiştir. Medyan yaş 64,5 (dağılım: 30 ila 87) oluphastaların %70'i erkektir. Hastaların çoğunluğu beyaz (%84) ve Asyalıdır (%14). Hastalarınçoğu halen sigara kullanmaktadır veya öncesinde kullanmıştır (%87) ve hastalarda başlangıçECOG performans durumu 0 (%36) veya 1'dir (%64). Genel olarak, hastaların %69'unda skuamoz olmayan hastalık ve hastaların %31 'inde skuamoz hastalık vardır. EGFR mutasyonları veya ALK yeniden düzenlemeleri olmayan (n=205) yüksekPD-L1 ekspresyonu olan (PD-L1 > %50 TC veya > %10 IC) hastalarda demografik ve çalışmabaşlangıcı hastalık özellikleri genellikle daha geniş çalışma popülasyonunu temsil etmiş vetedavi kolları arasında dengeli olmuştur. Birincil sonlanım noktası, genel sağkalım (GS) olmuştur. Geçici genel sağkalım analizi sırasında, EGFR mutasyonları veya ALK yeniden düzenlemeleri olanlar (n=205) hariç olmaküzere yüksek PD-L1 ekspresyonu olan hastalar, kemoterapiye kıyasla atezolizumaba (Kol A)randomize edilen hastalar için genel sağkalımda istatistiksel olarak anlamlı iyileşme göstermiş(B kolu) (risk oranı 0,59, %95 GA: 0,4, 0,89; medyan genel sağkalım 20,2 aya karşı 13,1 ay)olup iki taraflı p değeri 0,0106 bulunmuştur. Yüksek PD-L1 ekspresyonu olan hastalardaortanca sağkalım takip süresi 15,7 ay olmuştur. Bu hastalarda daha uzun bir takip süresi (medyan: 31,3 ay) içeren bir keşifsel genel sağkalım analizinde atezolizumab kolu için medyan genel sağkalım, birincil genel sağkalım ara analizinegöre (20,2 ay) değişmemiş ve kemoterapi kolu için 14,7 olmuştur (risk oranı 0,76, %95 GA:0,54, 1,09). Keşifsel analizdeki anahtar sonuçlar Tablo 8'de özetlenmiştir. Yüksek PD-L1ekspresyonu olan hastalarda genel sağkalım ve progresyonsuz sağkalım için Kaplan-Meiereğrileri Şekil 6 ve 7'de sunulmaktadır. Atezolizumab kolunda (16/107, %15) kemoterapi kolunakıyasla (10/98, %10,2) hastaların daha yüksek bir oranı ilk 2,5 ay içinde ölüm yaşamıştır. Erkenölümlerle ilişkili herhangi bir spesifik faktör/faktörler tanımlanamamıştır. 34
* Cinsiyet ve ECOG performans statüsüne (0'a karşı 1) göre sınıflandırılmıştır PS=progresyonsuz sağkalım; RECIST=Solid Tümörlerde Yanıt Değerlendirme Kriterleri v1.1.;GA= Güven Aralığı; OYO=objektif yanıt oranı; YS=yanıt süresi; GS=genel sağkalım,TE=Tahmin edilemiyor 35Şekil 6: Yüksek PD-L1 ekspresyonu >%50 TC veya >%10 IC olan hastalarda genel sağkalım için Kaplan-Meier eğrisi (IMpower110)1 edavı koluMedyan GS HO (%95 GA)A Kolu: Tecentriq 20.2 ayB Kolu: Kemoterapi 14.7 ay0.76 (0.54, 1.09)14.7 ay
¦a 0.7- 0.5-»2P 0.40.2A Kolu: Tecentriq B Kolu: Kemoterapi0.10.0-/.aıııaıı (ay)Riskli hasta sayısıA Kolu: Tecentriq 107 95 86 817774 70 64 645954 51 43B Kolu: Kemoterapi 98 90 76 665653 49 48 443634 33 30Şekil 7: Yüksek PD-L1 ekspresyonu >%50 TC veya >%10 IC olan hastalarda progresyonsuz sağkalım için Kaplan-Meier eğrisi (IMpower110)
Kemoterapi koluna kıyasla atezolizumab kolunda gözlemlenen genel sağkalım iyileşmesi, hem skuamoz olmayan küçük hücreli dışı akciğer kanseri hastaları HR 0,62, %95 GA: 0,4, 0,96;medyan genel sağkalım 20,2'ye karşı 10,5 ay) hem de skuamoz küçük hücreli dışı akciğerkanseri hastalarını (HR 0,56, %95 GA: 0,23, 1,37; 15,3 aya karşı medyan genel sağkalımaulaşılamaması) içeren alt gruplarda tutarlı bir şekilde gösterilmiştir. 75 yaş ve üzeri hastalarave hiç sigara içmemiş hastalara ilişkin veriler, bu alt gruplarda sonuç çıkarmak için çok sınırlıdır. İkinci basamak küçük hücreli dışı akciğer kanseri OAK (GO28915): kemoterapi tedavisi almış lokal ileri veya metastatik KHDAK hastalarında yapılan randomize Faz III çalışmaPlatin içeren bir rejim uygulanırken veya sonrasında progresyon görülmüş, lokal ileri veya metastatik KHDAK olan hastalarda, atezolizumab ile dosetakselin etkililik ve güvenlilik 36karşılaştırılmasının yapıldığı Faz III, açık etiketli, çok merkezli, uluslararası, randomize bir çalışma olan GO28915 (OAK) yürütülmüştür. Bu çalışma, otoimmün hastalık öyküsü olan,aktif veya kortikosteroid-bağımlı beyin metastazı öyküsü olan, başlangıçtan önceki 28 güniçinde canlı, attenüe aşı olmuş, başlangıçtan önceki 4 hafta içinde sistemik immunostimulatörajan uygulanmış veya başlangıçtan önceki 2 hafta içinde sistemik immunosupresif tıbbi ürünkullanmış hastalar çalışmaya alınmamıştır. Tümör değerlendirmeleri ilk 36 hafta boyunca her6 haftada bir ve sonra her 9 haftada bir gerçekleştirilmiştir. Tümör örnekleri, tümör hücrelerinde(TC) ve tümör sızdıran bağışıklık hücrelerinde (IC) PD-L1 ifadesi için prospektif olarakdeğerlendirilmiştir. Toplamda 1225 hasta kayıt edilmiştir ve analiz planına göre randomize edilen ilk 850 hasta primer etki analizine dahil edilmiştir. Randomizasyon PD-L1 IC ekspresyon durumu, öncekikemoterapi rejimlerinin sayısı ve histolojiye göre tabakalandırılmıştır. Hastalar 1:1 oranındaatezolizumab veya dosetaksel almak üzere randomize edilmiştir. Atezolizumab üç haftada bir intravenöz infüzyon yoluyla 1200 mg sabit dozda uygulanmıştır. Doz azaltımına izin verilmemiştir. Hastalar, araştırmacı tarafından değerlendirilecek klinikfayda kaybına kadar tedavi edilmiştir. Dosetaksel, progresyona kadar her üç haftalık siklusun1. gününde intravenöz infüzyon yoluyla 75 mg/m2 uygulanmıştır. Tedavi edilen tüm hastalariçin, medyan tedavi süresi dosetaksel kolu için 2,1 ay ve atezolizumab kolu için 3,4 aydır. Primer analiz hasta popülasyonunun demografik ve başlangıç özellikleri tedavi kolları arasında genel olarak iyi değerlendirilmiştir. Medyan yaş 64 yaştır (aralık: 33-85) ve hastaların %61'ierkektir. Hastaların çoğu beyaz ırktandır (%70). Hastaların yaklaşık olarak üçte birinde non-skuamoz hastalık vardır (%74). %10'unda EGFR mutasyonu, %0,2'sinde ALK yenidendüzenlenmesi ve %10'unda başlangıçta santral sinir sistemi metastazları vardır. Hastaların çoğuhalen veya önceden tütün kullanıcısıdır (%82). Başlangıçta ECOG performans skoru 0 (%37)veya 1'dir (%63). Hastaların %75'i önceden yalnızca bir platin bazlı tedavi rejimi almıştır. Birincil etkililik sonlanım noktası, randomizasyon tarihinden herhangi bir nedenle ölüme kadar geçen süre olarak tanımlanan GS'dir (genel sağkalım). 21 aylık medyan sağkalım takip süresiniiçeren bu çalışmanın anahtar sonuçları Tablo 9'da özetlenmiştir. ITT popülasyonu (tedaviamaçlı) için Kaplan-Meier eğrileri Şekil 8'de sunulmuştur. Şekil 9, ITT ve PD-L1 altgruplarındaki GS sonuçlarını özetlemekte olup , TC ve IC'de % 1'in altında PD-L1 ekspresyonuolanları da içeren tüm alt gruplarda atezolizumab ile GS faydasını göstermektedir.
