Elocta 1000 I.u Enjeksiyonluk Çözelti Hazırlamak İçin Toz ve Çözücü Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ'VBU ILAÇ EK IZLEMEYE TABIDIR. BU ÜÇGEN YENI GÜVENLILIK BILGISININ HıZLı OLARAK BELIRLENMESINI SAĞLAYACAKTıR. SAĞLıK MESLEĞI MENSUPLARıNıN ŞÜPHELI ADVERS REAKSIYONLARı TÜFAM'ABILDIRMELERI BEKLENMEKTEDIR. BAKıNıZ BÖLÜM 4.8 ADVERS REAKSIYONLAR NASıL RAPORLANıR?1. BEŞERİ TIBBİ ÜRÜNÜN ADIELOCTA 1000 I.U. enjeksiyonluk çözelti hazırlamak için toz ve çözücü Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir flakon nominal olarak 1000 I.U. efmoroktokog alfa içerir. Rekonstitüye edildikten sonra, enjeksiyonluk çözeltinin her mL'si yaklaşık 333 I.U. efmoroktokog alfa içerir. Potens (I.U.), Avrupa Farmakopesinin kromojenik tayini kullanılarak, Dünya Sağlık Örgütü (DSÖ) Faktör VIII standardını referans alan bir dahili standardına karşı belirlenmektedir.ELOCTA'nın spesifik aktivitesi 4000-10200 I.U./mg proteindir. Efmoroktokog alfa (rekombinant insan pıhtılaşma faktörü VIII, Fc füzyon proteini (rFVIIIFc)) 1890 amino asit içerir. Hücre kültürü prosesinde, saflaştırma ya da son formülasyonda insanveya hayvan kaynaklı herhangi bir ekzojen protein ilavesi olmaksızın insan embriyonik böbrek(HEK) hücre hattında rekombinant DNA teknolojisi ile üretilmektedir. Yardımcı madde(ler):Sodyum 0,6 mmol (14 mg)/flakon Yardımcı maddelerin tam listesi için 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti için toz ve çözücü. Toz: liyofilize, beyaz ile kırık beyaz arası toz veya topaklaşmış toz. Çözücü: enjeksiyonluk su, berrak, renksiz çözelti. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarELOCTA, hemofili A (konjenital faktör VIII eksikliği) hastalarında kanamanın tedavisinde ve profilaksisinde endikedir. ELOCTA, tüm yaş gruplarında kullanılabilir. 4.2. Pozoloji ve uygulama şekliTedavi hemofili tedavisinde deneyimli bir hekimin gözetimi altında başlatılmalıdır. Pozoloji/uygulama sıklığı ve süresi:Yerine koyma tedavisinin dozu ve süresi faktör VIII eksikliğinin şiddetine, kanamanın yerine ve boyutuna ve hastanın klinik durumuna bağlıdır. Uygulanan rekombinant faktör VIII Fc ünite sayısı, faktör VIII ürünleri için geçerli DSÖ standardına bağlı olan Uluslararası Ünite (I.U.) şeklinde ifade edilmektedir. Plazmadaki faktörVIII aktivitesi ya yüzde (normal insan plazmasına göre) ya da Uluslararası Ünite (plazmadakifaktör VIII için Uluslararası Standarta göre) şeklinde ifade edilmektedir. Bir Uluslararası Ünite (I.U.) rekombinant faktör VIII Fc aktivitesi, 1 mL normal insan plazmasındaki VIII'in miktarına eşdeğerdir. Gerektiğinde uygulanan tedaviGereken rekombinant faktör VIII Fc dozunun hesaplaması, vücut ağırlığının kg'ı başına 1 Uluslararası Ünite (I.U.) faktör VIII'in, plazma faktör VIII aktivitesini 2 I.U./dL yükselttiğiampirik bulguya dayanmaktadır. Gereken doz, aşağıdaki formül kullanılarak belirlenmektedir: Gereken ünite = vücut ağırlığı (kg) x istenen faktör VIII artışı (%) (I.U./dL) x 0,5 (I.U./dL başına I.U./kg) Uygulanacak miktarın ve uygulama sıklığının her zaman bireysel vakadaki klinik etkililiğe göre ayarlanması gerekmektedir. Aşağıdaki hemorajik durumlarda faktör VIII aktivitesi, karşılık gelen süreler içinde belirtilen plazma aktivitesi seviyesinin (normalin yüzdesi veya I.U./dL) altına düşmemelidir. Kanamaepizotlarında ve cerrahide doz uygulamasına yol göstermesi için Tablo 1 kullanılabilir. Tablo 1: Kanama epizotlarının tedavisi ve cerrahi için ELOCTA doz uygulama kılavuzu
