Ratebir 250 Mg Tablet Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'a bildirmeleribeklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır.1. BEŞERİ TIBBİ ÜRÜNÜN ADIRATEBİR 250 mg tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir tablet 250 mg abirateron asetat içerir. Yardımcı maddeler:Her bir tablet 256,05 mg laktoz monohidrat (inek sütü kaynaklı), 42,9 mg kroskarmelloz sodyum ve 28,6 mg sodyum lauril sülfat içerir. Yardımcı maddeler için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMOval, beyaz-gri beyaz renkli, tek taraf baskılı (Baskı: AA250) tabletler. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarRATEBİR, prednizolon ile birlikte aşağıdaki durumların tedavisinde endikedir: - Hormonal tedaviye duyarlı ancak kemoterapi için uygun olmayan* metastatik prostatkanserli hastalarda progresyona kadar; - Hormonal tedaviye ve sonrasında dosetaksel temelli kemoterapiyle progresyonun PSA veradyolojik görüntüleme yöntemleriyle gösterildiği, ECOG performans skorunun (0-1)olduğu ve testosteron düzeyinin kastrasyon seviyesinde olduğu gösterilmiş metastatikprostat kanserli hastalarda - Kastrasyona dirençli progresyonun PSA ve/veya görüntüleme yöntemleriyle gösterildiği vetestosteron düzeyi kastrasyon seviyesinde olan (<50 ng/dl), kemoterapi için uygunolmayan* metastatik prostat kanserli hastalarda progresyona kadar endikedir. * Kemoterapi için uygun olmayan hastalar: - ECOG performans durumu >1 olan hastalar, - Kreatinin klerensi <45 ml/dk olan hastalar, - Nötrofil sayısı <1500/mm3 ve/veya trombosit düzeyi <100.000/mm3 olan hastalar, - Karaciğer rezervi düşük olan hastalar. 4.2. Pozoloji ve uygulama şekliPozoloji, uygulama sıklığı ve süresi:Bu tıbbi ürün uygun bir sağlık mesleği mensubu tarafından reçete edilmelidir. 1 / 28 RATEBİR günde tek seferde 1.000 mg (dört adet 250 mg tablet) olarak yutulmalı ve yiyeceklerle birlikte alınmamalıdır (uygulama şekli bölümüne bakınız). RATEBİR'inyiyeceklerle birlikte alınması ilaca sistemik maruziyeti arttırır (bkz. Bölüm 4.5 ve 5.2). Prednizolon dozu RATEBİR, metastatik hormona duyarlı prostat kanserli hastalarda günde 5 mg prednizolon ile birlikte kullanılır. RATEBİR, metastatik kastrasyona dirençli prostat kanserli hastalarda günde 10 mg prednizolon ile birlikte kullanılır. Cerrahi kastrasyon uygulanmamış hastalarda tedavi sırasında luteinizan hormon serbestleyici hormon (LHRH) analoğu ile tıbbi kastrasyona devam edilmelidir. Önerilen izlem RATEBİRile tedaviye başlamadan önce serum transaminaz düzeyleri ölçülmelidir; tedavinin ilk üç ayında iki haftada bir, daha sonra ayda bir bu testler tekrarlanmalıdır. Hastalar kan basıncı,serum potasyumu ve sıvı retansiyonu açısından aylık olarak izlenmelidir. Bununla birlikte,konjestif kalp yetmezliği açısından anlamlı bir risk taşıyan hastalar tedavinin ilk 3 ayı boyuncaiki haftada bir, daha sonra ise ayda bir kere izlenmelidir (bkz. Bölüm 4.4). Daha önceden hipokalemisi olan ya da RATEBİR tedavisi sırasında hipokalemi gelişen hastalarda, hastanın potasyum düzeyinin 4.0 mM ve üzerinde tutulması değerlendirilmelidir. Hipertansiyon, hipokalemi, ödem ve diğer mineralokortikoid dışı toksisiteler dahil olmak üzere, Derece 3 ve üzerinde toksisite gelişen hastalarda, tedavi durdurulmalı ve uygun tıbbi tedaviyebaşlanmalıdır. Toksisite semptomları Derece 1 ya da başlangıç düzeyine dönünceye kadarRATEBİR tedavisine yeniden başlanmamalıdır. RATEBİR veya prednizolonun günlük dozunun alınmasının unutulması durumunda, tedaviye ertesi gün olağan günlük dozla devam edilmelidir. Uygulama şekli:RATEBİR ağızdan alınır. Tabletler günde bir kez aç karnına tek doz olarak alınmalıdır. RATEBİR, yemek yedikten en az iki saat sonra alınmalı ve RATEBİR'i aldıktan sonra en az bir saat boyunca yemekyenmemelidir. RATEBİR tabletleri bütün olarak suyla yutulmalıdır. Özel popülasyonlara ilişkin ek bilgiler:HepatotoksisiteRATEBİR tedavisi sırasında hepatotoksisite gelişen hastalarda (alanin aminotransferaz [ALT] veya aspartat aminotransferaz [AST] düzeylerinin normal kabul edilen üst sınırın 5 katındanfazla yükselmesi) tedavi hemen durdurulmalıdır (bkz. Bölüm 4.4). Karaciğer fonksiyon testleri tedaviye başlamadan önceki başlangıç değerlerine döndükten sonra tedaviye azaltılmış dozla 2 / 28 günde 500 mg (iki tablet) olarak başlanabilir. Tedaviye yeniden başlanan bu hastalarda serum transaminaz düzeyleri, tedavinin ilk üç ayında iki haftada bir, daha sonra ise ayda birölçülmelidir. Azaltılmış 500 mg'lık günlük dozla da hepatotoksisite tekrarlarsa, tedavi tümüylekesilmelidir. Tedavinin herhangi bir döneminde ağır hepatotoksisite gelişmesi durumunda (ALT ya da AST düzeylerinin normal kabul edilen üst sınırın 20 katı kadar yükselmesi) tedaviye hemen sonverilmeli ve RATEBİR ile yeniden tedavi uygulanmamalıdır. Karaciğer yetmezliği:Önceden hafif şiddette karaciğer bozukluğu olan hastalarda (Child-Pugh Sınıf A) doz ayarlamasına gerek yoktur. Orta şiddette karaciğer yetmezliğinin (Child Pugh sınıf B), abirateron asetatın oral yoldan tek doz halinde 1.000 mg alınması sonrası abiraterona sistemik maruziyeti yaklaşık dört katarttırdığı gösterilmiştir (bkz. Bölüm 5.2). Orta şiddette veya ağır karaciğer yetmezliği (Child-Pugh sınıf B veya C) olan hastalarda abirateron asetatın birden fazla dozunun klinik güvenlilikve etkililiğini gösteren bir veri bulunmamaktadır. Bu tür hastalarda doz ayarlamasıöngörülemez. RATEBİR kullanımı faydanın olası riskten açıkça ağır bastığı, orta şiddettekaraciğer yetmezliğine sahip hastalarda dikkatle değerlendirilmelidir (bkz. Bölüm 4.2 ve 5.2).RATEBİR şiddetli karaciğer yetmezliği olan hastalarda kullanılmamalıdır (bkz. Bölüm 4.3, 4.4ve 5.2). Böbrek yetmezliği:Ancak . Bu tür Böbrek yetmezliği olan hastalarda doz ayarlamasına gerek yoktur (bkz. Bölüm 5.2). prostat kanserli ağır böbrek yetmezliği olan hastalarda klinik deneyim bulunmamaktadırhastalarda dikkatli olunması tavsiye edilmektedir (bkz. Bölüm 4.4). Pediyatrik popülasyon:Pediyatrik popülasyonda prostat kanseri görülmediği için RATEBİR'in çocuklarda ve adolesanlarda kullanımı bulunmamaktadır. Geriyatrik popülasyon:Yaşlı hastalarda herhangi bir doz ayarlaması gerekmemektedir. 4.3. Kontrendikasyonlar- Abirateron asetata veya Bölüm 6.1'de listelenen herhangi bir yardımcı maddeye karşı aşırıduyarlılık varsa, - Gebe olan ya da gebe olma olasılığı bulunan kadınlarda (bkz. Bölüm 4.6), - Ağır karaciğer yetmezliği olanlarda [Child-Pugh sınıf C (bkz. Bölüm 4.2, 4.4 ve 5.2)]kontrendikedir. - RATEBİR ile prednizolon Ra-223 ile kombine kullanımda kontrendikedir. 3 / 28 4.4. Özel kullanım uyarıları ve önlemleriMineralokortikoid fazlalığına bağlı hipertansiyon, hipokalemi, sıvı retansiyonu ve kalp yetmezliğiRATEBİR, CYP17 inhibisyonu sonucunda (bkz. Bölüm 5.1) mineralokortikoid düzeylerindeki artışa bağlı olarak hipertansiyon, hipokalemi ve sıvı retansiyonuna (bkz. Bölüm 4.8) yol açabilir.RATEBİR ile birlikte kortikosteroid uygulanması, adrenokortikotropin hormon (ACTH)salgılanmasını baskılayarak bu advers etkilerin görülme sıklığı ve şiddetinde bir azalma sağlar.Kan basıncının yükselmesi, hipokalemi (örneğin kardiyak glikozidleri kullanan hastalar) veyasıvı retansiyonu (örneğin kalp yetmezliği olan hastalar), ağır veya stabil olmayan anjinapektoris, yakın zamanda miyokard enfarktüsü veya ventriküler aritmi ve ağır böbrek yetmezliğinedeniyle altta yatan tıbbi durumu risk altına girebilecek hastalar tedavi edilirken dikkatliolunmalıdır. RATEBİR kardiyovasküler hastalık öyküsü olan hastalarda dikkatle kullanılmalıdır. Hipertansiyonu kontrol altına alınamayan, miyokard enfarktüsüyle ortaya çıkmış klinik açıdananlamlı kalp hastalığı olan, son 6 ayda arteriyel trombotik olay geçirmiş olan, ağır ya da unstabilanjinası olan, New York Kalp Cemiyeti (NYHA) Sınıf III ve IV kalp yetmezliği olan (Çalışma301) ya da Sınıf II'den IV'e kalp yetmezliği (çalışma 3011 ve 302) olan ya da kardiyakejeksiyon fraksiyonu %50'nin altında olan hastalar Faz 3 çalışmalara dahil edilmemiştir.Medikal terapi gerektiren atriyal fibrilasyonu