Etkililik sonlanım noktasıAtezolizumab Dosetaksel(n=425) (n=425)12 aylık GS*** 218 (%55)151 (%41) 18 aylık GS*** 157 (%40)98 (%27) İkincil Etkililik Sonlanım NoktalarıAraştırmacı tarafından değerlendirilen PS (RECIST v1.1)Olay sayısı (%) 380 (%89)375 (%88) Medyan PS süresi (ay) 2,84 %95 GA (2,6; 3)(3,3; 4,2) Sınıflandırılmış risk oranı (%95 GA) 0,95 (0,82; 1,1) Araştırmacı Tarafından Değerlendirilen OYO (RECIST v1.1)Yanıt verenlerin sayısı (%) 58 (%14)57 (%13) %95 GA (10,5;17,3)(10,3;17) Araştırmacı Tarafından Değerlendirilen YS (RECIST v1.1)n=58 n=57 Medyan (ay) 16,36,2 %95 GA (10;TE)(4,9; 7,6) GA=güven aralığı; YS=yanıt süresi; TE=Tahmin edilemiyor; OYO=objektif yanıt oranı; GS=genel sağkalım; PS=progresyonsuz sağkalım; RECIST-STCDK=Solid Tümörlerde CevapDeğerlendirme Kriterleri v1.1. *Primer analiz popülasyonu randomize edilen ilk 850 hastayı içerir |IC düzeyleri, tümör sızdıran bağışıklık hücrelerindeki PD-L1 ekspresyonuna, önceki kemoterapi rejimi sayısı ve histolojiye göre sınıflandırılmıştır. ** sınıflandırılmış log-sıra sıralamasına göre *** Kaplan-Meier hesaplamalarına göre Şekil 8: Primer analiz popülasyonunda genel sağkalım için Kaplan-Meier eğrisi (tüm gelenler) (OAK)
Risk altındaki hasta sayısıRisk oranı sınıflandırılmış bir Cox modeli üzerinden; p-değeri sınıflandırılmış bir log-rank testi üzerinden tahmin edilmiştir38
a TC ve IC > % liçin sınıflandırılmış HR. Diğer açıklayıcı altgruplar içi sınıflandırılmamış HRTC: Tümör hücreleri; IC: İmmün hücreleri; HR: Risk oranıHem non-skuamoz KHDAK hastalarında (atezolizumab and dosetaksel için sırasıyla; risk oranı [HR]:>5% GA: 0,6, 0,89; medyan GS< 15,6'ya karşı 11,2 ay) hem de skuamoz KHDAK hastalarında (atezolizumab and dosetaksel için sırasıyla HR: 0,73, 95% GA: 0,54, 0,98; medyan GS: 8,9 vs. 7,7 ay) dosetaksel ile karşılaştırılan GS değerlerinde gelişme görülmüştür.Gözlemlenen GS gelişimi, başlangıçta beyin metastazı olanlar (atezolizumab and dosetakseliçin sırasıyla; risk oranı [HR]: 0,54, 95% GA: 0,31, 0,94; medyan GS< 20,1 vs. 11,9 ay) ve hiçtütün tüketmemiş olanlar (atezolizumab and dosetaksel için sırasıyla; risk oranı [HR]: 0,71,95% GA: 0,47, 1,08; medyan GS< 16,3 vs. 12,6 ay) dahil olmak üzere altgruplar arasında tutarlıbir biçimde gösterilmiştir. Bununla birlikte, EGFR mutasyonlu hastalar dosetaksele karşılıkatezolizumab kullanımında gelişmiş GS göstermemişlerdir (atezolizumab ve dosetaksel içinsırasıyla; risk oranı [HR]: 1,24, 95% GA: 0,71, 2,18; medyan GS< 10,5 'e karşı 16,2 ay). EORTC QLQ-LC13 ile ölçüldüğünde, hasta bildirimli göğüs ağrısındaki kötüleşmeye kadar zamanın, dosetaksele karşılık atezolizumab için (HR: 0,71, 95% GA: 0,49, 1,05; iki kolda damedyana ulaşılmamıştır) uzadığı görülmüştür. EORTC QLQ-LC13 ile ölçülen diğer akciğerkanseri semptomları (örn. öksürük, dispne ve kol/omuz ağrısı) için kötüleşme süreleriatezolizumab ve dosetaksel için benzerdir. Bu sonuçlar çalışmanın açık etiketli dizaynınadayanarak dikkatle yorumlanmıştır. POPLAR (GO28753): kemoterapi tedavisi almış lokal ileri veya metastatik KHDAK hastalarında yapılan randomize Faz II çalışmaPD-L1 ekspresyonundan bağımsız olarak platin içeren rejim uygulanırken ya da sonrasında progresyon görülmüş, lokal ileri veya metastatik KHDAK olan hastalarda Faz II, çok merkezli,uluslararası, randomize, açık etiketli, kontrollü bir çalışma olan GO28753 (POPLAR) çalışmasıda yürütülmüştür. Birincil etkililik sonlanım noktası, genel sağkalımdır. Toplam 287 hasta 1:1oranında TECENTRIQ (klinik fayda kaybına kadar üç haftada bir intravenöz infüzyon yoluyla1200 mg) ya da dosetaksel (progresyona kadar her üç haftalık siklusun 1. (birinci) günündeintravenöz infüzyon yoluyla 75 mg/m2) almak üzere randomize edilmiştir. Randomizasyon,PD-L1 IC ekspresyon durumu, önceki kemoterapi rejimlerinin sayısı ve histolojiye göre 39tabakalandırılmıştır. Gözlemlenen toplam 200 ölüm ve 22 aylık medyan sağkalım takibi ile güncelleştirilmiş analiz; dosetaksel ile tedavi edilen hastalarda medyan sağkalım 9,7 ay ve atezolizumab ile tedavi edilenhastalarda medyan sağkalım 12,6 ay (HR: 0,69% 95 GA: 0,52, 0,92) olarak göstermiştir.Atezolizumab ve dosetaksel için Objektif Yanıt Oranı (OYO) sırasıyla %15,3'e karşı %14,7,medyan Yanıt Süresi (YS) ise 18,6 ay ve 7,2 aydır. Küçük hücreli akciğer kanseri: IMpower133 (GO30081): kemoterapi tedavisi almamış yaygın evre KHAK hastalarında karboplatin ve etoposidle kombinasyon halinde yapılan randomize Faz I/III çalışmaKemoterapi kullanmamış, yaygın evre KHAK hastalarında atezolizumabın karboplatin ve etoposid ile kombinasyon halinde kullanıldığında etkililiğini ve güvenliliğini değerlendirmekiçin Faz I/III, randomize, çok merkezli, çift-kör, plasebo kontrollü bir çalışma olan IMpower133yürütülmüştür. Aktif veya tedavi edilmemiş beyin metastazı olan, otoimmün hastalık öyküsü olan, randomizasyondan önceki 4 hafta içinde canlı, attenüe aşı olmuş, randomizasyondan önceki 1(bir) hafta içinde sistemik immunostimulatör ajan uygulanmış hastalar çalışmaya alınmamıştır. Tümör değerlendirmeleri 1. (birinci) siklusun 1. (birinci) gününü takiben ilk 48 hafta boyunca her 6 haftada bir ve sonra her 9 haftada bir gerçekleştirilmiştir. Hastalık progresyonundan sonratedavi edilen hastaların tümör değerlendirmeleri kullanımın bırakılmasına kadar her 6 haftadabir gerçekleştirilmiştir. Toplamda 403 hasta kayıt edilmiştir ve 1:1 oranında Tablo 10'daki tedavi rejimlerinden birini almak üzere randomize edilmiştir. Randomizasyon cinsiyet, ECOG performans skoru ve beyinmetastaz varlığına göre sınıflandırılmıştır.
uygulanmıştır. b Karboplatin ve etoposid, 4 (dört) siklusun tamamlanmasına kadar, veya progresif hastalığa veya kabul edilemez toksisiteye kadar, herhangi biri ilk defa olana kadar uygulanmıştır.c Etoposid her siklusun 1. (brinci), 2. (ikinci) ve 3. (üçüncü) gününde uygulanmıştır. Hasta popülasyonunun demografik ve başlangıç özellikleri tedavi kolları arasında genel olarak benzer değerlendirilmiştir. Hastaların %10'u 75 yaş ve üzeri olmakla beraber medyan yaş 64yıldır (aralık: 26-90 yaş). Hastaların çoğunluğu erkektir (%65), beyaz ırktandır (%80), %9'undabeyin metastazı vardır ve çoğu halen veya önceden tütün kullanıcısıdır (%97). Başlangıçta 40ECOG performans skoru 0 (%35) veya 1 (bir)'dir (%65). Primer analizde, hastaların medyan sağkalım takip süresi 13,9 aydır. Kontrol koluna kıyasla atezolizumab ile karboplatin ve etoposid kombinasyonu ile genel sağkalımda istatistiksel olarakanlamlı bir gelişme gözlendi (risk oranı (HR) 0,7 %95 GA: 0,54, 0,91; ortalama genelsağkalım : 12,3 aya 10,3 ay). Uzun dönem takipli araştırma genel sağkalım analizinde(ortalama: 22,9 ay), her iki koldaki ortalama genel sağkalım, başlangıç genel sağkalım araanalizine göre değişmemiştir. Araştırma analizi yanında başlangıç analiziden elde edilenProgresyonsuz Sağkalım (PS), Objektif Yanıt Oranı (OYO)ve Yanıt Süresi (YS) sonuçlarıTablo 11'de özetlenmiştir. Genel sağkalım ve progresyonsuz sağkalım sonuçlarını gösterenKaplan-Meier eğrileri Şekil 10'da ve 11'de sunulmuştur. Beyin metastazı olan hastalar içinveriler bu popülasyonla ilgili sonuç çıkarmak için çok sınırlıdır.