ProfilaksiUzun süreli profilaksi için önerilen doz her 3 ila 5 günde bir 50 I.U./kg'dır. Doz, hastanın yanıtına göre, 25 ila 65 I.U./kg aralığında ayarlanabilir (bkz. Bölüm 5.1 ve 5.2). Bazıdurumlarda, özellikle daha genç hastalarda, daha kısa doz uygulama aralıkları veya daha yüksekdozlar gerekli olabilir. Tedavinin izlenmesiTedavi süresince, uygulanacak dozun ve tekrarlanan enjeksiyonların sıklığına yön vermesi için faktör VIII seviyelerinin uygun şekilde tayin edilmesi (tek aşamalı pıhtılaşma veya kromojenikölçümlerle) tavsiye edilmektedir. Her bir hasta farklı yarılanma ömrü ve toparlanma zamanınedeniyle FVIII'e yanıtta da farklılık gösterebilir. Vücut ağırlığına bağlı doz için az kilolu veyaaşırı kilolu hastalarda ayarlama yapılması gerekebilir. Özellikle majör cerrahi müdahalelerde,pıhtılaşma analizi (plazma faktör VIII aktivitesi) ile yerine koyma tedavisinin dikkatli birşekilde izlenmesi zorunludur. Hastaların kan örneklerinde faktör VIII aktivitesini belirlemek için in vitroUygulama şekli:ELOCTA, intravenöz kullanım içindir. ELOCTA birkaç dakika içinde intravenöz yolla enjekte edilmelidir. Uygulama hızı, hastanın rahatlık düzeyine göre belirlenir ve 10 mL/dakikayı geçmemelidir. Uygulama öncesinde tıbbi ürünün rekonstitüsyonuna ilişkin talimatlar için bkz. Bölüm 6.6. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:ELOCTA, böbrek yetmezliği olan hastalarda çalışılmamıştır. Karaciğer yetmezliği:ELOCTA, karaciğer yetmezliği olan hastalarda çalışılmamıştır. Pediyatrik popülasyon:12 yaş altı çocuklar için daha sık veya daha yüksek dozlar gerekebilir (bkz. Bölüm 5.1). 12 yaş ve üzeri ergenler için doz önerisi, erişkinlerdeki ile aynıdır. Geriyatrik popülasyon:65 yaş ve üzeri hastalarda sınırlı deneyim mevcuttur. 4.3. KontrendikasyonlarELOCTA, etkin maddeye (rekombinant insan pıhtılaşma faktör VIII ve/veya Fc bölgesine) veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine aşırı duyarlılığı olanhastalarda kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleriAşırı duyarlılıkELOCTA ile alerjik tip aşırı duyarlılık reaksiyonları olasıdır. Eğer aşırı duyarlılık reaksiyonları ortaya çıkarsa, hastalara bu tıbbi ürünün kullanımını derhal bırakmaları ve hekimleriyleiletişime geçmeleri önerilmelidir. Hastalar kurdeşen, yaygın ürtiker, göğüs sıkışması, hırıltı,hipotansiyon ve anafilaksi gibi aşırı duyarlılık belirtileri konusunda bilgilendirilmelidir. Anafilaktik şok durumunda, şok için standart tıbbi tedavi uygulanmalıdır. İnhibitörlerFaktör VIII'e karşı nötralizan antikorların (inhibitörler) oluşumu, hemofili A hastalarının tedavisinde bilinen bir komplikasyondur. Modifiye tayin yöntemi kullanılarak mL plazmabaşına Bethesda Birimi (BU) ile ölçülen bu inhibitörler genellikle, faktör VIII prokoagülanaktivitesine karşı olan IgG immünoglobulinlerdir. İnhibitör geliştirme riski, faktör VIII'emaruziyet kadar hastalığın şiddeti ile ilişkili olup, bu risk maruziyetin ilk 50 gününde en yüksekdüzeydedir. Ancak risk nadir olmasına rağmen yaşam boyunca devam eder. İnhibitör gelişiminin klinik önemi inhibitör titresine bağlı olacaktır; düşük titrenin teşkil ettiği yetersiz klinik yanıt riski, yüksek titreli inhibitörlere kıyasla daha az olacaktır. Genel olarak, pıhtılaşma faktör VIII ürünleri ile tedavi edilen tüm hastalar, uygun klinik gözlemler ve laboratuvar testleri ile inhibitörlerin gelişimi açısından dikkatle izlenmelidir. Eğerbeklenen faktör VIII aktivitesi plazma seviyeleri elde edilmezse veya kanama uygun bir dozlakontrol edilmezse, faktör VIII inhibitörü varlığına yönelik testler yapılmalıdır. Yüksek inhibitörseviyelerine sahip hastalarda, faktör VIII tedavisi etkili olmayabilir ve diğer terapötikseçenekler değerlendirilmelidir. Bu gibi hastaların tedavisi, hemofili tedavisi ve faktör VIIIinhibitörleri konusunda deneyimli bir hekim tarafından yönetilmelidir. Kardiyovasküler olaylarKardiyovasküler risk faktörleri bulunan hastalarda, FVIII ile yerine koyma tedavisi kardiyovasküler riski artırabilir. Kateter ile ilişkili komplikasyonlarEğer bir santral venöz damar yolu cihazı (CVAD) gerekli ise, lokal enfeksiyonlar, bakteremi ve kateter yeri trombozunu içeren CVAD ile ilişkili komplikasyon riskleri göz önündebulundurulmalıdır. Pediatrik popülasyonBelirtilen uyarılar ve önlemler hem erişkinler hem çocuklar hem de ergenler için geçerlidir. Bu tıbbi ürün her flakonunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında sodyum içermez. Bununla birlikte, vücut ağırlığına ve pozolojiye bağlı olarak, hasta bir flakondan daha fazla kullanabilir (flakon başına miktar hakkında bilgi için Bölüm 2'ye bakınız). Bu durum kontrollüsodyum diyetinde olan hastalar tarafından göz önünde bulundurulmalıdır. Biyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriİnsan pıhtılaşma faktör VIII (rDNA) ile diğer tıbbi ürünler arasında herhangi bir etkileşim bildirilmemiştir. ELOCTA ile herhangi bir etkileşim çalışması gerçekleştirilmemiştir. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin herhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:Pediyatrik popülasyona ilişkin herhangi bir etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. Bölüm5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. ELOCTA, açıkça belirtilmedikçe gebelik döneminde kullanılmamalıdır. Gebelik dönemiHayvanlar üzerinde ELOCTA ile üreme çalışması yapılmamıştır. Farelerde plasental geçiş çalışması yapılmıştır (bkz. Bölüm 5.3). Kadınlarda hemofili A'nın seyrek görülmesine bağlıolarak, gebelik süresince faktör VIII kullanımı ile ilgili deneyim bulunmamaktadır. Bu nedenle,faktör VIII gebelik süresinde yalnızca açıkça belirtilmişse kullanılmalıdır. Laktasyon dönemiEfmoroktokog alfa'nın insan sütüyle atılıp atılmadığı bilinmemektedir. Efmoroktokog alfa'nın süt ile atılımı hayvanlar üzerinde araştırılmamıştır. Emzirmenin durdurulupdurdurulmayacağına ya da ELOCTA tedavisinin durdurulup durdurulmayacağına/tedavidenkaçınılıp kaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası veELOCTA tedavisinin emziren anne açısından faydası dikkate alınmalıdır ve emzirme süresindeyalnızca açıkça belirtilmişse kullanılmalıdır. Üreme yeteneği/FertiliteHerhangi bir fertilite verisi bulunmamaktadır. Hayvanlar üzerinde ELOCTA ile herhangi bir fertilite çalışması yapılmamıştır. 4.7. Araç ve makine kullanımı üzerindeki etkilerELOCTA'nın araç ve makine kullanımı üzerinde etkisi bulunmamaktadır. 4.8. İstenmeyen etkilerGüvenlilik profilinin özetiAşırı duyarlılık reaksiyonları veya alerjik reaksiyonlar (anjiyoödem, infüzyon bölgesinde yanma ve batma hissi, üşüme, yüz kızarıklığı, yaygın ürtiker, baş ağrısı, kurdeşen, hipotansiyon,letarji, bulantı, huzursuzluk, taşikardi, göğüs darlığı, karıncalanma, kusma, hırıltılı solunum)seyrek olarak gözlenmiştir ve bazı durumlarda şiddetli anafilaksiye (şok dahil) ilerleyebilir. ELOCTA dahil faktör VIII ile tedavi edilen hemofili A hastalarında nötralizan antikorların (inhibitörler) gelişimi görülebilir. Bu tür inhibitörler ortaya çıkarsa, durum yetersiz klinik yanıtolarak kendini gösterecektir. Bu gibi durumlarda uzman bir hemofili merkezi ile irtibatageçilmesi önerilmektedir. Advers reaksiyonların tablolaştırılmış listesiAşağıda sunulan Tablo 2, MedDRA sistem organ sınıflandırmasına göredir (SOC ve Tercih edilen Terime göre). Advers reaksiyonların sıklığı, 276'sı daha önce tedavi görmüş (TGH'ler)ve 103'ü daha önce hiç tedavi görmemiş (HTGH'ler) olmak üzere toplam 379 şiddetli hemofiliA hastasıyla yapılan klinik çalışmalara dayanmaktadır. Klinik çalışmalarla ilgili ek ayrıntılariçin Bölüm 5.1'e bakınız. Advers reaksiyonların sıklığı şu sisteme göre değerlendirilmiştir: çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çokseyrek (<1/10.000) ve bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubunda advers reaksiyonlar, azalan ciddiyet sırasına göre sunulmaktadır. Tablo 2: Klinik çalışmalarda ELOCTA için bildirilen advers reaksiyonlar