veya diğer kardiyak aritmisi olan hastalar çalışma3011 ve 302'den çıkarılmıştır. Sol ventrikül ejeksiyon fraksiyonu (LVEF) %50'nin altında olanhastalar ya da New York Kalp Cemiyeti (NYHA) Sınıf III ve IV kalp yetmezliği olan (Çalışma301'de) ya da NYHA Sınıf II'den IV'e kalp yetmezliği (çalışma 3011 ve 302) olan hastalardagüvenlilik değerlendirilmemiştir (bkz. Bölüm 4.8 ve 5.1). Konjestif kalp yetmezliği açısından anlamlı risk taşıyan hastaların tedavi edilmesinden önce (örneğin, kalp yetmezliği, kontrol altına alınamayan hipertansiyon ya da iskemik kalp hastalığıgibi kalp olayları), kalp fonksiyonunun bir değerlendirmesinin yapılması düşünülmelidir(örneğin, ekokardiyogram). RATEBİR ile tedaviden önce, kalp yetmezliği tedavi edilmeli vekalp fonksiyonu mümkün olan en iyi düzeye çıkarılmalıdır. Hipertansiyon, hipokalemi ve sıvıretansiyonu düzeltilmeli ve kontrol altına alınmalıdır. Tedavi sırasında, 3 ay süreyle 2 haftadabir ve daha sonra ayda bir kere olmak üzere kan basıncı, serum potasyum, sıvı retansiyonu (kiloartışı, periferik ödem) ve diğer konjestif kalp yetmezliği bulgu ve belirtileri izlenmeli veanormallikler düzeltilmelidir. Abirateron asetat ile ilişkili hipokalemi görülen hastalarda QTuzaması gözlenmiştir. Kalp fonksiyonunda klinik olarak anlamlı bir azalma olması durumunda,kalp fonksiyonu klinik endikasyona göre değerlendirilmeli, uygun tedavi başlatılmalı veRATEBİR tedavisinin kesilmesi değerlendirilmelidir (bkz. Bölüm 4.2). Hepatotoksisite ve karaciğer yetmezliğiKontrollü klinik çalışmalarda karaciğer enzimlerinde ilacın kesilmesine ya da doz değişikliğine neden olan belirgin artışlar bildirilmiştir (bkz. Bölüm 4.8). RATEBİR ile tedaviye başlamadanönce serum transaminaz düzeyleri ölçülmelidir; tedavinin ilk üç ayında iki haftada bir, dahasonra ayda bir bu testler tekrarlanmalıdır. Hepatotoksisitenin klinik belirti veya bulguları görülürgörülmez, hemen serum transaminaz düzeyleri ölçülmelidir. Tedavinin herhangi bir yerindeALT ya da AST düzeyleri normal kabul edilen üst sınırın (NÜS) 5 katından fazla yükselen 4 / 28 hastalarda RATEBİR tedavisine hemen ara verilmeli ve karaciğer fonksiyonları yakından takip edilmelidir. RATEBİR ile yeniden tedaviye ancak karaciğer fonksiyon testleri başlangıç değerlerine döndüğünde ve azaltılmış dozlarla başlanabilir (bkz. Bölüm 4.2). Tedavinin herhangi bir döneminde ağır hepatotoksisite gelişmesi durumunda (ALT ya da AST düzeylerinin normal kabul edilen üst sınırın 20 katı kadar yükselmesi) RATEBİR tedavisikesilmeli ve bir daha başlatılmamalıdır. Klinik çalışmalara aktif veya semptomatik viral hepatiti olan hastalar dahil edilmemiştir; bu nedenle abirateron asetatın bu popülasyonda kullanımını destekleyen bir veri bulunmamaktadır. Orta şiddette veya ağır karaciğer yetmezliği (Child-Pugh sınıf B veya C) olan hastalarda abirateron asetatın birden fazla dozunun klinik güvenlilik ve etkililiğini gösteren bir veribulunmamaktadır. RATEBİR kullanımı faydanın olası riskten açıkça ağır bastığı, orta şiddettekaraciğer yetmezliğine sahip hastalarda dikkatle değerlendirilmelidir (bkz. Bölüm 4.2 ve 5.2).RATEBİR şiddetli karaciğer yetmezliği olan hastalarda kullanılmamalıdır (bkz. Bölüm 4.2, 4.3ve 5.2). Akut karaciğer yetmezliği ve fulminant hepatit tanımlayan az sayıda pazarlama sonrası rapor mevcuttur. Bunların bazıları ölümle sonuçlanmıştır (bkz. Bölüm 4.8) Kortikosteroidin geri çekilmesi ve stresli durumların karşılanmasıPrednizolon tedavisinin kesilmesi durumunda dikkatli olunması ve adrenokortikal yetmezlik gelişmemesi için hastaların izlenmesi önerilir. Kortikosteroidler kesildikten sonra RATEBİRtedavisine devam edilecekse, hastalar mineralokortikoid fazlalığına bağlı semptomlar açısındanizlenmelidir (bkz. Bölüm 4.4 Mineralokortikoid fazlalığına bağlı hipertansiyon, hipokalemi, sıvıretansiyonu ve kalp yetmezliği başlığı). Prednizolon kullanan hastalarda olağan dışı stres ortaya çıktığında, bu stresli durum öncesinde, sırasında ve sonrasında kortikosteroid dozunun arttırılması gerekebilir. Kemik dansitesiİleri evre metastatik prostat kanseri olan erkeklerde kemik dansitesinde azalma görülebilir. RATEBİR'in bir glukokortikoid ile birlikte kullanımı bu etkiyi arttırabilir. Daha önceden ketokonazol kullanımıDaha önceden prostat kanseri için ketokonazol kullanmış olan hastalarda daha düşük yanıt oranları beklenebilir. HiperglisemiGlukokortikoid kullanımı hiperglisemiyi artırabileceği için, diyabetli hastalarda kan şekeri sıklıkla ölçülmelidir. Diyabetik hastalarda abirateron ile birlikte kullanılacak kortikosteroidlerkan şekeri regülasyonunu bozabileceğinden dikkatli kullanılmalıdır. 5 / 28 HipoglisemiÖnceden diyabeti olup pioglitazon veya repaglinid alan hastalara abirateron asetat ve prednizolon uygulandığında hipoglisemi vakaları bildirilmiştir (bkz. Bölüm 4.5); bu nedenlediyabetli hastalarda kan şekeri izlenmelidir. Kemoterapi ile kullanımAbirateron asetatın sitotoksik kemoterapi ile eşzamanlı olarak kullanılmasının güvenliliği ve etkililiği gösterilmemiştir (bkz. Bölüm 5.1). Potansiyel risklerRATEBİR ile tedavi görenler de dahil, prostat kanseri olan erkeklerde anemi ve cinsel işlev bozukluğu görülebilir. ilk 6 ayındaİskelet kası etkileriAbirateron asetat ile tedavi gören hastalarda miyopati ve rabdomiyoliz vakaları bildirilmiştir. Bu olayların çoğu tedavinin ilk ayında ortaya çıkmış ve abirateron asetatın kesilmesinden sonradüzelmiştir. Miyopati/rabdomiyoliz ile ilişkili olduğu bilinen ilaçlarla eş zamanlı tedavi görenhastalarda dikkatli olunması önerilir. Diğer tıbbi ürünler ile etkileşimAzalmış RATEBİR maruziyeti riski nedeniyle, başka bir tedavi seçeneği olmadığı sürece tedavi sırasında güçlü CYP3A4 indükleyicilerinden kaçınılmalıdır (bkz. Bölüm 4.5). Abirateron ve prednizolonun Ra-223 ile kombinasyonuRa-223 ile kombinasyon halinde abirateron ve prednizolon ile tedavi, asemptomatik veya hafif semptomatik prostat kanseri hastalarda yürütülen klinik çalışmalarda gözlendiği üzere artmışkırık riski ve artmış mortalite eğilimi nedeniyle kontrendikedir (bkz. Bölüm 4.3). RATEBİR'in prednizolon ile kombine kullanıldığı son dozunu takiben en az 5 gün süreyle ardışık tedavide Ra-223 başlanması önerilmemektedir. Yardımcı maddelere karşı intoleransBu tıbbi ürün laktoz ihtiva eder. Nadir kalıtımsal galaktoz intoleransı, totalg laktaz yetmezliği ya da glukoz-galaktoz malabsorbsiyon problemi olan hastaların bu ilacı kullanmamaları gerekir. Bu tıbbi ürünün dört tabletlik her bir dozu 27,2 mg (1,18 mmol) fazla sodyum ihtiva eder. Bu DSÖ'nün bir yetişkin için önerdiği günlük maksimum 2 g sodyum alımının %4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriYiyeceklerin abirateron üzerindeki etkisiRATEBİR'in yiyeceklerle birlikte alınması abirateron emilimini anlamlı derecede arttırır. RATEBİR'in yiyeceklerle birlikte alınması halindeki etkililik ve güvenliliği gösterilmemiştir bunedenle RATEBİR yiyeceklerle birlikte alınmamalıdır (bkz. Bölüm 4.2 ve 5.2). 