°S=progresyonsuz sağkalım; RECIST-STCDK=Solid Tümörlerde Cevap Değerlendirme Kriterleri v1.1.; GA=güven aralığı; OYO=objektif yanıt oranı; YS=objektif yanıt süresi;GS=genel sağkalım * Cinsiyet ve ECOG performans skoruna göre tabakalandırılmıştır * Araştırma GS final analizi klinik kesim tarihi 21 Ocak 2019**PS, OYO ve YS analizi klinik kesim tarihi 24 Nisan 2018*** Sadece tanımlayıcı amaçlar için A Onaylanan OYO ve YS araştırma sonlanım noktasıdır Şekil 10: Genel sağkalım için Kaplan-Meier eğrisi (IMpower133)
1009080
e s-SM>bD C3
S
(DÜ706050403020 10 0 202194 139186183171 160146131 114 97 38 74 63 58 55 49 45 39 36 33 26 20 12 8 6 3222242Şekil 11: Progresyonsuz sağkalım için Kaplan-Meier eğrisi (IMpower133)
Risk altındaki hasta sayısı TECENTRIQ+ karboplatin + etoposidPlasebo + karboplatin + etoposid Ürotelyal Kanser: IMvigor211 (GO29294): Daha önce kemoterapi ile tedavi edilmiş lokal ileri veya metastatik evre ürotelyal kanserli hastalar üzerinde yapılan randomize çalışmaPlatin içeren bir rejim sırasında veya sonrasında hastalık ilerlemesi görülen lokal ileri veya metastatik evre ürotelyal kanserli hastalarda kemoterapiye (araştırmacının seçimine bağlıolarak vinflunin, dosetaksel veya paklitaksel) kıyasla atezolizumabın etkililiğini vegüvenliliğini değerlendirmek için faz III, açık etiketli, çok merkezli, uluslararası, randomize birçalışma (IMvigor2n) yürütülmüştür. Otoimmün hastalık öyküsü olan, aktif veyakortikosteroid-bağımlı beyin metastazı olan, kayıttan önceki 28 gün içinde canlı, attenüe aşıuygulanmış ve kayıttan önceki 4 (dört) hafta içinde sistemik immunostimulatör ajan veya 2 (iki)hafta içinde sistemik immunosupresif tıbbi ürün uygulanmış hastalar bu çalışmadandışlanmıştır. Tümör değerlendirmeleri ilk 54 hafta boyunca her 9 (dokuz) haftada bir vesonrasında her 12 haftada bir gerçekleştirilmiştir. Tümör örnekleri, tümörü infiltre eden immünhücrelerde (IC) PD-L1 ekspresyonu için prospektif olarak değerlendirilmiş ve sonuçlar aşağıdaaçıklanan analizler için PD-L1 ekspresyon alt gruplarını tanımlamak için kullanılmıştır. Toplam 931 hasta kaydedilmiştir. Hastalar atezolizumab ya da kemoterapi almak üzere randomize edilmiştir (1 (bir):1(bir)). Randomizasyon kemoterapi (vinflunin ya da taksan),IC'de PD-L1 ekspresyon durumu (<%5 (beş)'e karşı >%5 (beş)), prognostik risk faktörlerininsayısı (0'a karşı 1(bir)-3 (üç)) ve karaciğer metastazına (evete karşı hayır) göresınıflandırılmıştır. Prognostik risk faktörleri arasında önceki kemoterapiden itibaren <3 (üç) aygeçmesi, > 0 ECOG performans durumu ve < 10 g/dL hemoglobin yer almıştır. Atezolizumab, 3 (üç) haftada bir intravenöz infüzyon yoluyla 1200 mg sabit dozda 43uygulanmıştır. Atezolizumab dozunun azaltımına izin verilmemiştir. Hastalar, araştırmacı tarafından değerlendirildiği şekilde klinik faydanın kaybedilmesine veya kabul edilemeztoksisite görülene kadar tedavi edilmiştir. Vinflunin, hastalığın ilerlemesine veya kabuledilemez toksisite görülene kadar her 3 (üç) haftalık döngünün 1. (birinci) gününde intravenözinfüzyon yoluyla 320 mg/m2 dozunda uygulanmıştır. Paklitaksel, hastalığın ilerlemesine veyakabul edilemez toksisite görülene kadar her 3 (üç) haftalık döngünün 1. (birinci) gününde 3 (üç)saat boyunca intravenöz infüzyon yoluyla 175 mg/m2 dozunda uygulanmıştır. Dosetaksel,hastalığın ilerlemesine veya kabul edilemez toksisite görülene kadar her 3 (üç) haftalıkdöngünün 1. (birinci) gününde intravenöz infüzyon yoluyla 75 mg/m2 dozunda uygulanmıştır.Tedavi edilen tüm hastalarda medyan tedavi süresi atezolizumab kolu için 2,8 ay, vinflunin vepaklitaksel kolları için 2,1 ay ve dosetaksel kolu için 1,6 ay olmuştur. Primer analiz popülasyonunun demografik ve başlangıç hastalık özellikleri tedavi kolları arasında dengeli olmuştur. Medyan yaş 67 (aralık: 31-88) olup hastaların %77,1'i erkektir.Hastaların çoğunluğu (%72,1) beyaz ırktan olup kemoterapi kolundaki hastaların %53,9'uvinflunin almış, başlangıçta hastaların %71,4'ünde en az bir kötü prognostik risk faktörüve %28,8'inde karaciğer metastazı mevcuttur. Başlangıç ECOG performans durumu 0 (%45,6)veya 1 (%54,4) olmuştur. Hastaların %71,1'inde primer tümör yeri mesanedir vehastaların %25,4'ünde üst sistem ürotelyal karsinomu mevcuttur. Hastaların %24,2'si daha öncesadece platin içeren adjuvan veya neoadjuvan tedavi almış ve bu hastalarda 12 ay içinde hastalıkilerlemesi görülmüştür. IMvigor211 için primer etkililik sonlanım noktası genel sağkalımdır (GS). Araştırmacı tarafından değerlendirilen Solid Tümörlerde Yanıt Değerlendirme Kriterleri (RECIST) v1.1'egöre değerlendirilen sekonder etkililik sonlanım noktaları objektif yanıt oranı (OYO),progresyonsuz sağkalım (PS) ve yanıt süresidir (YS). IC2/3, IC1/2/3ve ITT (tedavisi amaçlanan,yani tüm katılanlar) popülasyonlarında tedavi kolu ve kontrol kolu arasındaki GSkarşılaştırmaları, aşağıdaki şekilde %5'lik iki taraflı seviyede sınıflandırılmış bir log-sıra testinedayalı hiyerarşik, sabit sıralı bir prosedür kullanılarak test edilmiştir: aşama 1)IC2/3popülasyonu; aşama 2) IC1/2/3 popülasyonu; aşama 3) tüm katılanlar popülasyonu.Aşama 2 ve 3'ün her birine ait GS sonuçları, ancak önceki aşamaya ait sonuç istatistiksel olarakanlamlıysa, istatistiksel anlamlılık açısından resmi olarak test edilebilmiştir. Medyan sağkalım takibi 17 aydır. IMvigor2n çalışmasının primer analizi primer genel sağkalım sonlanım noktasını karşılamamıştır. Atezolizumab, daha önce tedavi edilmiş, lokalileri evre veya metastatik ürotelyal karsinomlu hastalarda kemoterapiye kıyasla istatistikselolarak anlamlı bir sağkalım faydası göstermemiştir. Önceden belirlenmiş hiyerarşik test sırasınagöre, 0,87'lik bir genel sağkalım risk oranı ile önce IC 2/3popülasyonu test edilmiştir (%95 GA:0,63, 1,21; atezolizumab ve kemoterapi için medyan genel sağkalım sırasıyla 11,1 aya karşı10,6 ay). Sınıflandırılmış log-sıra p değeri 0,41'dir ve bu nedenle sonuçlar bu popülasyondaistatistiksel olarak anlamlı kabul edilmemiştir. Sonuç olarak, IC 1/2/3 veya tüm katılanlarpopülasyonunda genel sağkalım için herhangi bir istatistiksel anlamlılık testigerçekleştirilememiş ve bu analizlerin sonuçları açıklayıcı olarak değerlendirilmiştir. Tümkatılanlar popülasyonundaki kilit sonuçlar Tablo 12'de özetlenmiştir. Tüm katılanlarpopülasyonunda Kaplan Meier genel sağkalım eğrisi Şekil 12'de sunulmaktadır. ITT popülasyonunda 34 aylık bir medyan sağkalım takip süresi ile açıklayıcı bir güncellenmiş sağkalım analizi gerçekleştirilmiştir. Medyan genel sağkalım, 0,82'lik bir risk oranı (%95 GA:0,71, 0,94) ile atezolizumab kolunda 8,6 ay (%95 GA: 7,8, 9,6) ve kemoterapi kolunda 8 ay(%95 GA: 7,2, 8,6) olmuştur. 12 aylık genel sağkalım oranları için primer analizde gözlemleneneğilim ile uyumlu olarak, atezolizumab kolundaki hastalarda, ITT popülasyonundaki 44kemoterapi koluna kıyasla sayısal olarak daha yüksek 24 ve 30 aylık genel sağkalım oranları gözlemlenmiştir. 24. ayda sağ olan hastaların yüzdesi (KM tahmini) kemoterapi kolunda %12,7ve atezolizumab kolunda %22,5; 30. ayda sağ olan hastaların yüzdesi (KM tahmini) isekemoterapi kolunda %9,8 ve atezolizumab kolunda %18,1 olmuştur.