HTGH (Daha önce hiç tedavi görmemiş hastalar) TGH (Daha önce tedavi görmüş hastalar) 1 Tersi belirtilmediği sürece, advers ilaç reaksiyonları ve sıklık sadece TGH'lerde meydana gelenleredayanmaktadır. 2 Sıklık, tüm FVIII ürünleriyle ilgili, şiddetli hemofili A hastalarının yer aldığı çalışmalara dayanır. 3 Advers ilaç reaksiyonları ve sıklık sadece HTGH'lerde meydana gelenlere dayanmaktadır. 4 Araştırmacı terimi: ELOCTA enjeksiyonundan sonra vasküler ağrıÖzel popülasyonlara ilişkin ek bilgiler:Pediyatrik popülasyon:Pediyatrik ile erişkin hastalar arasında advers reaksiyonlarda yaşa özgü farklılıklar gözlenmemiştir. Çocuklarda görülen advers reaksiyonların sıklığının, tipinin ve ciddiyetininerişkinlerle aynı olması beklenmektedir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir ([email protected];4.9. Doz aşımı ve tedavisiHerhangi bir doz aşımı semptomu bildirilmemiştir. 5. FARMAKOLOJİK ÖZELLİKLER5.1.Farmakodinamik özelliklerFarmakoterapötik grup: Antihemorajikler, kan koagülasyon faktörü VIII ATC kodu: B02BD02 Etki mekanizmasıFaktör VIII/von Willebrand faktörü, farklı fizyolojik fonksiyonlara sahip 2 molekülden (faktör VIII ve von Willebrand faktörü) oluşur. Faktör VIII hemofili hastasına infüzyon ileverildiğinde, hastanın dolaşımındaki von Willebrand faktörüne bağlanır. Aktive edilmiş faktörVIII, faktör X'un aktive edilmiş faktör X'a dönüşümünü hızlandıran aktive edilmiş faktör IXiçin kofaktör olarak görev yapar. Aktive edilmiş faktör X, protrombini trombine dönüştürür.Trombin daha sonra fibrinojeni fibrine dönüştürür ve pıhtı oluşur. Hemofili A, fonksiyonel faktör VIIIFc seviyelerindeki azalmaya bağlı olarak ortaya çıkan X'e bağlı kalıtımsal bir kan pıhtılaşma bozukluğudur ve kendiliğinden veya kaza ya da cerrahitravmalar sonucunda eklemlerde, kaslarda veya iç organlarda kanamayla sonuçlanır.Replasman tedavisi ile plazma faktör VIII seviyeleri artar ve böylelikle faktör eksikliğiningeçici olarak düzeltilmesi ve kanama eğilimlerinin düzeltilmesi sağlanır. Önemli olarak, yıllık kanama oranları (ABR) farklı faktör konsatrasyonları arasında ve farklı klinik çalışmalar arasında karşılaştırılabilir değildir. ELOCTA (efmoroktokog alfa) uzun yarılanma ömrüne sahip tamamen rekombinant bir füzyon proteinidir. ELOCTA insan immünoglobulini G1'in Fc domenine kovalent bağlı, rekombinantB domeni çıkarılmış insan pıhtılaşma faktör VIII'den oluşur. İnsan immünoglobulini G1'in Fcbölgesi, neonatal Fc reseptörüne bağlanır. Bu reseptör yaşam süresince eksprese edilir ve buimmünoglobulinleri dolaşıma geri döndürerek bu proteinleri lizozomal degradasyondankoruyan doğal bir yolağın parçasıdır; bu da proteinlerin yarı ömürlerinin uzaması ile sonuçlanır.Efmoroktokog alfa neonatal Fc reseptörüne bağlanarak lizozomal degradasyonu geciktirmekve endojen faktör VIII'den daha uzun bir plazma yarılanma ömrüne olanak vermek üzere aynıdoğal yolağı kullanır. Klinik etkililik ve güvenlilikDaha önce tedavi görmüş hastalarda (TGH) ELOCTA'nın güvenliliği, etkililiği ve farmakokinetiği 2 çok uluslu, açık etiketli, pivotal faz 3 çalışmada, yani Çalışma I ve ÇalışmaII'de (bkz. Pediatrik popülasyon) ve bir uzatma çalışmasında (Çalışma III) dört yıla kadar süreile değerlendirilmiştir. Toplamda 276 TGH, hasta başına ortalama 294 (1-735) maruziyet günüile toplam 80848 maruziyet günü boyunca takip edilmiştir. Ek olarak, ELOCTA'nın daha öncehiç tedavi görmemiş hastalarda (HTGH) güvenliliğini ve etkililiğini değerlendirmek için bir faz3 çalışma (Çalışma IV) gerçekleştirilmiştir (bkz. Pediatrik popülasyon). Çalışma I'e şiddetli hemofili A'sı olan, önceden hiç tedavi görmemiş toplam 165 erkek hasta (12 ila 65 yaş aralığında) kaydedilmiştir. Çalışmaya girmelerinden önce profilaksi rejimlerindeolan gönüllüler bireyselleştirilmiş profilaksi koluna alınmıştır. Giriş öncesinde gerektiğindeuygulanan tedavide olan gönüllüler ise ya bireyselleştirilmiş profilaksi koluna alınmış ya dahaftalık profilaksi veya gerektiğinde uygulanan tedavi kollarına randomize edilmiştir. Profilaksi rejimi: Bireyselleştirilmiş profilaksi: Her 3 ila 5 günde bir 25 ila 65 I.U./kg'dır. Haftalık profilaksi: 65 I.U./kg Çalışma I'i tamamlayan 153 gönüllüden 150'si Çalışma III'e kaydedilmiştir (uzatma çalışması). Çalışma I + III'de toplam medyan zaman 4,2 yıldı ve maruziyet gününün medyansayısı 