6 / 28 Diğer tıbbi ürünler ile etkileşimlerDiğer ilaçların abirateron maruziyetlerini etkileme potansiyeli Güçlü bir CYP3A4 indükleyicisi olan rifampisin ile önce 6 gün boyunca günde bir kere 600 mg dozunda tedavi edilen ve takiben tek bir 1.000 mg'lık abirateron asetat dozu verilen sağlıklıgönüllülerde yürütülen bir klinik farmakokinetik etkileşim çalışmasında, ortalama abirateronplazma EAAro değeri %55 azalmıştır. Başka bir tedavi seçeneği olmadığı sürece tedavi sırasında güçlü CYP3A4 indükleyicilerinden kaçınılmalıdır (örneğin; fenitoin, karbamazepin, rifampisin, rifabutin, rifapentin, fenobarbital,St. John's wort (sarı kantaron - Hypericum perforatum).Sağlıklı gönüllülerde yürütülen ayrı bir klinik farmakokinetik etkileşim çalışmasında, güçlü bir CYP3A4 inhibitörü olan ketokonazolün eşzamanlı uygulanması abirateronun farmakokinetiğiüzerinde klinik olarak anlamlı bir etki yaratmamıştır. Abirateronun diğer ilaçların maruziyetini etkileme potansiyeliAbirateron, hepatik ilaç metabolize eden CYP2D6 ve CYP2C8 enzimlerinin bir inhibitörüdür. Abirateron asetatın (artı prednizolon) tek dozda alınan CYP2D6 substratı dekstrometorfanüzerindeki etkisini belirlemek için yapılan bir çalışmada, dekstrometorfanın sistemikmaruziyetinin (EAA) yaklaşık 2,9 kat arttığı bildirilmiştir. Dekstrometorfanın aktif metabolitidekstrorfanın EAA24'ü ise yaklaşık %33 artmıştır. RATEBİR, özellikle dar terapötik indekse sahip ilaçlar olmak üzere, CYP2D6 tarafından aktive veya metabolize edilen ilaçlarla birlikte alındığında dikkatli olunmalıdır. CYP2D6 tarafındanmetabolize edilen dar terapötik indekse sahip ilaçların dozunun azaltılması değerlendirilmelidir.CYP2D6 tarafından metabolize edilen ilaçlara örnek olarak metoprolol, propranolol, desipramin,venlafaksin, haloperidol, risperidon, propafenon, flekanid, kodein, oksikodon ve tramadolgösterilebilir (kodein, oksikodon ve tramadolun aktif analjezik metabolitlerinin oluşabilmesi içinCYP2D6 gereklidir). Sağlıklı gönüllüler üzerinde yapılan bir CYP2C8 ilaç etkileşim çalışmasında, pioglitazon 1,000 mg'lık tek doz abirateron asetat ile birlikte verildiğinde, pioglitazonun EAA değeri %46 artmışve pioglitazonun aktif metabolitleri olan M-III ve M-IV için EAA değerleri %10 azalmıştır.CYP2C8 substratı olan dar terapötik aralıklı ilaçlar ile birlikte RATEBİR kullanılmasıdurumunda hastalar toksisite semptomları açısından izlenmelidir. CYP2C8 tarafındanmetabolize edilen tıbbi ürün örnekleri arasında pioglitazon ve repaglinid bulunmaktadır (bkz.Bölüm 4.4). İn vitroortamda, abirateronun majör metabolitleri olan abirateron sülfat ve N-oksit abirateron sulfatın karaciğer alım taşıyıcısı OATP1B1'i inhibe ettiği ve sonuç olarak OATP1B1 tarafındanelimine edilen ilaçların konsantrasyonlarını artırabileceği gösterilmiştir. Taşıyıcı bağlı etkileşimidoğrulayacak klinik veri yoktur.QT aralığını uzattığı bilinen ilaçlar ile kullanımıAndrojen azaltma tedavisi QT aralığını uzattığı için, QT aralığını uzattığı bilinen ilaçlar ile 7 / 28 RATEBİR verirken veya sınıf IA (örn. Kinidin, disopiramid) gibi Torsade de pointes'i indükleyebilecek ya da sınıf III (örn. Amiadoron, sotalol, dofetilid, ibutilid) gibi antiaritmikürünler, metadon, moksifloksasin, antipsikotikler gibi ürünler ile verilirken dikkatli olunmasıönerilir. Spironolakton ile kullanımSpironolakton androjen reseptörüne bağlanır ve prostat spesifik antijen (PSA) seviyelerini artırabilir. Abirateron asetat ile kullanımı önerilmez (bkz. Bölüm 5.1). Özel popülasyonlara ilişkin ek bilgilerHerhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:Herhangi bir etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: X RATEBİR kadınlarda kullanımı olan bir ilaç değildir. RATEBİR gebe olan ya da gebe olma olasılığı bulunan kadınlarda kontrendikedir (bkz. Bölüm 4.3 ve 5.3). Çocuk doğurma potansiyeli bulunan kadınlarGebelikte abirateron asetat kullanımına ilişkin klinik bir veri mevcut değildir ve RATEBİR çocuk doğurma potansiyeli bulunan kadınlarda kullanım için uygun bir ilaç değildir. Doğum kontrolü (kontrasepsiyon)RATEBİR ya da metabolitlerinin semende bulunup bulunmadığı bilinmemektedir. Hastanın gebe bir kadınla cinsel ilişkiye girmesi durumunda kondom kullanması gerekir. Hastanın çocukdoğurma potansiyeli bulunan bir kadınla cinsel ilişkiye girmesi durumunda etkili bir doğumkontrol yöntemine ek olarak kondom kullanması gerekir. Hayvanlarda yapılan çalışmalar üremetoksisitesi olduğunu göstermiştir (bkz. Bölüm 5.3). Gebelik dönemiRATEBİR kadınlarda kullanımı olan bir ilaç değildir ve gebelik döneminde ya da gebelik potansiyeline sahip kadınlarda kontrendikedir (bkz. Bölüm 4.3 ve 5.3). Laktasyon dönemiRATEBİR kadınlarda kullanımı olan bir ilaç değildir. Üreme yeteneği / FertiliteAbirateron asetat erkek ve dişi sıçanlarda fertiliteyi etkilemiş olmakla birlikte bu etkiler tamamen geri dönüşümlü olmuştur (bkz. Bölüm 5.3). 8 / 28 4.7. Araç ve makine kullanımı üzerindeki etkilerRATEBİR'in araç ve makine kullanımı yeteneği üzerinde etkisi yoktur ya da ihmal edilebilir düzeydedir. 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti Birleştirilmiş Faz 3 çalışmalarının istenmeyen yan etkiler üzerindeki bir analizinde, abirateron asetat ile hastaların %10'unda veya fazlasında gözlemlenen istenmeyen etkiler; periferik ödem,hipokalemi, hipertansiyon, idrar yolu enfeksiyonu ve alanin aminotransferaz düzeylerindeyükselme ve/veya aspartat aminotransferaz düzeylerinde yükselmedir.Diğer önemli advers etkiler arasında kalp hastalıkları, hepatotoksisite, kırıklar ve alerjik alveolit bulunur. RATEBİR, etki mekanizmasının farmakodinamik sonucu olarak hipertansiyon, hipokalemi ve sıvı retansiyonuna neden olabilir. Faz 3 çalışmalarında, abirateron asetat ile tedavi edilenhastalarda, plasebo ile tedavi edilen hastalara oranla beklenen mineralokortikoid advers etkilerdaha yaygın olarak görülmüştür. Çalışmada hipokalemi abirateron asetat alanlarda %18 ikenplasebo alanlarda %8, hipertansiyon abirateron asetat alanlarda %22 iken plasebo alanlarda %16ve sıvı retansiyonu (periferik ödem) abirateron asetat alanlarda %23 iken plasebo alanlarda %17olarak bildirilmiştir. Abirateron asetat ile tedavi edilen hastalarda, Grade 3 ve 4 hipokalemi(CTCAE, versiyon 4.0 sınıflamasına göre), % 6 iken plasebo alanlarda % 1; abirateron asetat iletedavi edilen hastalarda Grade 3 ve 4 hipertansiyon (CTCAE, versiyon 4.0 sınıflamasına göre)% 7 iken plasebo alanlarda % 5; abirateron asetat ile tedavi edilen hastalarda Grade 3 ve 4 sıvıretansiyonu (periferal ödem) % 1 iken plasebo alanlarda % 1 olarak gözlemlenmiştir.Mineralokortikoid reaksiyonlar tıbbi tedaviyle genellikle başarıyla yönetilebilmiştir.Kortikosteroidlerin birlikte kullanılması bu advers ilaç reaksiyonlarının sıklık ve şiddetini azaltır(bkz. Bölüm 4.4). Advers reaksiyonların özeti Luteinizan hormon salgılayıcı hormon (LHRH) analoğunun kullanılmakta olduğu ya da daha önceden orşiektomi tedavisi uygulanmış ileri evre metastatik prostat kanserli hastalardakiçalışmalarda abirateron asetat, düşük doz prednizolon (endikasyona bağlı olarak günlük 5 veya10 mg) ile kombine olarak günde 1.000 mg dozunda kullanılmıştır. Klinik çalışmalarda ve pazarlama sonrası deneyimlerde gözlenen advers etkiler aşağıdaki sıklık derecelerine göre listelenmiştir. Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan(>1/1,000 ila <1/100); seyrek (>1/10,000 ila <1/1,000); çok seyrek (<1/10,000), bilinmiyor(eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubunda, istenmeyen etkiler azalan ciddiyet sırasına göre sunulmuştur. Enfeksiyonlar ve enfestasyonlarÇok yaygın: İdrar yolu enfeksiyonu Yaygın: Sepsis
9 / 28 Bağışıklık sistemi hastalıklarıBilinmiyor: Anafilaktik reaksiyonlar Endokrin hastalıklarıYaygın olmayan: Adrenal yetmezlik Metabolizma ve beslenme hastalıklarıÇok yaygın: Hipokalemi Yaygın: Hipertrigliseridemi Kardiyak hastalıklarYaygın: Kalp yetmezliği*, anjinapektoris, atrial fibrilasyon, taşikardi Yaygın olmayan: Diğer aritmiler Bilinmiyor: Miyokard enfarktüs, QT uzaması (bkz. Bölüm 4.4 ve 4.5) Vasküler hastalıklarÇok yaygın: Hipertansiyon Solunum, göğüs hastalıkları ve mediastinal hastalıklarSeyrek: Alerjik alveolita Gastrointestinal hastalıklarÇok yaygın: Diyare Yaygın: Dispepsi Hepato-bilier hastalıklarÇok yaygın: Alanin aminotransferazdüzeylerindeyükselme ve/veya aspartat aminotransferaz düzeylerinde yükselmeb Seyrek:Fulminant hepatit, akut karaciğeryetmezliği Deri ve deri altı doku hastalıklarıYaygın: Döküntü Kas-iskelet bozuklukları, bağ doku ve kemik hastalıklarıYaygın olmayan: Miyopati, rabdomiyoliz Böbrek ve idrar yolu hastalıklarıYaygın: Hematüri Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarÇok yaygın: Periferik ödem Yaralanma ve zehirlenmeYaygın: Kırıklar** 10 / 28 * Kalp yetmezliği aynı zamanda konjestif kalp yetmezliği, sol ventriküler disfonksiyon ve ejeksiyon fraksiyonunda azalmayı da içermektedir. **Kırıklar, osteoporozu ve patolojik kırık dışındaki tüm kırıkları içerir. a Pazarlama sonrası deneyimden spontan bildirimler b Alanin aminotransferaz düzeyinde yükselme ve/veya aspartat aminotransferaz düzeyinde yükselme, ALT