GA=güven aralığı; YS=yanıt süresi; OYO=objektif yanıt oranı; GS=genel sağkalım; PS=progresyonsuz sağkalım; RECIST = Katı Tümörlerde Yanıt Değerlendirme Kriterleri v1.1.* Tüm katılanlar popülasyonunda GS analizi sınıflandırılmış log-sıra testine göregerçekleştirilmiş ve sonuç sadece açıklama amaçlı olarak sunulmuştur (p = 0,0378); öncedenbelirlenmiş analiz hiyerarşisine göre, tüm katılanlar popülasyonunda GS analizi için p değeriistatistiksel olarak anlamlı kabul edilememektedir. fKemoterapi (vinflunin ya da taksan), IC'de durum (< %5 (beş)'e karşı > %5(beş)), prognostik risk faktörlerinin sayısı (0'a karşı 1(bir)-3(üç)) ve karaciğer metastazına (evete karşı hayır) göresınıflandırıldı.** Kaplan-Meier tahminine göre. *** Yanıtlar atezolizumab kolunda yanıt verenlerin %63'ünde ve kemoterapi kolunda yanıt verenlerin %21'inde devamlıydı. 45
IMvigor210 (GO29293): Sisplatin tedavisi için uygun bulunmayan, daha önceden tedavi edilmemiş ürotelyal kanserli hastalar üzerinde yapılan tek kollu çalışmaLokal ileri veya metastatik evre ürotelyal kanserli (ürotelyal mesane kanseri olarak da bilinir) hastalar üzerinde faz II, çok merkezli, uluslararası, iki kohortlu, tek kollu bir klinik çalışma(IMvigor210) yürütülmüştür. Çalışmada toplam 438 hasta ve iki hasta kohortu vardır. Kohort 1'e, sisplatin bazlı kemoterapiye uygun bulunmayan veya uygun olmayan ya da platin içeren bir neoadjuvan veyaadjuvan kemoterapi rejimi ile uygulanan tedaviden en az 12 ay sonra hastalık ilerlemesi görülen,daha önce tedavi görmemiş, lokal ileri veya metastatik evre ürotelyal kanserli hastalar dahiledilmiştir. Kohort 2'ye ise, lokal ileri veya metastatik evre ürotelyal kanserli için en az bir platinbazlı kemoterapi rejimi alan veya platin içeren bir neoadjuvan veya adjuvan kemoterapi rejimiile uygulanan tedaviden sonraki 12 ay içinde hastalık ilerlemesi görülen hastalar dahil edilmiştir. Kohort 1'de, 119 hasta hastalığın ilerlemesine kadar intravenöz infüzyon yoluyla 3 (üç) haftada bir 1200 mg atezolizumab ile tedavi edilmiştir. Medyan yaş 73'tür. Hastaların çoğu (%81) erkekve çoğunluğu (%91) beyaz ırktan olmuştur. Kohort 1'e, ECOG performans durumu 0 olan 45 hasta (%38), ECOG performans durumu 1 olan 50 hasta (%42) ve ECOG performans durumu 2 olan 24 hasta (%20), Bajorin risk faktörü(ECOG performans durumu > 2 ve viseral metastaz) olmayan 35 hasta (%29), bir Bajorin riskfaktörü olan 66 hasta (%56) ve iki Bajorin risk faktörü olan 18 hasta (%15), böbrek işlevbozukluğu olan (glomerüler filtrasyon hızı [GFR] < 60 mL/dk) 84 hasta (%71) ve karaciğermetastazı olan 25 hasta (%21) dahil edilmiştir. 46Kohort 1'in primer etkililik sonlanım noktası, bağımsız bir değerlendirme tesisi (IRF) tarafından RECIST v1.1 kullanılarak değerlendirilen objektif yanıt oranı (OYO) olarakdoğrulanmıştır. Primer analiz, tüm hastalar en az 24 hafta süreyle takip edildiğinde gerçekleştirilmiştir. Tüm katılanlarda medyan tedavi süresi 15 hafta ve medyan sağkalım takip süresi 8,5 ay olmuştur.RECIST v1.1'e göre IRF tarafından değerlendirilen klinik olarak anlamlı OYO'lar gösterilmiş;ancak önceden belirlenmiş %10'luk bir geçmiş kontrol yanıt oranı ile karşılaştırıldığında,anlamlı faydaya ulaşılamamıştır. IRF RECIST v1.1'e göre doğrulanmış OYO'lar PD-L1ekspresyonu >%5 olan hastalarda %21,9 (%95 GA: 9,3, 40), PD-L1 ekspresyonu >%1 olanhastalarda %18,8 (%95 GA: 10,9, 29) ve tüm katılanlarda %19,3 (%95 GA: 12,7, 27,6)olmuştur. Herhangi bir PD-L1 ekspresyon alt grubunda veya tüm katılanlarda medyan yanıtsüresine (YS) ulaşılmamıştır. Yaklaşık %40'lık bir olay-hasta oranı ile genel sağkalım matürdeğildir. Tüm hasta alt gruplarında (PD-L1 ekspresyonu > %5 ve >%1) ve tüm katılanlardamedyan genel sağkalım 10,6 ay olmuştur. Kohort 1 için gerçekleştirilen ve 17,2 aylık bir medyan sağkalım takip süresi ile güncellenmiş olan analiz Tablo 13'te özetlenmektedir. Herhangi bir PD-L1 ekspresyon alt grubunda veyatüm katılanlarda medyan YS'ye ulaşılmamıştır. Tablo 13: Güncellenmiş etkililik özeti (IMvigor210 Kohort 1)
TE) 1 yıllık GS oranı (%) %52,4%54,8%57,2 GA = güven aralığı; YS = yanıt süresi; IC = tümörü infiltre eden immün hücreler; IRF = bağımsız değerlendirme tesisi; TE = tahmin edilemiyor; OYO = objektif yanıt oranı;GS = genel sağkalım; PS = progresyonsuz sağkalım; RECIST = Katı Tümörlerde YanıtDeğerlendirme Kriterleri v1.1. Kohort 2'de, ko-primer etkililik sonlanım noktaları, bir IRF tarafından RECIST v1.1 kullanılarak değerlendirilen OYO ve Modifiye RECIST (mRECIST) kriterlerine görearaştırmacı tarafından değerlendirilen OYO olarak doğrulanmıştur. 310 hasta, klinik faydanınkaybedilmesine kadar 3 (üç) haftada bir intravenöz infüzyon yoluyla 1200 mg atezolizumab iletedavi edilmiştir. Kohort 2'nin primer analizi, tüm hastalar en az 24 hafta süreyle takipedildiğinde gerçekleştirilmiştir. Çalışma Kohort 2'deki ko-primer sonlanım noktalarınıkarşılamış; bu da önceden belirlenmiş %10'luk bir geçmiş kontrol yanıt oranı ilekarşılaştırıldığında, IRF tarafından RECIST v1.1 kullanılarak değerlendirilen ve araştırmacıtarafından mRECIST kullanılarak değerlendirilen istatistiksel olarak anlamlı OYO'largöstermiştir. Kohort 2'de 21,1 aylık bir medyan sağkalım takip süresi ile de analiz gerçekleştirilmiştir. IRF RECIST v1.1'e göre doğrulanmış OYO'lar, PD-L1 ekspresyonu >%5 olan hastalarda %28(%95 GA: 19,5, 37,9), PD-L1 ekspresyonu >%1 olan hastalarda %19,3 (%95 GA: 14,2, 25,4)ve tüm katılanlarda %15,8 (%95 GA: 11,9, 20,4) olmuştur. Araştırmacı tarafındandeğerlendirilen mRECIST'e göre doğrulanmış OYO ise PD-L1 ekspresyonu >%5 olanhastalarda %29 (%95 GA: 20,4, 38,9), PD-L1 ekspresyonu >%1 olan hastalarda %23,7 (%95GA: 18,1, 30,1) ve tüm katılanlarda %19,7 (%95 GA: 15,4, 24,6) olmuştur. Tüm katılanlarpopülasyonunda IRF RECIST v1.1'e göre tam yanıt oranı %6,1 (%95 GA: 3,7, 9,4) olmuştur.Kohort 2'de, herhangi bir PD-L1 ekspresyon alt grubunda veya tüm katılanlarda medyan YS'yeulaşılmamış; ancak PD-L1 ekspresyonu <%1 olan hastalarda ulaşılmıştır (13,3 ay; %95 GA 4,2,TE). 