309'du. Bireyselleştirilmiş profilaksi:Çalışma I'de yıllık medyan faktör tüketimi 4212 I.U./kg (min. 2877, maks. 7943) ve Çalışma III'de 4223 I.U./kg (min. 2668, maks. 8317) idi. İlgilimedyan Yıllık Kanama Oranı (ABR) 1,60 (min. 0, maks. 18,2) ve 0,74 (min. 0, maks. 15,6) idi.Haftalık profilaksi:Yıllık medyan faktör tüketimi Çalışma I'de 3805 I.U./kg (min. 3353, maks. 6196) Çalışma III'de 3510 I.U./kg (min. 2758, maks. 3984) idi. İlgili medyan ABR 3,59(min. 0, maks. 58,0) ve 2,24 (min 0, mak. 17,2) idi.Gerektiğinde uygulanan tedavi:Yıllık medyan faktör tüketimi, Çalışma I'deki gerektiğinde uygulanan tedavi koluna randomize edilen 23 hasta için 1039 I.U./kg (min. 280, maks. 3571)ve Çalışma III'de en az bir yıl boyunca gerektiğinde uygulanan tedavide kalan 6 hasta için671 I.U./kg (min. 286, maks. 913) idi.Gerektiğinde uygulanan tedaviden haftalık profilaksiye geçirilen gönüllülere ait medyan ABR Çalışma III boyunca 1,67 olmuştur. Kanamanın tedavisi:Her kanamayı kontrol etmek için Çalışma I ve III sırasında 43,8 I.U./kg (min. 13,0, maks. 172,8) medyan doz ile 2490 kanama olayı tedavi edilmiştir. İlkenjeksiyonların %79,2'si hastalar tarafından mükemmel veya iyi olarak değerlendirdi.Perioperatif tedavi (cerrahi profilaksi):Çalışma I ve Çalışma III'de toplam 48 majör cerrahi operasyon gerçekleştirilmiştir ve 34 gönüllüde değerlendirilmiştir. Hemostatik yanıt, doktorlartarafından 44 major cerrahinin 3'ünde iyi, 41'inde mükemmel olarak değerlendirilmiştir.Operasyon sırasında hemostazı korumak için medyan doz 60,6 I.U./kg (min. 38, maks. 158)olmuştur.Pediyatrik popülasyonÇalışma II'ye şiddetli hemofili A'sı olan, önceden tedavi görmüş 12 yaşından küçük toplam 71 pediyatrik erkek hasta kaydedilmiştir. Kaydedilen 71 hastanın 69'u en az bir ELOCTA dozualmıştır ve etkililik açısından değerlendirilebilir olmuştur (35'i 6 yaşından küçüktür ve 34'ü 6ila 12 yaş aralığındadır). Başlangıç profilaktik rejim, ilk gün 25 I.U./kg'dan, bunu takibendördüncü gün 50 I.U./kg'dan oluşmuştur. 80 I.U./kg'a kadarki dozlara ve 2 gün gibi kısa dozuygulama aralıklarına izin verilmiştir ve çalışmadaki sınırlı sayıda hastada kullanılmıştır.Çalışma II'yi tamamlayan 67 gönüllüden 61'i Çalışma III'e (uzatma çalışması) kaydedilmiştir.Çalışma II +III'de medyan toplam süre 3,4 yıl ve maruziyet gününün medyan sayısı 332olmuştur. 6yaşın altındaki çocuklar içinprofilaksi:Medyan doz aralığı Çalışma II ve Çalışma III'de 3,50 gündür. Yıllık medyan faktör tüketimi Çalışma II'de 5146 I.U./kg (min. 3695, maks. 8474) veÇalışma III'de 5418 I.U./kg (min. 3435, maks. 9564) olmuştur. İlgili medyan Yıllık KanamaOranı (ABR) 0,00 (min. 0, maks. 10,5) ve 1,18 (min. 0, maks. 9,2) olmuştur.6 ila 12 yaş arasındaki çocuklar için profilaksi:Medyan doz aralığı Çalışma II' de 3,49 gün ve Çalışma III'de 3,50 gündür. Yıllık medyan faktör tüketimi Çalışma II'de 4700 I.U./kg(min. 3819, maks. 8230 I.U./kg) ve Çalışma III'de 4990 I.U./kg (min. 3856, maks. 9527)olmuştur. İlgili medyan ABR 2,01 (min. 0, maks. 27,2) ve 1,59 (min. 0, maks. 8) olmuştur.12 ila 18yaş arasındaki 12 ergen gönüllüprofilaktik tedavide yetişkin çalışma popülasyonuna dahil edilmiştir. Yıllık medyan faktör tüketimi Çalışma I'de 5572 I.U./kg (min. 3849, maks.7035) ve Çalışma III'de 4456 I.U./kg (min. 3563, maks. 8011) olmuştur. İlgili medyan ABR1,92 (min. 0, maks. 7,1) ve 1,25 (min. 0, maks. 9,5) olmuştur.Kanama tedavisi:Her kanamayı kontrol etmek için 63 I.U./kg (min. 28, maks. 186) olan bir medyan doz ile Çalışma II ve III sırasında 447 kanama olayı tedavi edilmiştir. İlk enjeksiyonun%90,2'si hastalar ya da onların hasta bakıcıları tarafından mükemmel ya da iyi olarakdeğerlendirilmiştir.Çalışma IV'te, şiddetli hemofili A'sı olan 6 yaşından küçük, önceden hiç tedavi görmemiş (HTGH) 103 erkek hasta değerlendirmiştir. Hastalar, hasta başına ortanca 100 (aralık 0-649)maruziyet günü olmak üzere toplam 11255 maruziyet günü boyunca izlenmiştir. Çoğu gönüllü(N= 81) epizodik tedaviyle başlamış, ardından profilaksiye geçişmiştir (N= 69). 