düzeyinde yükselmeyi, AST düzeyinde yükselmeyi ve anormal karaciğerfonksiyonunu içerir. Abirateron asetat ile tedavi edilen hastalarda aşağıdaki Grade 3 advers ilaç reaksiyonları (CTCAE, versiyon 4.0 sınıflamasına göre) görülmüştür: %5 hipokalemi; %2 idrar yoluenfeksiyonu; %4 alanin aminotransferaz düzeyinde yükselme ve/veya aspartat aminotransferazdüzeyinde yükselme, % 6 hipertansiyon, % 2 kırık; %1 periferik ödem, kalp yetmezliği ve atrialfibrilasyon. CTCAE (versiyon 4.0) Grade 3 hipertrigliseridemi ve anjina pektoris hastaların%1'inden azında meydana gelmiştir. Hastaların %1'inden azında Grade 4 idrar yoluenfeksiyonu, alanin aminotransferaz düzeyinde yükselme ve/veya aspartat aminotransferazdüzeyinde yükselme, hipokalemi, kalp yetmezliği atriyal fibrilasyon ve kırıklar görülmüştür. Hormona duyarlı popülasyonda (çalışma 3011) hipertansiyon ve hipokalemi daha yüksek bir insidansta gözlemlenmiştir. Hormona duyarlı popülasyonda (çalışma 3011) hastaların %36,7'sinde hipertansiyon rapor edilmişken, çalışma 301 ve 302'de sırasıyla % 11,8 ve %20,2'idi. Hormona duyarlı popülasyonda hastaların % 20,4'ünde hipokalemi gözlemlenmişken(çalışma 3011), çalışma 301 ve 302'de sırasıyla % 19,2 ve % 14,9'idi. ECOG2 performans durum skoru temel olan alt grup hastalarda ve ayrıca yaşlı hastalarda (>75 yaş), yan etkilerin görülme sıklığı ve ciddiyeti daha yüksekti. Seçilmiş advers reaksiyonların tanımlanması Kardiyovasküler etkiler Abirateron asetat ile yürütülen üç Faz 3 çalışmasında da, hipertansiyonu kontrol altına alınamayan, miyokard enfarktüsüyle ortaya çıkmış klinik açıdan anlamlı kalp hastalığı olan, son6 ayda arteriyel trombotik olay geçirmiş olan, ağır ya da unstabil anjinası olan, New York KalpCemiyeti (NYHA) Sınıf III ve IV kalp yetmezliği (Çalışma 301) ya da Sınıf II ila IV kalpyetmezliği (Çalışma 3011 ve 302) olan ya da kardiyak ejeksiyon fraksiyon ölçümü %50'dendüşük olan hastalar çalışmaya dahil edilmemiştir. Çalışmaya alınan diyabet, miyokardenfarktüsü, serebrovasküler olay ve ani kardiyak ölüm riski olan tüm hastalara (hem aktif ilaçhem de plasebo alanlar) aynı zamanda öncelikle LHRH analogları kullanılarak androjen azaltmatedavisi uygulanmıştır. Faz 3 çalışmasında kardiyovasküler advers reaksiyonların sıklığıAbirateron asetat alanlarda ve plasebo alanlarda aşağıdaki gibi bulunmuştur: Atriyal fibrilasyon%2,6 ve %2; taşikardi, %1,9 ve %1; anjina pektoris %1,7 ve %0,8; kalp yetmezliği %0,7 ve%0,2 ve aritmi, %0,7 ve %0,5. Hepatotoksisite Abirateron asetat ile tedavi edilen hastalarda ALT, AST ve total bilirubin düzeylerinde yükselmeyle seyreden hepatotoksisite bildirilmiştir. Faz 3 klinik çalışmalarında, Abirateron asetat alan hastaların yaklaşık %6'sında tipik olarak tedaviye başladıktan sonraki ilk 3 ayda 11 / 28 Grade 3 ve 4 karaciğer toksisitesi (örneğin, ALT ve AST düzeylerinde normal kabul edilen üst sınırın 5 katından fazla yükselme veya bilirubin düzeylerinde normal kabul edilen üst sınırın 1,5katından fazla yükselme) bildirilmiştir. Çalışma 3011'de, abirateron asetat ile tedavi edilenhastaların %8,4'ünde Grade 3 ve 4 hepatotoksisite gözlemlenmiştir. Abirateron asetat alanhastalardan 10'unda hepatotoksisite sebebiyle tedavi sonlandırılmıştır; bunlardan ikisi Grade 2hepatotoksisite, altı tanesi Grade 3 hepatotoksisite ve iki tanesi Grade 4 hepatotoksisitedir.Çalışma 3011'de hepatotoksisite sebebiyle ölen hasta olmamıştır. Faz 3 klinik çalışmalarında,başlangıç ALT veya AST düzeyleri yüksek olan hastalarda, başlangıç değerleri normal olanlaragöre karaciğer fonksiyon testlerinde artış olasılığı daha yüksek olmuştur.ALT veya AST düzeylerinde normal kabul edilen üst sınırın 5 katından fazla yükselme olduğunda ya da bilirubin düzeylerinde normal kabul edilen üst sınırın 3 katından fazlayükselme olduğunda Abirateron asetat tedavisine ara verilmiş ya da kesilmiştir. İki olgudakaraciğer fonksiyon testlerinde belirgin yükselmeler görülmüştür (bkz. Bölüm 4.4). Başlangıçdeğerleri normal olan bu iki hastada ALT veya AST düzeyleri normal üst sınır değerlerin 15 ila40 katı ve bilirubin düzeyleri ise normal üst sınır değerlerin 2 ila 6 katı yükselmiştir. RATEBİRtedavisinin kesilmesinden sonra, her iki hastada da karaciğer fonksiyon testleri normale dönmüşve hastalardan birinde bu defa yükselme olmaksızın Abirateron asetat ile yeniden tedaviuygulanabilmiştir. Çalışma 302'deki abirateron asetat ile tedavi edilen 35 (% 6,5) hastada,Grade 3 veya 4 ALT ya da AST yükselmeleri gözlemlenmiştir. Aminotransferaz yükselmeleri 3hasta (son abirateron asetat dozundan yaklaşık 3 hafta sonra yeni çoklu karaciğer metastazı olan2 ve AST yükselmesi olan 1 hasta) hariç tüm hastalarda normale dönmüştür. Faz 3 klinikçalışmalarında, ALT ve AST artışları veya karaciğer fonksiyonlarında anormallik nedeniyletedaviyi kesme oranları abirateron asetat alan hastalar için % 1,1, plasebo alan hastalar için ise% 0,6 olarak bildirilmiştir; hepatotoksisite olaylarına bağlı hiçbir ölüm vakası bildirilmemiştir. Klinik çalışmalarda başlangıçta hepatiti ya da karaciğer fonksiyon testlerinde anlamlı anormallikleri olan hastalar hariç tutulmak suretiyle hepatotoksisite riski azaltılmıştır. Çalışma3011'de, başlangıç ALT ve AST değerleri NÜS'ün 2,5 katından fazla olan hastalar, bilirubindeğerleri normal kabul edilen üst sınırın 1,5 katından fazla olan hastalar ve aktif veyasemptomatik viral hepatit veya kronik karaciğer hastaları; karaciğer fonksiyon bozukluğunabağlı asit veya kanama bozukluğu olan hastalar çalışmaya alınmamıştır. Çalışma 301'de,başlangıç ALT ve AST düzeyleri karaciğer metastazının olmadığı durumlarda normal kabuledilen üst sınırın >2.5 katı, metastaz olanlarda ise normal kabul edilen üst sınırın >5 katı olanhastalar çalışmaya dahil edilmemiştir. Çalışma 302'de, karaciğer metastazı olanlar ve başlangıçALT ve AST düzeyleri > 2,5 x NÜS olan hastalar çalışmaya alınmamıştır. Klinik çalışmayaalınan hastalarda karaciğer fonksiyon testlerinde anormalleşme olduğunda ise, bu hastalarıntedavileri kesilmiş ve ancak karaciğer fonksiyon testleri tedavinin başlangıcındaki düzeylerinedöndükten sonra yeniden tedavi almalarına izin verilmiştir (bkz. Bölüm 4.2). ALT ve ASTdüzeyleri normal kabul edilen üst sınırın 20 katından fazla yükselen hastalarda yeniden tedaviuygulanmamıştır. Bu tür hastalarda tedaviye yeniden başlamanın güvenliliği bilinmemektedir.Abirateron asetat tedavisi sırasında gelişen hepatotoksisitenin mekanizması bilinmemektedir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem 12 / 28 taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta:[email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9. Doz aşımı ve tedavisiAbirateron asetat ile doz aşımına dair insan deneyimi sınırlıdır. Spesifik antidotu yoktur. Doz aşımı durumunda RATEBİR uygulaması durdurularak aritmilerin, hipokalemi ve sıvı retansiyonunun bulgu ve belirtilerinin izlenmesi de dahil olmak üzere geneldestekleyici önlemler alınmalıdır. Karaciğer fonksiyonları da değerlendirilmelidir. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Endokrin tedavisi, diğer hormon antagonistleri ve ilişkili ajanlar ATC kodu: L02BX03 Etki mekanizması Abirateron asetat (RATEBİR) in vivoolarak bir androjen biyosentez inhibitörü olan abiraterona dönüşür. Spesifik olarak abirateron 17a-hidroksilaz/C17,20-liyaz (CYP17) enzimini seçiciolarak inhibe eder. Bu enzim testiküler, adrenal ve prostatik tümör dokularında eksprese olur veandrojenin biyosentezi için gereklidir. CYP17 enzimi, sırasıyla 17a-hidroksilasyon ve C17,20bağının kırılmasıyla pregnenolon ve progesteronun testesteron prekürsörleri olan DHEA veandrostenediona dönüşümünü katalize eder. CYP17 inhibisyonu aynı zamanda adrenallertarafından mineralokortikoid üretiminde artışa da yol açar (bkz. Bölüm 4.4).Androjene duyarlı prostat karsinomu, androjen düzeylerini azaltan tedaviye yanıt verir. LHRH analogları ya da orşiektomi gibi androjen azaltıcı tedaviler, testislerdeki androjen üretiminiazaltmalarına rağmen, adrenaller ya da tümör dokusundaki androjen üretimini etkilemezler.LHRH analogları (ya da orşiektomi) ile birlikte Abirateron asetat tedavisi uygulandığında serumtestesteron düzeyleri (ticari testlerle ölçüldüğünde) saptanabilir düzeylerin altına düşer. Farmakodinamik etkiler RATEBİR, serum testesteron ve diğer androjen seviyelerini, tek başına LHRH analogları ya da orşiektomi ile elde edilen seviyelerin altına düşürür. Bu, androjen biyosentezi için gerekli olanCYP17 enziminin selektif olarak inhibe edilmesinin bir sonucudur. Prostat spesifik antijen(PSA) prostat kanserli hastalarda bir biyogösterge olarak kullanılır. Daha önce taksanlarlayapılan kemoterapiden fayda görmeyen hastalarda gerçekleştirilen bir Faz 3 klinik çalışmada,Abirateron asetat ile tedavi edilen hastaların %38'inde başlangıç PSA değerlerine göre en az%50 azalma sağlanabilmişken, bu azalma oranı plasebo ile tedavi edilenlerin ancak %10'undasağlanabilmiştir. Klinik etkililik ve güvenlilik Abirateron asetatın etkililiği metastatik hormona duyarlı prostat kanseri (mHDPK) ve metastatik kastrasyona dirençli prostat kanseri (mKDPK) olan hastalarda gerçekleştirilen plasebo kontrollüçok merkezli randomize üç Faz 3 çalışmayla (çalışma 3011, 302 ve 301) gösterilmiştir. Çalışma 13 / 28 3011'e, yüksek riskli prognostik faktörlere sahip yeni tanı almış (randomizasyondan önceki 3 ay içerisinde) mHDPK'li hastalar dahil edilmiştir. Yüksek riskli prognoz aşağıdaki 3 riskfaktöründen en az 2'sine sahip olmak olarak tanımlanmıştır: (1) Gleason skorunun >8 olması;(2) kemik taramasında 3 veya daha fazla lezyon olması; (3) ölçülebilir visseral (lenf noduhastalığı hariç) metastaz olması. Aktif tedavi kolunda, standart tedavi olan ADT'ye (LHRHanaloğu veya orşiektomi) ilave olarak, abirateron asetat günde 1.000 mg dozunda, günde tek doz5 mg düşük doz prednizon ile kombine olarak uygulandı. Kontrol kolundaki hastalara abirateronasetat ve prednizon yerine ADT ve plasebo verildi. Çalışma 301'e daha önceden dosetakselkullanmış hastalar, çalışma 302'ye ise daha önce dosetaksel kullanmamış hastalar dahiledilmiştir. Hastalar bir LHRH analoğu kullanıyorlardı ya da daha önce orşiektomi olmuşlardı.Aktif tedavi uygulanan kolda, abirateron asetat, günde iki defa 5 mg düşük doz prednizon ya daprednizolonla kombine olarak günde 1.000 mg dozunda kullanılmıştır. Kontrol grubundaysaplaseboya ek olarak günde iki defa 5 mg düşük doz prednizon ya da prednizolon uygulanmıştır. Serum PSA konsantrasyonlarındaki değişiklikler bağımsız olarak her zaman klinik faydayı göstermeyebilir. Bu nedenle, tüm çalışmalarda hastaların aşağıda verilen tedavi kesilmekriterlerini karşılamalarına kadar tedaviye devam etmeleri önerilir. Spironolakton androjen reseptörüne bağlandığı ve PSA düzeylerini artırabileceği için, tüm çalışmalarda spironolakton kullanımına izin verilmemiştir. Çalışma 3011 (yeni tanı almış yüksek riskli mHDPK hastaları)Çalışma 3011'e (n=1.199) dahil edilen hastaların medyan yaşı 67 idi. Abirateron asetat ile tedavi edilen hastaların 832'si (% 69,4) beyaz ırka mensup, 246'sı (% 20,5) Asyalı, 25'i (% 2,1) Siyahiveya Afro Amerikan, 80'i (% 6,7) diğer, 13'ü (% 1,1) bilinmeyen/raporlanmamış ve 3'ü (% 0,3)Amerikan yerlisi veya Alaska yerlisi idi. Hastaların % 97'si için ECOG performans durumu 0veya 1 idi. Bilinen beyin metastazı, kontrol altına alınamayan hipertansiyonu, önemli kalphastalığı olan veya NYHA Sınıf II-IV kalp yetmezliği olan hastalar çalışmaya alınmamıştır.Metastatik hastalıktan kaynaklanan semptomların tedavisi için 3 aya kadar ADT veya 1 kürpalyatif radyasyon veya operasyon tedavisi alan hastalar haricinde daha önce farmakoterapi,radyasyon terapisi veya metastatik prostat kanseri operasyonu geçirerek tedavi edilen hastalarçalışmaya alınmamıştır. Ortak birincil etkililik sonlanım noktaları genel sağkalım (OS) veradyografik progresyonsuz sağkalım (rPFS) idi. Kısa Ağrı Envanteri Kısa Form (BPI-SF) ileölçülen medyan başlangıç ağrı skoru hem tedavi kolunda, hem de plasebo gruplarında 2.0 idi.Ortak birincil sonlanım noktaları ölçümlerine ilave olarak; iskeletle ilişkili olaya (SRE) kadargeçen süre, prostat tedavisi için sonraki tedaviye kadar geçen süre, kemoterapi başlangıcınakadar geçen süre, ağrı progresyonuna kadar geçen süre ve PSA progresyonuna kadar geçen sürekullanılarak tedavi faydası da değerlendirildi. Tedavi hastalık progresyonuna, onamın geriçekilmesine, kabul edilemez toksisite veya ölüme kadar devam etti. Radyografik progresyonsuz sağkalım, randomizasyondan radyografik progresyon görülmesine veya herhangi bir nedene bağlı ölüme kadar geçen süre olarak tanımlandı. Radyografikprogresyon, kemik taramasıyla progresyonu (modifiye PCWG2'ye göre) veya BT veya MRG ileyumuşak doku lezyonlarındaki progresyonu (RECIST 1.1'e göre) kapsıyordu. 14 / 28 Tedavi grupları arasında rPFS bakımından anlamlı bir farklılık gözlendi (bkz. Tablo 1 ve Şekil 1).
OS bakımından Plasebo artı ADT (TO=0,66; %%34'lük bir azalma ile AA-P artı ADT lehine istatistikselolarak anlamlı bir iyileşme gözlenmiştir (bkz. Tablo 2 ve Şekil 2).15 / 28
Alt grup analizleri RATEBİRile tedaviyi sürekli olarak desteklemektedir. Önceden belirlenmiş alt gruplarda AA-P'nin rPFS ve OS üzerindeki tedavi etkisi genel çalışma popülasyonunda dahaüstün ve tutarlı olurken, ECOG skoru 2 olan alt grupta lehde bir yarar gözlenmemiştir, ancakörneklem büyüklüğünün küçük olması (n=40) anlamlı bir sonuç çıkarılmasını kısıtlamıştır. Genel sağkalım ve rPFS'de gözlenen artışlara ilave olarak, prospektif olarak tanımlanan tüm ikincil sonlanım noktaları için plasebo karşısında abirateron astetat lehine faydalargösterilmiştir. Çalışma 302 (daha önce kemoterapi almamış hastalar)Bu çalışmaya asemptomatik veya hafif düzeyde semptomatik olan ve henüz kemoterapi endikasyonu bulunmayan kemoterapi almamış hastalar dahil edilmiştir. Kısa Ağrı Envanteri -Kısa Formunun (BPI-SF) son 24 saat içindeki en kötü ağrı maddesinin puanının 0-1 olmasıasemptomatik, puanın 2-3 olması ise hafif semptomatik olarak değerlendirilmiştir. 16 / 28 Çalışma 302'de (n= 1.088) yer alan hastaların medyan yaşı abirateron asetat ile birlikte prednizon veya prednizolon alan hastalar için 71, plasebo ile birlikte prednizolon alan hastalariçin 70 idi. Abirateron asetat ile tedavi edilen hastaların ırklarına göre dağılımı şöyleydi: 520 (%95,4) beyaz ırk, 15 (% 2,8) siyah ırk, 4 (% 0,7) sarı ırk ve 6 (% 1,1) diğer ırklar. Doğu OrtakOnkoloji Grubu (ECOG) performans durumu, her iki koldaki hastaların % 76'sı için 0 ve %24'ü için 1 idi. Hastaların % 50'sinde yalnızca kemik metastazları, % 31'inde kemik veyumuşak doku veya lenf nodu metastazları ve % 19'unda yalnızca yumuşak doku veya lenfnodu metastazları meydana gelmiştir. Visseral metastazı olan hastalar çalışmaya alınmamıştır.Ortak birincil etkinlik sonlanım noktaları genel sağkalım ve radyografik progresyonsuz sağkalım(rPFS) idi. Ortak birincil sonlanım noktaları ölçümlerine ilave olarak; kanser ağrısı için opiatkullanımına kadar geçen süre, sitotoksik kemoterapi başlangıcına kadar geçen süre, ECOGperformans durumunda > 1 puanlık kötüleşmeye kadar geçen süre ve Prostat Kanseri ÇalışmaGrubu 2 (PCWG2) kriterlerine göre PSA progresyonuna kadar geçen süre kullanılarak tedavifaydası da değerlendirilmiştir. Bariz klinik progresyon durumunda çalışma tedavilerine sonverilmiştir. Araştırmacı kararıyla, doğrulanmış radyografik progresyon ile de tedavikesilebilecekti. Radyografik progresyonsuz sağkalım (rPFS) PCWG2 kriterlerinde (kemik lezyonları için) tanımlanan seri görüntüleme çalışmalarına ve Solid Tümörlerde Yanıt DeğerlendirmeKriterleri'ne (RECIST) göre (yumuşak doku lezyonları için) değerlendirildi. rPFS analizinde,radyografik progresyon değerlendirmesi merkezi inceleme ile yapılmıştır. Planlı rPFS analizi 401 olayı kapsamış, abirateron asetat ile tedavi edilen hastaların 150'sinde (% 28) ve plasebo ile tedavi edilen hastaların 251'inde (% 46) ya radyografik progresyonbulgusu görülmüş ya da hastalar ölmüştür. Tedavi grupları arasında rPFS bakımından anlamlıbir farklılık gözlenmiştir (bkz. Tablo 3 ve Şekil 3).