12. ayda GS oranı tüm katılanlarda %37 olmuştur. IMvigor130 (WO30070): Tedavi edilmemiş lokal ileri evre veya metastatik ürotelyalkarsinomlu hastalarda monoterapi olarak ve platin bazlı kemoterapi ile kombinasyonhalinde atezolizumabın değerlendirildiği faz III çok merkezli, randomize, plasebo kontrollüçalışma.Sağkalım verilerinin ilk aşamada gözden geçirilmesini takiben bağımsız bir Veri İzleme Kurulu (İDMC) tarafından verilen tavsiyeye dayanılarak, tümörlerinde PD-L1 ekspresyonu düşük olan(PD-L1 ile boyanmış, tümörü infiltre eden immün hücrelerin [IC], tümör alanının <%5'inikaplaması) hastaların atezolizumab monoterapisi koluna eklenmesi, bu alt grubun genelsağkalımında azalma gözlemlendikten sonra durdurulmuştur. İDMC, halihazırda monoterapikoluna randomize edilmiş ve monoterapi kolunda tedavi görmekte olan hastalar için herhangibir tedavi değişikliği tavsiye etmemiştir. Başka bir değişiklik tavsiye edilmemiştir. Hepatoselüler kanser: IMbrave150 (YO40245): Bevacizumab ile kombinasyon halinde, daha önce sistemik tedavialmamış rezeke edilemeyen hepatoselüler kanserli hastalarda randomize faz III çalışmaDaha önce sistemik tedavi görmemiş lokal ileri veya metastatik evre ve/veya rezeke edilemeyen hepatoselüler kanserli hastalarda bevacizumab ile kombinasyon halinde atezolizumabın 48etkililiği ve güvenliliğini değerlendirmek üzere, faz III, randomize, çok merkezli, uluslararası, açık etiketli bir çalışma olan IMbrave150 yürütülmüştür. Toplamda 501 hasta (2:1) oranında yaintravenöz infüzyon ile her 3 haftada bir uygulanan atezolizumab (1200 mg) ve 15 mg/kgbevacizumaba ya da günde iki kez oral yolla uygulanan sorafenib 400 mg'a randomizeedilmiştir. Randomizasyon coğrafi bölge (geri kalan dünya ülkelerine karşı Japonya hariçAsya), makrovasküler invazyon ve/veya ekstrahepatik yayılma (yoka karşı var), başlangıç a-fetoprotein (AFP) (>400 ng/mL'ye karşı <400 ng/mL) ve ECOG performans durumuna (1'ekarşı 0) göre sınıflandırılmıştır. Her iki koldaki hastalar klinik fayda kaybına veya kabuledilemez toksisiteye kadar tedavi almıştır. Hastalar atezolizumab veya bevacizumabı bırakıp(örn. advers olaylardan dolayı) klinik fayda kaybı veya tekli ajan ile ilişkili kabul edilemeztoksisiteye kadar tekli ajan tedavisine devam edebilmiştir. Çalışmaya hastalığı cerrahi ve/veya bölgesel tedavilere uygun olmayan veya cerrahi ve/veya bölgesel tedavilerden sonra ilerleyen Child-Pugh A, ECOG 0/1'e sahip ve daha önce sistemiktedavi almamış yetişkinler kaydedeilmiştir. Kanama (ölümcül olaylar dahil) bevacizumab ilebilinen bir advers reaksiyondur ve üst gastrointestinal kanama HSK'li hastalarda yaygın vehayati risk taşıyan bir komplikasyondur. Bu nedenle, hastaların tedaviden önceki 6 ay içindevaris varlığı açısından değerlendirilmesi zorunlu tutulmuş ve tedaviden önceki 6 ay içindevarise bağlı kanama geçirmişlerse, kanama veya yüksek kanama riski olan tedavi edilmemiş yada yeterince tedavi edilmemiş varisleri varsa çalışmadan hariç tutulmuştur. Aktif hepatit B'lihastalar için çalışma tedavisine başlamadan önce 28 gün içinde HBV DNA< 500 IU/mL veçalışmaya dahil olmadan önce ve çalışma boyunca minimum 14 günlük standart anti-HBVtedavisi gerekli olmuştur. Hastalar ayrıca, orta şiddette veya şiddetli asitleri; hepatik ensefalopati öyküleri, hepatik ensefalopati öyküsü, bilinen fibrolameller HSKsi, sarkomatoid HSK'si, mikstkolanjiokarsinomu ve HSK'si, HBV ve HCV'nin aktif koenfeksiyonu, otoimmün hastalıköyküleri varsa, randomizasyondan önceki 4 (dört) hafta içinde canlı, attenüe aşı uygulanmışsa;randomizasyondan önceki 4 (dört) hafta içinde sistemik immunostimulatör ajan veya 2 (iki)hafta içinde immunsupresif ilaçlar uygulanmışsa; tedavi edilmemiş veya kortikosteroidebağımlı beyin metastazları varsa da hariç tutulmuştur. Tümör değerlendirmeleri 1. (birinci)Döngü, 1. (birinci) Günden sonraki ilk 54 hafta boyunca her 6 (altı) haftada bir, ardından her 9(dokuz) haftada bir yürütülmüştür. Çalışma popülasyonunun demografik ve başlangıç hastalık özellikleri tedavi kolları arasında iyi dengelenmiştir. Medyan yaş 65 olup (aralık: 26 ila 88), hastaların %83'ü erkektir. Hastalarınçoğu Asyalı (%57) ve beyazdır (%35). 0%40'ı Asya'dan (Japonya hariç), %60'ı geri kalan dünyaülkelerindendir. Hastaların yaklaşık %75'i makrovasküler invazyon ve/veya esktrahepatikyayılma ile başvururken, %37'sinde başlangıç AFP değeri >400 ng/mL'dir. Başlangıç ECOGperformans durumu 0 (%62) veya 1 (bir)'dir (%38). HSK gelişimi açısından birincil riskfaktörleri hastaların %48'inde Hepatit B virüsü enfeksiyonu, hastaların %22'sinde Hepatit Cvirüsü enfeksiyonu ve hastaların %31'inde viral olmayan hastalıktır. HSK, hastaların%82'sinde Barselona Kliniği Karaciğer Kanseri (BCLC) evre C, hastaların %16'sında evre Bve hastaların %3 (üç)'ünde evre A olarak sınıflandırılmıştır. Eş-primer etkililik sonlanım noktaları RECIST vl.l'e göre GS (genel sağkalım) ve bağımsız değerlendirme merkezi (IRF) tarafından değerlendirilen PS'dir (progresyonsuz sağkalım).Birincil analiz zamanında hastalar 8,6 aylık medyan sağkalım takibi süresine sahiptir. Verilersorafenibe kıyasla atezolizumab + bevacizumab ile RECIST vl.l'e göre IRF tarafındandeğerlendirildiği üzere PS ve GS'de istatistiksel anlamlı bir iyileşme göstermiştir. İstatistikselanlamlı bir iyileşme, ayrıca RECIST vl.l'e ve HSK değiştirilmiş RECIST'e (mRECIST) göre 49IRF tarafından doğrulanmış objektif yanıt oranında da (OYO) gözlenmiştir. Primer analize ait kritik etkililik bulguları Tablo 14'te özetlenmektedir. 15,6 aylık ortanca sağkalım takip süresi ile tanımlayıcı bir güncellenmiş etkililik analizi yürütülmüştür. Medyan GS, atezolizumab + bevacizumab kolunda 19,2 ay (%95 GA: 17, 23,7)iken sorafenib kolunda 13,4 ay (%95 GA: 11,4, 16,9) olup risk oranı 0,66'dır (%95 GA: 0,52,0,85). RECIST v1.1 doğrultusunda IRF değerlendirmesine göre medyan progresyonsuzsağkalım, atezolizumab + bevacizumab kolunda 6,9 ay (%95 GA: 5,8, 8,6) iken sorafenibkolunda 4,3 ay (%95 GA: 4, 5,6) olup risk oranı değeri 0,65'tir (%95 GA: 0,53, 0,81). RECIST v1.1'e göre IRF ile değerlendirilen genel yanıt oranı, atezolizumab + bevacizumab kolunda %29,8 (%95 GA: 24,8, 35) iken sorafenib kolunda %11,3 (%95 GA: 6,9, 17,3)olmuştur. Doğrulanmış yanıt verenlerde RECIST v1.1 doğrultusunda IRF değerlendirmesinegöre ortanca yanıt süresi (YS), atezolizumab + bevacizumab kolunda 18,1 aya (%95 GA: 14,6,tahmin edilemiyor (TE)) kıyasla sorafenib kolunda 14,9 ay bulunmuştur (%95 GA: 4,9, 17). Genel sağkalım (güncellenmiş analiz) ve progresyonsuz sağkalım (primer analiz) için Kaplan-Meier eğrileri Şekil 13 ve 14'te sunulmaktadır.