89 HTGHçalışma sırasında herhangi bir zamanda profilaksi almıştır. Profilakside önerilen başlangıç dozu3-5 günlük aralıklarla 25-80 IU/kg'dır. Profilaksi alan gönüllüler için, ortanca ortalama haftalıkdoz 101,4 IU/ g (aralık: 28,5-776,3 IU/kg) ve ortanca doz aralığı 3,87 gün (aralık 1,1 ila 7 gün)olmuştur. Ortanca yıllık faktör tüketimi 3971,4 IU/kg olmuştur. Yıllık Kanama Oranı 1,49(min. 0,0, maks. 18,7) bulunmuştur. 5.2. Farmakokinetik özelliklerGenel özelliklerELOCTA ile tüm farmakokinetik çalışmalar, önceden tedavi görmüş şiddetli hemofili A hastalarında yürütülmüştür. Bu bölümde sunulan veriler, kromojenik yöntem ve tek aşamalıpıhtılaşma testiyle elde edilmiştir. Kromojenik yöntem verilerinden elde edilen farmakokinetikparametreler, tek aşamalı yöntemle elde edilenlere benzerdir. Farmakokinetik özellikler, ELOCTA (rFVIIIFc) kullanan 28 gönüllüde (15 yaş ve üzeri) değerlendirilmiştir. En az 96 saatlik (4 gün) bir arınma periyodunu takiben, gönüllüler tek bir50 I.U./kg ELOCTA dozu almıştır. Dozdan önce ve dozdan sonraki 120 saate (5 gün) kadar 7zaman noktasında farmakokinetik örnekleri toplanmıştır. ELOCTA'nın 50 I.U./kg dozundansonraki farmakokinetik parametreler Tablo 3 ve 4'te sunulmaktadır.
Farmakokinetik veriler ELOCTA'nın dolaşımda uzun bir yarılanma ömrü olduğunu göstermektedir. Hastalardaki karakteristik özelliklerPediyatrik popülasyonÇalışma I'de ergenler (dozdan önce farmakokinetik örnekleme yapılmış, ardından dozdan sonra 120 saate (5 gün) kadarki çoklu zaman noktalarında değerlendirme yapılmıştır) ve çalışma II'deçocuklar (dozdan önce farmakokinetik örnekleme yapılmış, ardından dozdan sonra 72 saate (3gün) kadar çoklu zaman noktalarında değerlendirme yapılmıştır) için ELOCTA'nınfarmakokinetik parametreleri belirlenmiştir. Tablo 5 ve 6'da 18 yaşın altındaki gönüllülerinpediyatrik verilerinden hesaplanan farmakokinetik parametreler sunulmaktadır.
Ergenler ve erişkinler ile karşılaştırıldığında, 12 yaşın altındaki çocuklarda, diğer pıhtılaşma faktörlerine ait gözlemlerle uyumlu şekilde, klerens daha yüksek ve yarılanma ömrü daha kısaolabilir. Doz uygulanırken yapılırken bu farklılıklar göz önünde bulundurulmalıdır. 5.3. Klinik öncesi güvenlilik verileriKlinik dışı veriler, akut ve tekrarlı doz toksisitesi (lokal toksisite ve güvenlilik farmakolojisinin değerlendirmelerini içeren) çalışmalarına dayanarak insanlar için özel bir tehlike olmadığınıortaya koymaktadır. Genotoksisite, karsinojenite, üreme toksisitesi ve embriyofetal gelişimaraştırmaları yapılmamıştır. Plasental transfer çalışmasında, ELOCTA'nın farelerde plasentayıaz miktarda geçtiği gösterilmiştir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiTozSükroz Sodyum klorür Histidin Kalsiyum klorür dihidrat Polisorbat 20 Sodyum hidroksit (pH ayarlayıcı) Hidroklorik asit (pH ayarlayıcı) ÇözücüEnjeksiyonluk su 6.2. GeçimsizliklerGeçimlilik çalışmaları bulunmadığından bu tıbbi ürün, diğer tıbbi ürünler ile karıştırılmamalıdır. Bazı enjeksiyon ekipmanlarının iç yüzeyine pıhtılaşma faktörü VIII'in adsorpsiyonunun sonucu olarak tedavi başarısız olabileceğinden sadece verilen infüzyon seti kullanılmalıdır. 6.3. Raf ömrüAçılmamış flakon4 yılRaf ömrü boyunca ürün, oda sıcaklığında (30°C'ye kadar) yalnızca bir kez 6 ayı geçmeyen bir süre için saklanabilir. Ürünün buzdolabından çıkarıldığı tarih kutu üzerine kaydedilmelidir.Oda sıcaklığında saklandıktan sonra ürün buzdolabına geri konulmamalıdır. Flakon üzerindeyazan son kullanma tarihinden sonra veya karton kutu buzdolabından çıkarıldıktan 6 ay sonra(hangisi daha erken ise) kullanmayınız. Rekonstitüsyon sonrasıRekonstitüsyondan sonra, oda sıcaklığında (30°C'ye kadar) saklandığında 6 saate kadar kimyasal ve fiziksel stabilitesi gösterilmiştir. Ürünü doğrudan güneş ışığından koruyunuz.Rekonstitüsyondan sonra, eğer 6 saat içinde kullanılmazsa ürün atılmalıdır. Mikrobiyolojikaçıdan, ürün sulandırıldıktan hemen sonra kullanılmalıdır. Hemen kullanılmadığı takdirde,kullanım içi saklama süreleri ve koşulları kullanıcının sorumluluğundadır. 