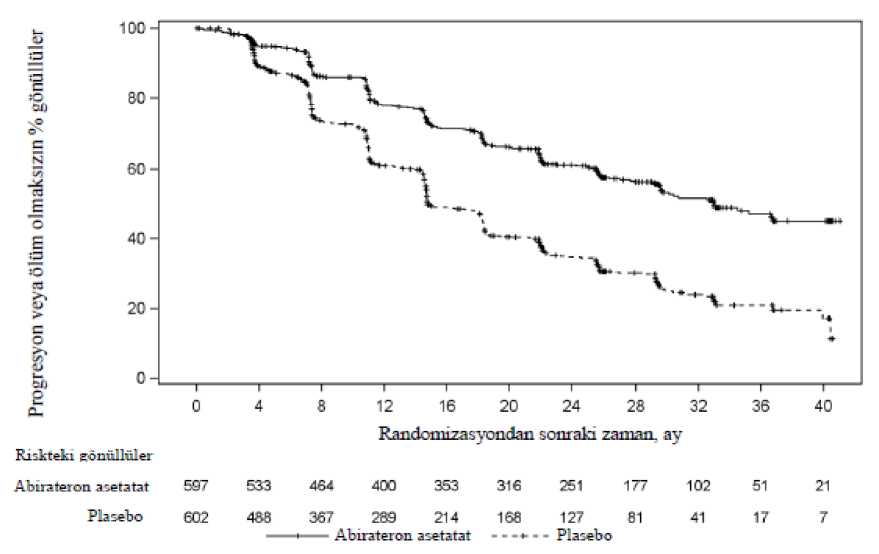
17 / 28Şekil 3: Kaplan Meier grafiğinde abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH agonistleri ya da öncesinde orşiektomi olan hastalardaki radyografikprogresyonsuz sağkalım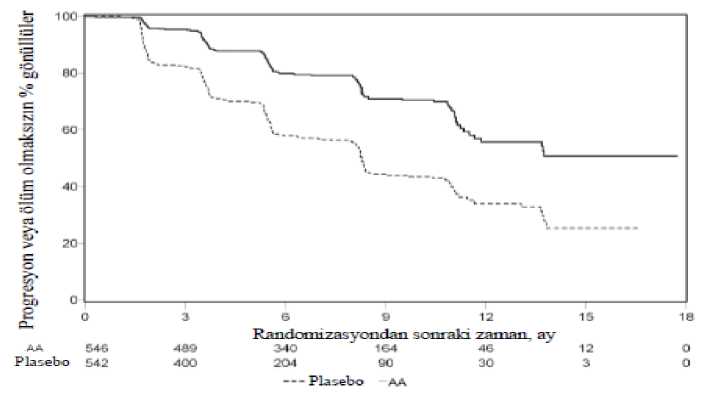
AA=abirateron asetatAncak, Genel Sağkalıma (OS) ilişkin ikinci ara analiz tarihine kadar hasta verisi toplanmaya devam edilmiştir. Araştırmacının rPFS üzerinde bir takip duyarlılık analizi olarak yaptığıradyografik inceleme Tablo 4 ve Şekil 4'te sunulmuştur. 271'i (% 50) abirateron asetat grubundan, 336'sı (% 62) plasebo grubundan olmak üzere toplam 607 hasta radyografik progresyon göstermiş veya ölmüştür. Abirateron asetat ile tedavi,plaseboya kıyasla radyografik progresyon riskini % 47'ye kadar azaltmıştır (TO = 0,530; % 95GA: [0,451; 0,623], p < 0,0001). Medyan rPFS, abirateron asetat grubunda 16,5 ay, plasebogrubunda ise 8,3 ay olmuştur.
18 / 28Şekil 4: Kaplan Meier grafiğinde abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH agonistleri ya da öncesinde orşiektomi olmuş hastalardaki radyografikprogresyonsuz sağkalım (İkinci ara analizdeki GS - Araştırıcı İncelemesi)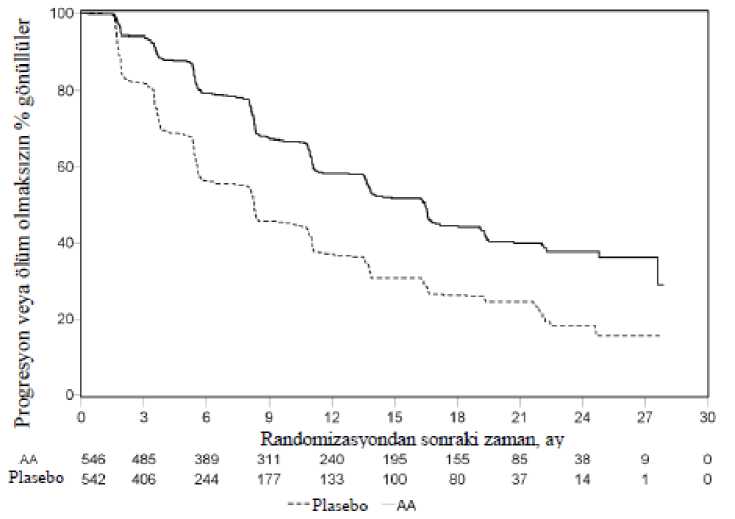
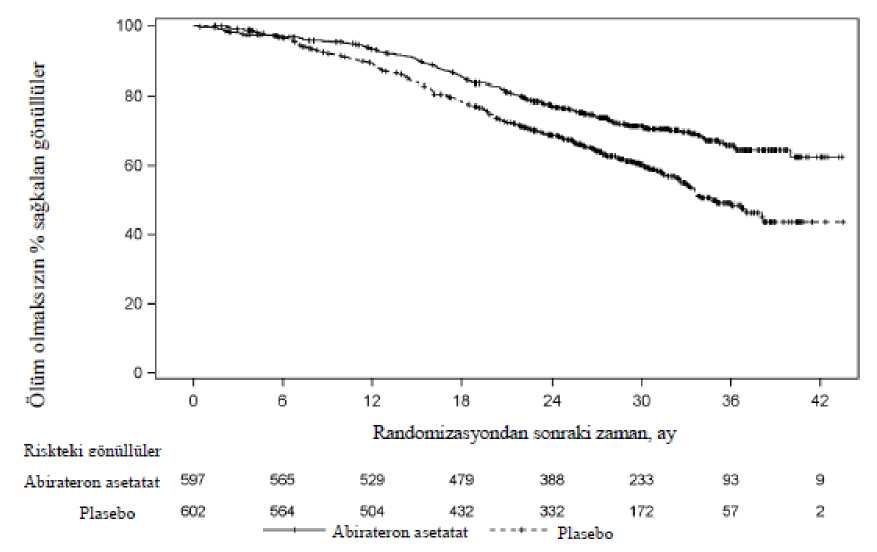
AA= abirateron asetat%25azaltarak plaseboya kıyasla daha uzun bir genel sağkalım sağladı (TO = 0,752; % 95 GA:[0,606; 0,934], p = 0,0097), ancak OS yeterli düzeye erişmemişti ve ara analiz sonuçlarıistatistiksel anlamlılık için önceden belirlenmiş sonlandırma sınırını karşılamıyordu (bkz. Tablo3). Bu ara analiz sonrasında, sağkalım takibine devam edildi.OS için planlanan nihai analiz 741 ölüm gözlendikten sonra yapıldı (medyan takip süresi 49 ay). Abirateron asetat ile tedavi edilen hastaların % 65'inin (354/546), plasebo ile tedavi edilenhastaların ise % 71'inin (387/542) öldüğü saptandı. Ölüm riskinde % 19,4'lük bir azalma ile, OSbakımından abiraterone astetat ile tedavi edilen grup lehine istatistiksel olarak anlamlı bir fayda(TO = 0,806; % 95 GA: [0,697; 0,931], p = 0,0033) ve 4,4 aylık medyan OS artışı sağlanmıştır(abirateron asetat 34,7 ay, plasebo 30,3 ay) (bkz. Tablo 5 ve Şekil 5). Bu iyileşme, plasebokolundaki hastaların % 44'ü müteakip tedavi olarak abiraterone asetat almış olmalarına rağmengösterilmiştir.