51Şekil 13: ITT popülasyonunda genel sağkalım için Kaplan-Meier eğrisi (IMbrave150 güncellenmiş analiz)
Şekil 14: ITT popülasyonunda RECIST v1.1'e göre IRF-PS için Kaplan-Meier eğrisi (IMbrave150 primer analiz)
Pediyatrik popülasyon:Atezolizumabın güvenlilik ve farmakokinetiğini değerlendirmek için relaps veya progresif solid tümörlü ve Hodgkin ile non-Hodgkin lenfomalı pediyatrik (<18 yaş, n= 69) ve gençerişkin (18-30 yaş, n=18) hastalarda bir erken faz, çok merkezli, açık etiketli çalışma yapılmıştır. 52Hastalar 3 haftada 1 kez 15 mg/kg vücut ağırlığı intravenöz atezolizumab ile tedavi edilmiştir (bkz. Bölüm 5.2). Geriyatrik hastalarda etkililik:Atezolizumab monoterapisi alan > 65 yaş hastalar ve daha genç hastalar arasında etkililikte genel bir farklılık gözlenmemiştir. Impower150, IMpower133 ve rMpower110 çalışmalarından elde edilen veriler, 75 yaş ve üzeri hasta grubu hakkında değerlendirme yapılması için çok sınırlıdır. 5.2. Farmakokinetik özelliklerGenel özelliklerAtezolizumaba maruziyet 1 (bir) mg/kg vücut ağırlığı - 20 mg/kg vücut ağırlığı doz aralığında 3 (üç) haftada bir uygulanan sabit doz 1200 mg doz ile orantılı olarak artmıştır. 472 hastayıiçeren bir popülasyon analizi, aşağıdaki doz aralığı için atezolizumab farmakokinetiğiniaçıklamıştır: Birinci derece eliminasyonla bir doğrusal iki bölmeli dağılım modeli ile 1 - 20mg/kg vücut ağırlığı. Bir popülasyon farmakokinetik analizi, 6 (altı)-9 (dokuz) hafta tekrarlıdozlamadan sonra kararlı durumun elde edildiğini öne sürmektedir. Eğri altındaki alanda,maksimum konsantrasyon ve en düşük konsantrasyonda sistemik birikim sırasıyla 1,91; 1,46ve 2,75 kat olmuştur. Emilim:Atezolizumab intravenöz infüzyon şeklinde uygulanır. Diğer uygulama yollarıyla yapılan çalışmalar olmamıştır. Dağılım:Popülasyon farmakokinetik analizi, bir hastada merkezi kompartman dağılım hacminin 3,28 L ve kararlı durumunda hacmin 6,91 L olduğunu göstermektedir. Biyotransformasyon:Atezolizumabın metabolizması doğrudan araştırılmamıştır. Antikor klerensi esas olarak katabolizmayla gerçekleşir. Eliminasyon:Popülasyon farmakokinetik analizi, atezolizumabın klerensinin 0,2 L/gün ve tipik terminal eliminasyon yarı ömrünün 27 gün olduğunu göstermektedir. Özel popülasyonlara ilişkin ek bilgiler:Popülasyon farmakokinetiği ve maruziyet-yanıt analizlerine göre aşağıdaki faktörlerin atezolizumabın farmakokinetiği üzerinde bir etkisi yoktur: yaş, (21-89 yaş), bölge, etnik köken,böbrek bozukluğu, hafif karaciğer bozukluğu, PD-L1 ekspresyonu düzeyi veya ECOG durumu.Vücut ağırlığı, cinsiyet, pozitif ADA durumu, albümin seviyeleri ve tümör yükününatezolizumab farmakokinetiği üzerindeki etkisi istatistiki olarak anlamlı ancak klinik olarak 53anlamlı değildir. Doz ayarlanması önerilmemektedir. Pediyatrik popülasyon:Pediyatrik (<18 yaş, n= 69) ve genç erişkin (18-30 yaş, n=18) hastalarda yapılan bir erken faz, çok merkezli, açık etiketli çalışmadan elde edilen farmakokinetik sonuçlar, atezolizumabklerensi ve dağılım hacminin, normal vücut ağırlığında her 3 (üç) hafta 15 mg/kg vücut ağırlığıalan pediyatrik hastalar ve 1200 mg atezolizumab alan genç erişkin hastalar arasındakarşılaştırılabilir olduğunu göstermiştir. Maruziyetin ise, vücut ağırlığı düştükçe pediyatrikhastalarda arttığı gözlenmiştir. Bu değişiklikler, atezolizumab konsantrasyonunun terapötikhedef maruziyetinin altına düşmesi ile bağlantılı değildir. 