6.4. Saklamaya yönelik özel tedbirler2°C - 8°C arasında buzdolabında saklayınız. Dondurmayınız. Flakonu ışıktan korumak için orijinal ambalajında saklayınız. Tıbbi ürünün rekonstitüsyonundan sonraki saklama koşulları için Bölüm 6.3'e bakınız. 6.5. Ambalajın niteliği ve içeriğiHer ambalaj; - klorobütil kauçuk tıpalı 1 adet tip 1 cam flakon içinde toz - bromobütil lastik piston tıpalı kullanıma hazır tip 1 cam enjektör içinde 3 mL çözücü - 1 adet piston çubuğu - rekonstitüsyon için 1 adet steril flakon adaptörü - 1 adet steril infüzyon seti - 2 adet alkollü mendil - 2 adet flaster - 1 adet gazlı beziçermektedir. Tek kullanımlık 1 flakon ve 1 kullanıma hazır enjektörlük ambalaj büyüklüğü 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerFlakondaki enjeksiyonluk liyofilize toz, rekonstitüsyon için steril flakon adaptörü kullanılarak kullanıma hazır enjektörde bulunan çözücü (enjeksiyonluk su) ile rekonstitüye edilmelidir.Flakon tozun tamamı çözününceye kadar yavaşça döndürülmelidir. Rekonstitüye tıbbi ürün uygulamadan önce partikül madde ve renk değişikliği açısından gözle incelenmelidir. Çözelti, berrak ile hafif opalesan ve renksiz olmalıdır. Bulanık olan ya da tortuiçeren çözeltileri kullanmayınız. Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİProceutica İlaç Pazarlama ve Danışmanlık A.Ş. Huzur Mah. Metin Oktay Cad. Nurol Life Blok No:3 İç Kapı No: 49 34396 Sarıyer/İstanbulTel : +90 212 843 47 44Faks : +90 212 843 47 45 8. RUHSAT NUMARASI2022/185 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 03.04.2022 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ
<
> Hazırlama ve Uygulama TalimatlarıELOCTA; enjeksiyonluk toz, kullanıma hazır enjektörde temin edilen çözücü ile çözüldükten sonra intravenöz (IV) enjeksiyon ile uygulanır. ELOCTA ambalajı şunları içerir:  A) 1 adet toz içeren flakon B) Kullanıma hazır enjektör içinde 3 mLçözücü C) 1 adet piston çubuğu D) 1 adet flakon adaptörü E) 1 adet infüzyon seti F) 2 adet alkollü mendil G) 2 adet flaster H) 1 adet gazlı bez
ELOCTA, enjeksiyonluk veya infüzyonluk diğer çözeltilerle karıştırılmamalıdır.
Ambalajı açmadan önce ellerinizi yıkayın. Hazırlama:1. Doğru ilacı içerdiğinden emin olmak için paketin adını ve dozunu kontrol ediniz. ELOCTA kutusu üzerindeki son kullanma tarihini kontrol ediniz. Son kullanma tarihigeçmiş ise ürünü kullanmayınız. 2. Eğer ELOCTA buzdolabında saklanmış ise ELOCTA flakonunun (A) ve çözücü içeren enjektörün (B) kullanmadan önce oda sıcaklığına gelmesini bekleyiniz. Harici ısıkullanmayınız. 3. Flakonu temiz bir yüzey üzerine koyunuz. Plastik geçme kapağı ELOCTA flakonundan çıkarınız.  4. Flakonun üst kısmını, pakette verilen alkollü bezlerden (F) biriyle siliniz ve kurumaya bırakınız. Silindikten sonraflakonun üst kısmına dokunmayınız ya da başka herhangi birşeyin dokunmasına izin vermeyiniz.  5. Şeffaf plastik flakon adaptöründen koruyucu kağıdı (D) çıkarınız. Adaptörü koruyucu yuvasından çıkarmayınız. Flakon adaptörü paketinin iç kısmına dokunmayınız._6. Flakonu düz bir yüzeye yerleştiriniz. Flakon adaptörünü koruyucu yuvasından tutarak flakonun üstüne düz bir şekildeyerleştiriniz. Adaptörü, flakon tıpasını delen adaptörün sivriucu ile flakonun üst kısmına oturana kadar aşağı doğru sertçebastırınız.  7. Piston çubuğunun (C) ucunu, enjektör pistonundaki açıklığa sokarak piston çubuğunu çözücü enjektörüne takınız. Pistonçubuğunu, enjektör pistonuna sağlam bir şekilde oturana kadarsaat yönünde çeviriniz.  8. Beyaz, emniyet kilitli plastik kapağı, kopana kadar delikli kapaktan eğip çözücü enjektöründen kırarak çıkarınız. Kapağıdüz bir yüzey üzerine baş aşağı koyarak elinizden bırakınız.Kapağın iç kısmına ya da enjektör ucuna dokunmayınız. 