Şekil 5: Kaplan Meier grafiğinde abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH analogları ya da öncesinde orşiektomi olmuş hastalardaki genel sağkalım - nihaianaliz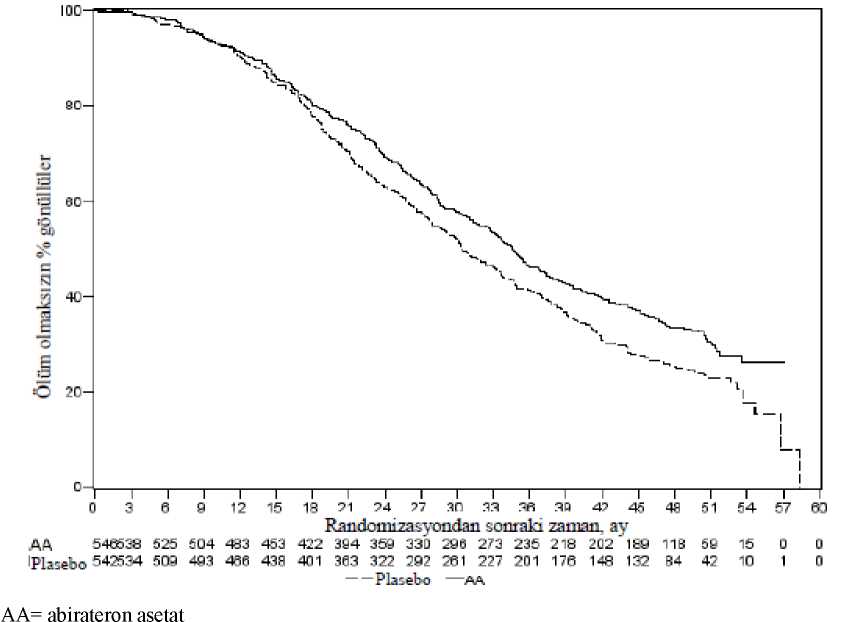
Genel sağkalım ve rPFS'de gözlenen artışlara ilave olarak, tüm sekonder sonlanım noktaları için plasebo karşısında abirateron asetat lehine aşağıdaki faydalar gösterilmiştir: PCWG2 kriterlerine göre PSA progresyonuna kadar geçen süre: PSA progresyonuna kadar geçen medyan süre abirateron asetat alan hastalar için 11,1 ay, plasebo alan hastalar için ise 5,6ay olmuştur (TO = 0,488; % 95 GA: [0,420; 0,568], p < 0,0001). PSA progresyonuna kadargeçen süre abirateron asetat tedavisi ile yaklaşık iki katına çıkmıştır (TO = 0,488). PSA yanıtıbelgelenmiş olan hastaların oranı abirateron asetat grubunda, plasebo grubuna oranla daha fazlaolmuştur (% 24 karşısında %62, p=0,0001). Yumuşak doku hastalığı ölçülebilir olan hastalarda,abirateron asetat ile anlamlı düzeyde artmış tam ve kısmı yanıt oranları görülmüştür.20 / 28Kanser ağrısı için opiat kullanımına kadar geçen süre: Nihai analiz sırasında, prostat kanseri ağrısı için opiat kullanımına kadar geçen medyan süre abirateron asetat alan hastalar için 33,4ay, plasebo alan hastalar için 23,4 ay olmuştur; (TO = 0,721; %95 GA: [0,614; 0,846], p <0,0001).Sitotoksik kemoterapi başlangıcına kadar geçen süre: Sitotoksik kemoterapi başlangıcına kadar geçen medyan süre abirateron asetat alan hastalar için 25,2 ay, plasebo alan hastalar için 16,8 ayolmuştur (TO = 0,580; % 95 GA: [0,487; 0,691], p < 0,0001). ECOG performans skorunda > 1 puan gerilemeye kadar geçen süre: ECOG performans skorunda > 1 puan gerilemeye kadar geçen medyan süre abirateron asetat alan hastalar için 12,3ay, plasebo alan hastalar için 10,9 ay olmuştur (TO = 0,821; % 95 GA: [0,714; 0,943], p =0,0053). Aşağıdaki çalışma sonlanım noktaları abirateron asetat tedavisi lehine istatistiksel olarak anlamlı bir üstünlük göstermiştir: Objektif yanıt: Objektif yanıt, RECIST kriterlerine göre ölçülebilir hastalığı olup tam veya kısmi yanıt elde eden hastaların oranı olarak tanımlanmıştır (hedef lezyon olarak değerlendirilebilmesiiçin başlangıçtaki lenf nodu büyüklüğü > 2 cm olması gereklidir). Başlangıçta ölçülebilirhastalığı olup objektif yanıt elde eden hastaların oranı abirateron asetat grubunda %36, plasebogrubunda ise % 16 olmuştur (p < 0,0001).Ağrı: Abirateron asetat tedavisi, plasebo ile karşılaştırıldığında, ortalama ağrı şiddeti progresyonunu anlamlı şekilde % 18'e kadar azaltmıştır (p = 0,0490). Progresyona kadar geçenmedyan süre abirateron asetat grubunda 26,7 ay, plasebo grubunda ise 18,4 ay olmuştur. FACT-P (Toplam Skor) anketinde kötüleşmeye kadar geçen süre: abirateron asetat tedavisi, plasebo ile karşılaştırıldığında, FACT-P (Toplam Skor) anketindeki kötüleşme riskini anlamlışekilde % 22'ye kadar azaltmıştır (p = 0,0028). FACT-P (Toplam Skor) anketinde kötüleşmeyekadar geçen süre abirateron asetat grubunda 12,7 ay, plasebo grubunda ise 8,3 ay olmuştur. Çalışma 301 (daha önce kemoterapi almış olan hastalar)Çalışma 301'e daha önceden dosetaksel kullanmış olan hastalar dahil edilmiştir. Bu kemoterapiden kaynaklanan toksisite tedavinin kesilmesine neden olmuş olabileceği için,hastaların dosetaksel tedavisi sırasında hastalık ilerlemesi göstermiş olması gerekmemiştir.Hastalar protokole tanımlanan radyografik ilerleme ve semptomatik ya da klinik ilerleme ilebirlikte PSA ilerlemesi oluncaya kadar (hastanın başlangıç/en düşük düzeyine göre doğrulanmış%25 artış) çalışma tedavilerine devam etmiştir. Daha önceden prostat kanseri için ketokonazoltedavisi gören hastalar bu çalışmaya alınmamıştır. Birincil etkililik sonlanım noktası genelsağkalım olmuştur. Çalışmaya alınan hastaların medyan yaşı 69 idi (yaş aralığı 39 - 95 yaş). Abirateron asetat ile tedavi edilen hastaların ırklarına göre dağılımı şöyleydi: 737'si (%93,2) beyaz ırktan, 28'i(%3,5) siyah ırktan, 11'i (%1,4) sarı ırktan ve geri kalan 14'ü (%1,8) ise diğer ırklardandı.Çalışmaya alınan hastaların %11'inin ECOG performans skoru 2 idi; %70'i için PSA ilerlemesi 21 / 28 olsun ya da olmasın, hastalık ilerlemesine dair radyografik kanıtlar mevcuttu; %70'i daha önce bir sitotoksik kemoterapi, %30'u ise iki sitotoksik kemoterapi almıştı. Abirateron asetat iletedavi edilen hastaların %11'inde karaciğer metastazı vardı. 552 ölüm gözlendikten sonra yapılan planlı bir analizde, abirateron asetat ile tedavi edilen hastaların %42'sinin (797 hastanın 333'ü), plasebo ile tedavi edilen hastaların ise %55'inin (398hastanın 219'u) öldüğü saptandı. Abirateron asetat ile tedavi edilen hastaların medyan genelsağkalımda istatistiksel olarak anlamlı bir artış görüldü (bkz. Tablo 6).
b Tehlike oranı tabakalandırılmış orantısal bir risk modeline göre hesaplanmıştır. Tehlike oranı < 1 abirateron asetat lehineTedavinin başlangıçtaki ilk birkaç ayından sonra her değerlendirme noktasında, plasebo ile tedavi edilip hayatta kalan hastaların oranıyla karşılaştırıldığında, Abirateron asetat ile tedaviedilen hastaların daha yüksek sağkalım oranına sahip olduğu belirlenmiştir (bkz. Şekil 6). 22 / 28Şekil 6: Kaplan Meier grafiğinde abirateron asetat veya plasebo ile birlikte prednizolon ve LHRH agonistleri ya da öncesinde orşiektomi olmuş hastalardaki genel sağkalım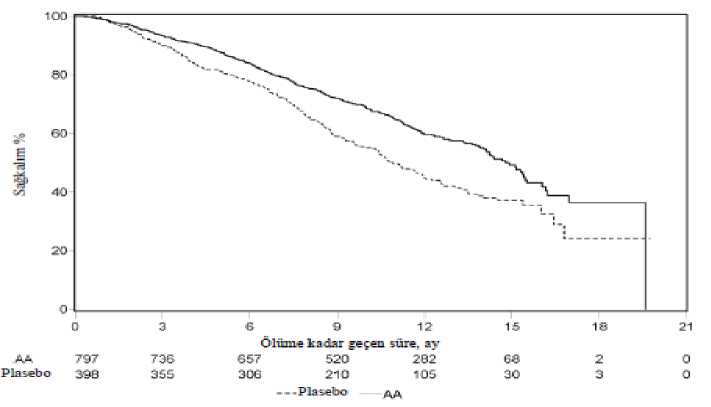 AA= abiraterone asetatAltgrup sağkalım analizleri abirateron asetat tedavisi için tutarlı bir sağkalım faydası gösterdi (bkz. Şekil 7). Şekil 7: Alt gruplara göre genel sağkalım: risk oranı ve %95'lik güven aralığı
Genel sağkalımda gözlenen artışa ek olarak, tüm ikincil sonlanım noktaları Abirateron asetat lehine idi ve aşağıda gösterildiği şekilde bu farklılıklar birden fazla test için ayarlandığındaistatistiksel olarak anlamlı idi: 23 / 28 Abirateron asetat alan hastalarda, plasebo alanlara göre istatistiksel olarak anlamlı düzeylerde daha yüksek PSA yanıt oranı (başlangıç değerinden >%50 olarak tanımlanmış) elde edildi,Abirateron asetat ile %38 iken, plaseboyla %10, p < 0,0001. Abirateron asetat ile tedavi edilen hastalarda, PSA ilerlemesine kadar olan medyan süre 10,2 ay iken, bu süre plasebo ile tedavi edilen hastalarda 6,6 ay idi (TO=0,580; %95 GA: [0,462; 0,728], p < 0,0001). Abirateron asetat ile tedavi edilen hastalarda medyan radyografik progresyonsuz sağkalım 5,6 ay iken, bu süre plasebo ile tedavi edilen hastalarda 3,6 ay idi (TO=0,673; %95 GA: [0,585; 0,776], p < 0,0001). Ağrı Ağrısında hafifleme olan hastaların oranı Abirateron asetat grubunda, plasebo grubuna oranla istatistiksel olarak anlamlı derecede daha fazlaydı (%44'e karşı %27, p=0,0002). Bir hastadadört hafta arayla yapılan iki ardışık değerlendirmede 24 saatlik bir sürede analjezik kullanımskorunda herhangi bir artış olmaksızın BPI-SF en kötü ağrı yoğunluğu skorunda başlangıca göreen az %30 azalma sağlanması ağrının hafiflemesi yanıtı olarak tanımlandı. Ağrı palyasyonu içinyalnızca başlangıç ağrı puanı > 4 olan hastalar değerlendirildi ve en az bir başlangıç-sonrası ağrıskoru analiz edildi (N=512). Abirateron asetat ile tedavi edilen hastalarda, plasebo ile tedavi edilenlere oranla daha düşük oranda ağrı artışı görüldü: 6. ayda Abirateron asetat tedavisi alanların %22'sine karşılık plaseboalanların %28'i, 12. ayda Abirateron asetat tedavisi alanların %30'una karşılık plasebo alanların%38'i ve 18. ayda Abirateron asetat tedavisi alanların %35'ine karşılık plasebo alanların%46'sında ağrıda artış vardı. Ağrı artışı boyunca, iki ardışık değerlendirmede son 24 saatlik birsürede analjezik kullanım skorunda herhangi bir azalma olmaksızın BPI-SF en kötü ağrıyoğunluğu skorunda başlangıca göre en az %30 veya daha fazla bir artış ya da iki ardışıkdeğerlendirmede analjezik kullanım skorunda %30 veya daha fazla bir artış olarak tanımlandı.25. persentilde ağrı ilerlemesine kadar geçen süre Abirateron asetat grubunda 7,4 ay iken,plasebo grubunda 4,7 ay idi. İskelet sistemiyle ilişkili olaylar 6. ayda, 12'inci ayda ve 18'inci ayda Abirateron asetat grubunda iskelet sistemiyle ilişkili olaygörülen hasta oranı plasebo ile karşılaştırıldığında daha düşüktü (sırasıyla 6. ayda %18'e karşı%28, 12. ayda %30'a karşı %40 ve 18. ayda %35'e karşı %40). 