2 yaş altındaki çocuklar için verilersınırlıdır, bu nedenle kesin sonuçlara varılamamaktadır. Geriyatrik popülasyon:Yaşlı hastalarda TECENTRIQ için özel bir çalışma yapılmamıştır. Yaşın atezolizumabın farmakokinetiği üzerindeki etkisi bir popülasyon farmakokinetik analizinde değerlendirilmiştir.Yaş, 21-89 yaş aralığındaki (n=472) ve medyan 62 yaşındaki hastalar temel alındığında,atezolizumabın farmakokinetiğini etkileyen önemli bir kovaryant olarak tanımlanmamıştır. <65yaşındaki (n=274), 65-75 yaşındaki (n=152) ve >75 yaşındaki (n=46) hastalardaatezolizumabın farmakokinetiğinde klinik açıdan anlamlı bir fark gözlenmemiştir (bkz. Bölüm4.2). Böbrek yetmezliği:Böbrek bozukluğu olan hastalarda TECENTRIQ için özel bir çalışma yapılmamıştır. Popülasyon farmakokinetik analizinde, böbrek fonksiyonu normal (90 mL/dk/1,73 m2 veyaüzeri eGFR; n=140) olan hastalarla karşılaştırıldığında, hafif (60 - 89 mL/dk/1,73 m2 eGFR;n=208) veya orta şiddetli (30 - 59 mL/dk/1,73 m2 eGFR; n=116) böbrek bozukluğu olanhastalarda atezolizumabın klerensinde klinik açıdan önemli farklar bulunmamıştır. Yalnızcabirkaç hastada şiddetli böbrek bozukluğu vardır (eGFR 15 - 29 mL/dk/1,73 m2; n=8) (bkz.Bölüm 4.2). Şiddetli böbrek yetmezliğinin atezolizumabın farmakokinetiği üzerindeki etkisibilinmemektedir. Karaciğer yetmezliği:Karaciğer bozukluğu olan hastalarda TECENTRIQ için özel bir çalışma yapılmamıştır. Popülasyon farmakokinetik analizinde hafif karaciğer bozukluğu (bilirubin <NÜS veAST>NÜS veya bilirubin <1 - 1,5 x NÜS ve herhangi bir AST) veya orta düzeyde karaciğerbozukluğu (bilirubin <1,5 - 3 x NÜS ve herhangi bir AST) olan hastalarda normal karaciğerfonksiyonu (bilirubin ve AST <NÜS) olan hastalara kıyasla atezolizumabın klerensibakımından klinik olarak önemli farklar bulunmamıştır. Şiddetli karaciğer bozukluğu olanhastalara ilişkin veri mevcut değildir (bilirubin > 3 x NÜS ve herhangi bir AST). Karaciğerbozukluğu, Ulusal Kanser Enstitüsü (NCI) karaciğer fonksiyon bozukluğu kriterlerine göretanımlanmıştır (bkz. Bölüm 4.2). Şiddetli karaciğer yetmezliğinin (bilirubin > 3 x NÜS veherhangi bir AST) atezolizumabın farmakokinetiği üzerindeki etkisi bilinmemektedir. 545.3. Klinik öncesi güvenlilik verileriKarsinojenisite:TECENTRIQ'in karsinojenik potansiyelini belirlemek için karsinojenisite çalışması yapılmamıştır. Mutajenisite:TECENTRIQ'in mutajenik potansiyelini belirlemek için mutajenisite çalışması yapılmamıştır. Bununla birlikte, monoklonal antikorların DNA veya kromozomları değiştirmesibeklenmemektedir. Fertilite:TECENTRIQ ile herhangi bir fertilite çalışması yapılmamıştır. Bununla birlikte, kronik toksisite çalışmasına sinomolgus maymunlarında erkek ve dişi üreme organlarınındeğerlendirilmesi dahil edilmiştir. Atezolizumabın dişi maymunlara tahmini EAA'dauygulanması (önerilen dozu alan hastalardaki EAA'nın yaklaşık 6 katı), geri dönüşümlü olarakdüzensiz adet döngüsü paternine ve yumurtalıklarda yeni oluşturulmuş korpus lutea eksikliğineneden olmuştur. Erkek üreme organları üzerinde herhangi bir etki olmamıştır. Teratojenisite:TECENTRIQ ile hayvanlarda üreme veya teratojenisite çalışmaları yapılmamıştır. Hayvan çalışmaları, PD-L1/PD-1 yolağının inhibisyonunun büyümekte olan fetüsün reddi riskindeimmünite ile ilişkili artışa ve böylelikle fetal ölüme yol açabileceğini ortaya koymuştur.TECENTRIQ uygulamasının gebelik üzerinde advers bir reaksiyona yol açması beklenir veembriyo-fetal letalitesi de dahil olmak üzere insan fetüsü için bir risk oluşturur. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiL-histidin Glasiyal asetik asitSükrozPolisorbat 20Enjeksiyonluk su 6.2. GeçimsizliklerGeçimsizlik çalışması yapılmadığından, bu tıbbi ürün, Bölüm 6.6'da sözü edilenler dışında başka tıbbi ürünlerle karıştırılmamalıdır. 6.3. Raf ömrüAçılmamış flakon: 36 ay Seyreltilmiş çözelti: Hazırlama zamanından sonra ortam sıcaklığında (<30°C) 24 saat ve 2-8°C'de 30 güne kadar kullanımdaki kimyasal ve fiziksel stabilite gösterilmiştir. 55Mikrobiyolojik açıdan, hazırlanan infüzyonluk çözelti hemen kullanılmalıdır. Hemen kullanılmazsa, kullanım sırasındaki saklama süreleri ve kullanım öncesi koşullar kullanıcınınsorumluluğundadır ve normalde, seyreltme kontrollü ve valide aseptik koşullarda yapılmadığısürece, 2-8°C'de 24 saatten fazla veya ortam sıcaklığında (<25°C) 8 saatten fazla olmamalıdır. 