9. Koruyucu yuvayı adaptörden çıkarınız ve atınız.  10. Enjektörün ucunu adaptör girişine yerleştirerek çözücü enjektörünü flakon adaptörüne takınız. Sertçe itiniz ve sağlambir şekilde takılana kadar enjektörü saat yönünde döndürünüz.  11. Çözücünün tamamını ELOCTA flakonuna enjekte etmek için piston çubuğunu yavaşça aşağıya doğru bastırınız.  12. Enjektör hala adaptöre bağlı ve piston çubuğu basılı haldeyken, toz çözülene kadar flakonu hafifçe ve daireselhareketlerle döndürünüz. Çalkalamayınız.  13. Nihai çözelti, uygulanmadan önce incelenmelidir. Çözelti berrak ile hafif bulanık arası ve renksiz görünmelidir. Bulanık ise veya gözle görülebilir partiküller içeriyorsa çözeltiyi kullanmayınız._14. Enjektörün piston çubuğunun tamamen basılı olduğundanemin olduktan sonra flakonu baş aşağı çeviriniz. Çözeltinintamamını flakon adaptöründen enjektöre çekmek için pistonçubuğunu yavaşça çekiniz.  15. Enj ektörü flakon adaptöründen hafifçe çekerek ve flakonu saat yönünün tersine çevirerek çıkarınız.  Not: Eğer her enjeksiyonda bir ELOCTA flakonundan daha fazla kullanıyorsanız her flakon, önceki basamaklara (basamaklar 1 ila 13) göre ayrı ayrı hazırlanmalıdır ve flakon adaptörüyerinde bırakılarak çözücü şırıngası çıkarılmalıdır. Ayrı flakonların her biri için hazırlananiçeriğin çekilmesi için tek bir büyük luer kilitli enjektör kullanılabilir. 16. Flakonu ve adaptörü atınız. Not: Eğer çözelti hemen kullanılmayacaksa enjektör kapağı dikkatle enjektör ucuna geri takılmalıdır. Enjektör ucuna veya kapağının iç kısmına dokunmayınız. Hazırlama sonrasında ELOCTA, uygulanmadan önce oda sıcaklığında 6 saate kadar saklanabilir. Bu süre sonunda hazırlanan ELOCTA atılmalıdır. Doğrudan güneş ışığındankoruyunuz. Uygulama (İntravenöz enjeksiyon):ELOCTA, bu ambalaj ile sağlanan infüzyon seti (E) kullanılarak uygulanmalıdır. 1. İnfüzyon setinin paketini açınız ve tüpün ucundaki kapağı çıkarınız. İçinde hazırlanan ELOCTA çözeltisi bulunanenjektörü, infüzyon setinin tüpünün ucuna, saat yönündeçevirerek takınız.  Eğer gerekirse turnike yapınız ve pakette verilen diğer alkollü bez ile deriyi iyice silerek enjeksiyon bölgesini hazırlayınız.  3. İnfüzyon setinin tüpünde hava varsa, sıvı infüzyon seti iğnesine ulaşana kadar pistonçubuğuna yavaşça bastırarak havayı çıkarınız. Çözeltiyi iğnenin içinden geçirmeyiniz.Şeffaf plastik koruyucu kapağı iğneden çıkarınız. 4. İnfüzyon setinin iğnesini, doktorunuzun veya hemşirenizin gösterdiği gibi bir damarabatırınız ve turnikeyi çıkarınız. Tercih ediyorsanız iğnenin plastik kanatlarını enjeksiyonbölgesinde tutmak için pakette verilen flasterlerden (G) birini kullanabilirsiniz.Hazırlanan çözelti birkaç dakika boyunca intravenöz yolla enjekte edilmelidir.Doktorunuz sizin için daha rahat hale getirmek amacıyla önerilen enjeksiyon hızınıdeğiştirebilir. 5. Enjeksiyonu tamamladıktan ve iğneyi çıkardıktan sonra iğne koruyucusunu katlamanız ve iğne üzerine geçirmeniz gerekir. 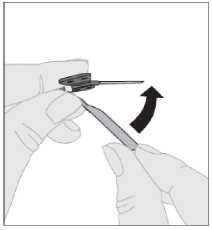 6. Lütfen kullanılmış iğneyi, varsa kullanılmamış çözeltiyi, enjektörü ve boş flakonu uygun bir tıbbi atık kutusu içinde güvenli bir şekilde atınız çünkü bu materyaller, doğru şekilde atılmadığında, başkalarına zarar verebilir. Ekipmanı tekrar kullanmayınız._1 Bazı hastalarda ve durumlarda, doz uygulama aralığı 36 saate kadar uzatılabilir. Farmakokinetik veriler için bkz.Bölüm 5.2. |
İlaç BilgileriElocta 1000 I.u Enjeksiyonluk Çözelti Hazırlamak İçin Toz ve ÇözücüEtken Maddesi: Efmoroktokog Alfa Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.