25. persentilde iskelet sistemiyleilişkili ilk olayın görülme süresi Abirateron asetat grubunda 9,9 ay ile, 4,9 ay olan kontrolgrubundan iki kat daha uzundu. Patolojik kırık, spinal kord basısı, kemiğe palyatif radyoterapiveya kemiğe yönelik cerrahi girişim iskelet sistemiyle ilişkili olay olarak tanımlandı. Pediyatrik popülasyon Pediyatrik popülasyonda prostat kanseri görülmediği için RATEBİR'inpediyatrik hastalarda kullanımı bulunmamaktadır. RATEBİR'e ait pediatrik popülasyonda klinik etkililik vegüvenlilik verisi yoktur (bkz. Bölüm 4.2). 24 / 28 5.2. Farmakokinetik özelliklerAbirateron asetat uygulamasından sonra, abirateron farmakokinetiği sağlıklı gönüllülerde, ilerlemiş metastatik prostat kanserli hastalarda ve kanser hastası olmayan karaciğer veya böbrekyetmezliği olan gönüllülerde çalışılmıştır. Abirateron asetat in vivoolarak hızla bir androjenbiyosentez inhibitörü olan abiraterona dönüşür (bkz. Bölüm 5.1).Emilim:Abirateron asetat açlık durumunda oral yoldan uygulandıktan sonra, en yüksek abirateron konsantrasyonlarına yaklaşık 2 saatte ulaşılır. Abirateron asetatın yemekle birlikte alınması, yemeğin yağ içeriğine bağlı olarak, aç karına alınmasına oranla 10 kata kadar (EAA açısından) ve 17 kata kadar (Cmaks açısından) daha fazlaortalama sistemik abirateron maruziyetine yol açar. Yemeklerin içerik ve bileşimindeki normalfarklılıklar göz önüne alındığında, RATEBİR'in yemeklerle birlikte alınması oldukça değişkenmaruziyetler ile sonuçlanma potansiyeline sahiptir. Bu nedenle, RATEBİRyemeklerle birliktealınmamalıdır. RATEBİR tabletler aç karnına günde bir kez tek doz olarak alınmalıdır.RATEBİRyemekten en az iki saat sonra alınmalıdır ve RATEBİR alındıktan sonra en az bir saatyemek yenmemelidir Tabletler bütün olarak suyla yutulmalıdır (bkz. Bölüm 4.2). Dağılım:İnsan plazmasında 14C-abirateronun proteine bağlanma oranı %99,8'dir. Görünür dağılım hacmi yaklaşık 5,630 litre olup, abirateronun periferik dokulara yoğun bir şekilde dağıldığını gösterir. Biyotransformasyon:14C-abirateron asetat kapsül şeklinde oral yoldan alındıktan sonra, abirateron asetat abiraterona hidrolize olur ve daha sonra birincil olarak karaciğerde olmak üzere sülfasyon, hidroksilasyonve oksidasyona uğrar. Dolaşımdaki radyoaktivitenin büyük çoğunluğu (yaklaşık %92)abirateronun metabolitleri halinde bulunur. Belirlenebilen 15 metabolitten iki temel metabolit,her biri toplam radyoaktivitenin yaklaşık %43'ünü temsil eden abirateron sülfat ve N-oksitabirateron sülfattır. Eliminasyon:Sağlıklı gönüllülerden elde edilen verilere göre, plazmadaki abirateronun yarılanma süresi yaklaşık 15 saattir. 1,000 mg 14C-abirateron asetatın oral yoldan alınmasından sonra radyoaktifdozun yaklaşık %88'i feçeste ve yaklaşık %5'i idrarda belirlenmiştir. Feçeste bulunan anabileşikler değişmemiş abirateron asetat ve abiraterondur (sırasıyla uygulanan dozun yaklaşık%55 ve %22'si). Hastalardaki karekteristik özelliklerKaraciğer yetmezliği olan hastalarAbirateron asetatın farmakokinetiği hafif ya da orta şiddette karaciğer yetmezliği olan hastalar (sırasıyla Child-Pugh Sınıf A ve B) ile sağlıklı kontrollerde çalışılmıştır. 1,000 mg'lık tek biroral doz sonrası abiraterona sistemik maruziyet hafif ve orta şiddette karaciğer yetmezliği olanhastalarda sırasıyla %11 ve %260 oranında artmıştır. Abirateronun ortalama yarılanma ömrü hafif karaciğer yetmezliği olan hastalarda yaklaşık 18 saate ve orta şiddette karaciğer yetmezliği 25 / 28 olan hastalarda ortalama 19 saate uzamaktadır. Bir başka çalışmada, abirateronun farmakokinetiği daha önceden ciddi karaciğer yetmezliği (Child-Pugh Sınıf C) olan (n=8) ve normal hepatik fonksiyonu olan 8 sağlıklı birey üzerindeincelenmiştir. Abirateronenin sistemik etkisi (EAA) yaklaşık %600 oranında artış göstermiş veserbest ilacın etkisi şiddetli karaciğer bozukluğu olanlarda normal karaciğer fonksiyonu olanlaragöre %80 oranında artmıştır. Daha önceden hafif karaciğer yetmezliği olan hastalarda doz ayarlamasına gerek yoktur. RATEBİR kullanımı faydanın olası riskten açıkça ağır bastığı, orta şiddette karaciğer yetmezliğiolan hastalarda dikkatle değerlendirilmelidir (bkz. Bölüm 4.2 ve 4.4). RATEBİR ağır karaciğeryetmezliği olan hastalarda kullanılmamalıdır (bkz. Bölüm 4.2, 4.3 ve 4.4). RATEBİR tedavisi sırasında hepatotoksisite gelişen hastalarda, tedavinin durdurulması ve doz ayarlaması gerekebilir (bkz. Bölüm 4.2 ve 4.4). Böbrek yetmezliği olan hastalarStabil bir hemodiyaliz programında olan son dönem böbrek yetmezliği olan hastalar ile böbrek fonksiyonları normal olan eşlenmiş kontrol hastalarında Abirateron asetatın farmakokinetiğikarşılaştırılmıştır. Diyaliz programında olan son dönem böbrek yetmezliği olan hastalarda 1,000mg'lık tek bir oral doz sonrası abiraterona sistemik maruziyette artış olmamıştır. Ağır böbrekyetmezliği dahil, böbrek yetmezliği olan hastalarda RATEBİR uygulanması sırasında dozuazaltmaya gerek yoktur (bkz. Bölüm 4.2). Ancak prostat kanseri ve ağır böbrek yetmezliği olanhastalarla ilgili klinik deneyim bulunmamaktadır. Bu tür hastalarda dikkatli olunması tavsiyeedilir. 5.3. Klinik öncesi güvenlilik verileriTüm hayvan toksisite çalışmalarında, dolaşımdaki testesteron düzeyleri anlamlı derecelerde azalmıştır. Buna bağlı olarak üreme organlarıyla adrenal, hipofiz ve meme bezlerininağırlıklarında azalma ile morfolojik ve/veya histopatolojik değişiklikler gözlenmiştir. Tümdeğişiklikler tamamen ya da kısmen geri döndürülebilir nitelikteydi. Üreme organları veandrojene duyarlı organlardaki değişiklikler abirateronun farmakolojisiyle uyumludur.Tedaviyle ilişkili tüm hormonal değişiklikler eski haline dönmüştür veya 4 haftalık birtoparlanma dönemi sonunda düzeldiği gösterilmiştir. Erkek ve dişi sıçanlarda yapılan fertilite çalışmalarında, abirateron asetat fertiliteyi azaltmış, fakat abirateron asetat kesildikten sonra 4 ila 16 haftada fertilite tamamen eski haline dönmüştür. Sıçanlarda yapılan bir gelişim toksisitesi çalışmasında, abirateron asetat azalmış fetüs ağırlığında azalma ve sağkalım da dahil olmak üzere gebeliği etkilemiştir. Abirateron asetatteratojenik olmasa da dış genital organlarda etkiler görülmüştür. Sıçanlarda yürütülen bu fertilite ve gelişim toksisitesi çalışmalarında, tüm etkiler abirateronun farmakolojik aktivitesine bağlı olmuştur. 26 / 28 Tüm hayvan toksikoloji çalışmalarında görülen üreme organlarındaki değişiklikler dışında, klasik güvenlilik farmakolojisi, tekrarlayan doz toksisitesi, genotoksisite ve karsinojenikpotansiyel çalışmalarından elde edilen klinik dışı veriler insanlar için özel bir tehlikeyigöstermemiştir. Transjenik farelerde (Tg.rasH2) yapılan 6 aylık bir çalışmada abirateron asetatkarsinojenik bulunmamıştır. Sıçanlarda yapılan 24 aylık bir karsinojenite çalışmasında,abirateron asetat testislerde interstisyel hücre neoplazmalarının insidansını arttırmıştır. Bu bulguabirateronun farmakolojik etkisine bağlı ve sıçanlara özgü olarak değerlendirilmiştir. Abirateronasetat dişi sıçanlarda karsinojenik değildir. Çevresel Risk Değerlendirmesi Abirateron etkin maddesi, sucul ortam (özellikle balıklar) için çevresel bir risk oluşturur. 6. FARMASÖTİK ÖZELLİKLERİ6.1. Yardımcı maddelerin listesiLaktoz monohidrat (inek sütü kaynaklı) Mikrokristalin selüloz Kroskarmelloz sodyumPovidon K-30Sodyum lauril sülfatKolloidal silikon dioksitMagnezyum stearat 6.2. GeçimsizliklerBilinen herhangi bir geçimsizliği bulunmamaktadır. 6.3. Raf ömrü24 ay 6.4. Saklamaya yönelik özel tedbirler25 °C'nin altındaki oda sıcaklığında saklanmalıdır. Çocukların göremeyeceği, erişemeyeceği yerlerde ve ambalajında saklayınız. 6.5. Ambalajın niteliği ve içeriğiAlu-PVC/PE/PVDC blister içeren karton kutularda 60 ve 120 tablet. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerEtki mekanizmasına dayanarak, bu tıbbi ürün gelişmekte olan fetüse zarar verebilir, bu yüzden gebe olan ya da gebe olma olasılığı bulunan kadınlar RATEBİR ile korunmasız temas etmemeli;örneğin eldiven kullanmalıdır (bkz. Bölüm 4.6). Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir.Bu tıbbi ürün akuatik ortam için bir risk oluşturabilir (bkz. Bölüm 5.3). 27 / 28 7. RUHSAT SAHİBİWorld Medicine İlaç San. ve Tic. A.Ş. Bağcılar /İstanbul 8. RUHSAT NUMARASI2022/499 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 01.09.2022 Ruhsat yenileme tarihi: - 10. KÜB'ÜN YENİLENME TARİHİ28 / 28 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.