6.4. Saklamaya yönelik özel tedbirlerBuzdolabında saklayınız (2°C-8°C). Işıktan korumak için flakonu karton kutusunda saklayınız. Dondurmayınız. Çalkalamayınız. Tıbbi ürünün seyreltme sonrasında saklama koşulları için bkz. Bölüm 6.3. 6.5. Ambalajın niteliği ve içeriği20 mL infüzyonluk konsantre çözelti içeren tapalı (butil kauçuk) flakon (Tip I cam). Bir flakonluk paket. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerTECENTRIQ herhangi bir antimikrobiyal koruyucu veya bakteriyostatik ajan içermemektedir ve hazırlanan çözeltinin steril olmasını sağlamak için bir sağlık mesleği mensubu tarafındanaseptik teknik ile hazırlanmalıdır. Aseptik hazırlama, taşıma ve saklama İnfüzyonun hazırlanması sırasında aseptik temas sağlanmalıdır. Hazırlama işlemi: Eğitilmiş personel tarafından aseptik koşullar altında, özellikle de parenteral ürünlerinaseptik hazırlanmasına ilişkin iyi uygulama kuralları doğrultusunda yapılmalıdır. İntravenöz ajanların güvenli teması için standart önlemleri kullanarak laminer havaakımı veya biyolojik güvenlik kabininde hazırlanmalıdır. Aseptik koşulların idamesini sağlamak için hazırlanan intravenöz infüzyon çözeltisineuygun saklama işlemi ile devam etmelidir. Çalkalamayınız. Seyreltme talimatları:20 mL TECENTRIQ konsantresi flakondan çekilmeli ve içinde sodyum klorür 9 mg/mL (% 0,9) enjeksiyonluk çözelti bulunan bir PVC, polietilen (PE) veya poliolefin infüzyon torbasıiçine seyreltilmelidir. Seyreltmeden sonra çözeltinin final konsantrasyonu 3,2 ila 16,8 mg/mLolmalıdır. Torba, köpük oluşumuna izin vermeden çözeltiyi karıştırmak için yavaşça alt üst edilmelidir. İnfüzyon hazırlandığında hemen uygulanmalıdır (bkz. Bölüm 6.3). Parenteral tıbbi ürünler uygulanmadan önce partiküller ve renk değişimi açısından çıplak gözle 56incelenmelidir. Partiküller veya renk değişimi gözlenirse çözelti kullanılmamalıdır. TECENTRIQ ile ürüne temas eden polivinil klorür (PVC), polietilen (PE) veya poliolefin (PO) yüzeyleri olan intravenöz torbalar arasında geçimsizlik gözlenmemiştir. İlave olarak,polietersülfon veya polisülfon içeren düz eksenli filtre membranları ve infüzyon setleri ile PVC,PE, polibutadien veya polieterüretan içeren diğer infüzyon yardımcıları ile de geçimsizlikgözlenmemiştir. Düz eksenli filtre membranlarının kullanılması seçime bağlıdır. Diğer ilaçları aynı infüzyon hattında birlikte uygulamayınız. Kullanılmamış/son kullanma tarihi geçmiş ilaçların imhası:Tıbbi ürünlerinin çevreye salınması en aza indirilmelidir. İlaçlar, atık suyla birlikte atılmamalıdır ve evsel atıklarla imhasından kaçınılmalıdır. Kullanılmamış olan ürünler ya da atık materyaller, Tıbbi Atıkların Kontrolü Yönetmeliği'' ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİRoche Müstahzarları Sanayi Anonim Şirketi Uniq İstanbul, Ayazağa Cad. No:4, D/101Maslak 34396, Sarıyer- İstanbul 8. RUHSAT NUMARASI(LARI)2018/316 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 14.06.2018 Ruhsat yenileme tarihi: 25.04.2023 10. KÜB'ÜN YENİLENME TARİHİXX.XX.XXXX 57 |
İlaç BilgileriTecentriq 1200 Mg/20 Ml İnfüzyonluk Çözelti Hazırlamak İçin KonsantreEtken Maddesi: Atezolizumab Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
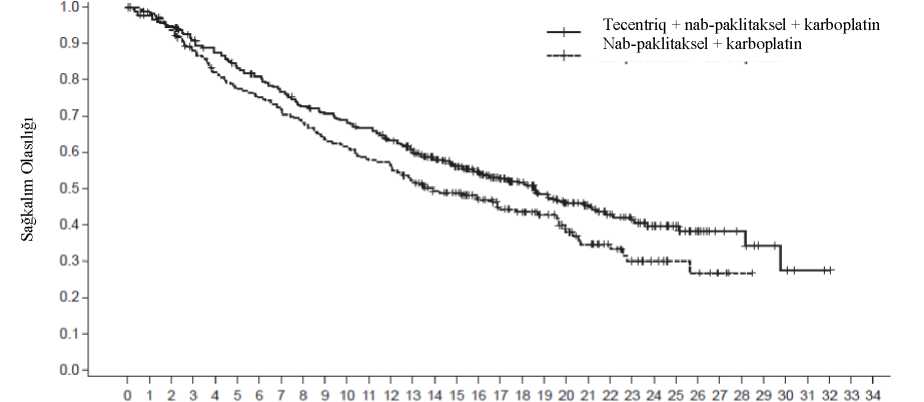
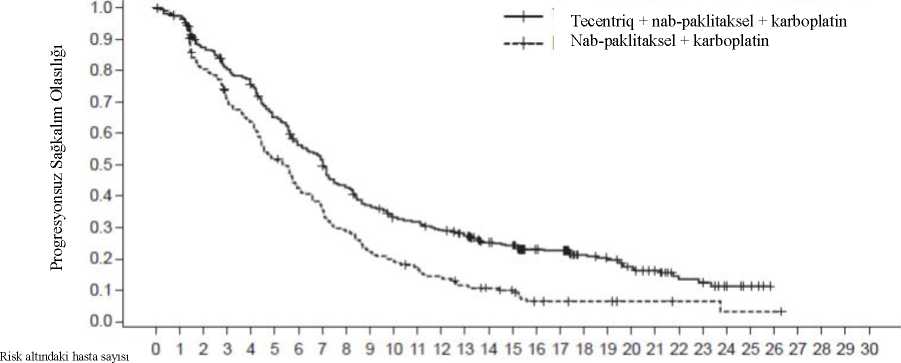
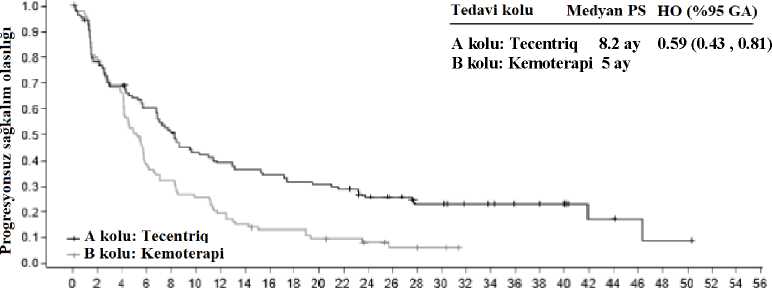
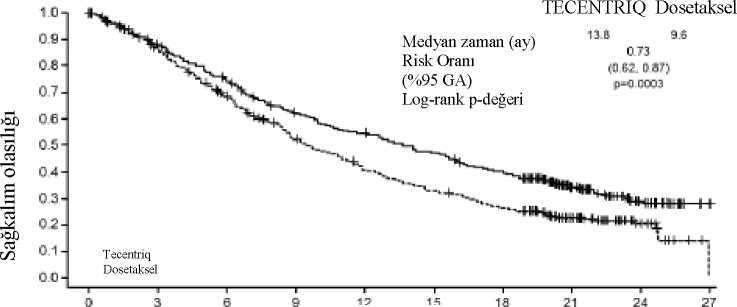
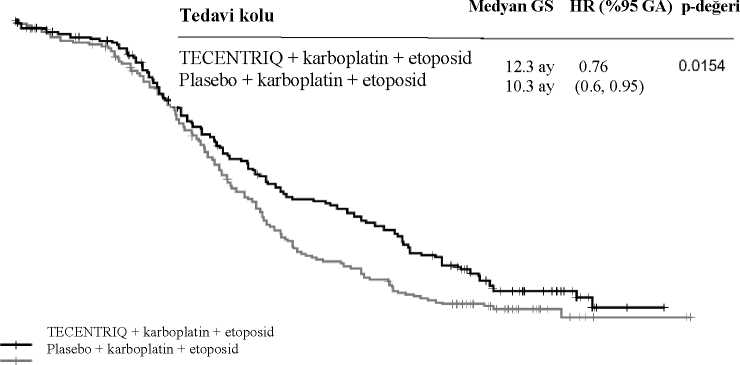
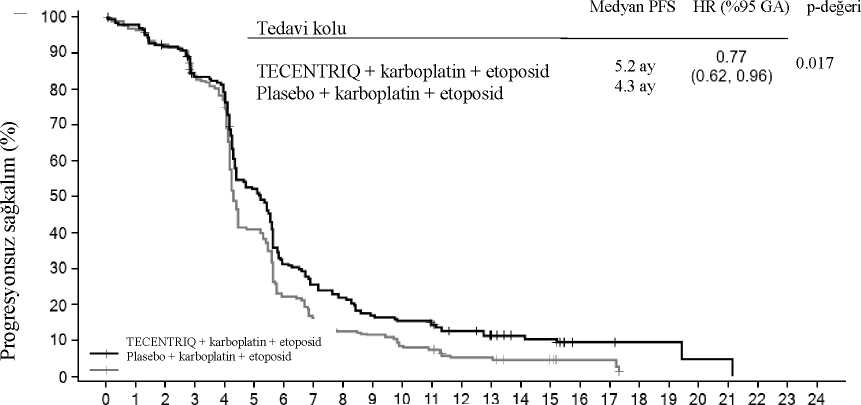
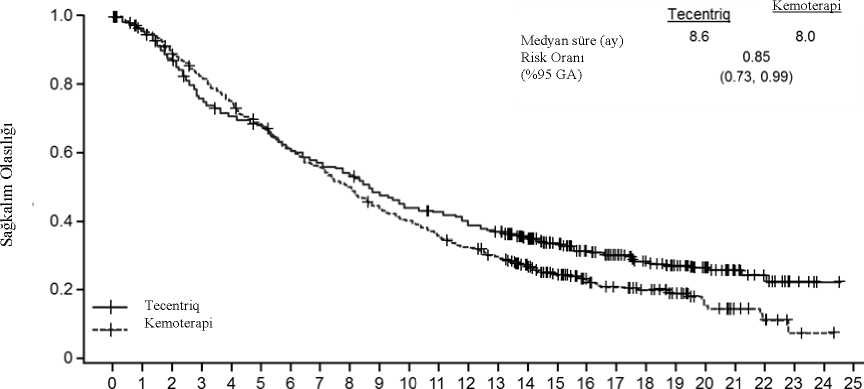
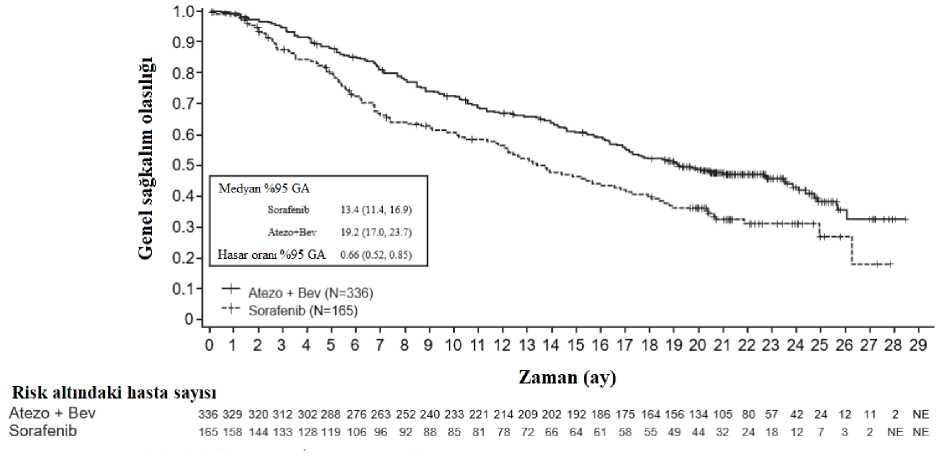
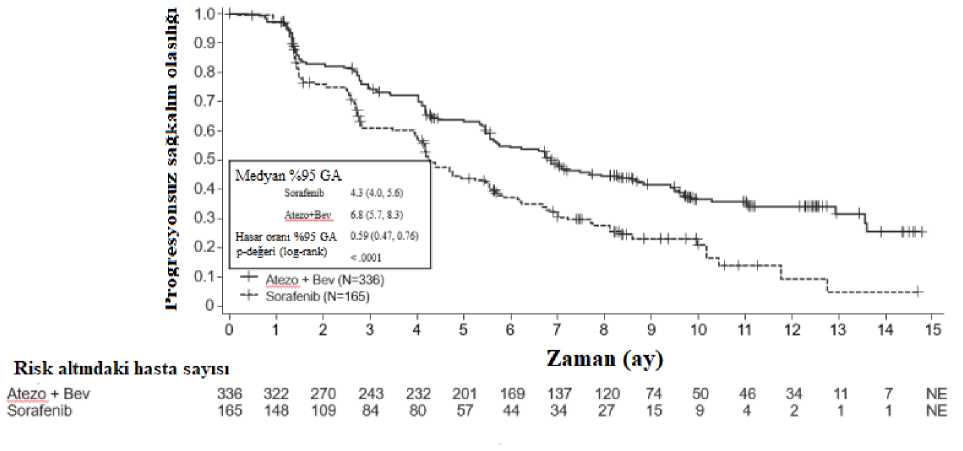
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.