Haemocomplettan P 1 G I.v. Enjeksiyonluk/infüzyonluk Çözelti Tozu Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ^ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIHAEMOCOMPLETTAN P 1 g I.V. enjeksiyonluk/infüzyonluk çözelti tozu Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her flakon 1 g konsantre insan fibrinojeni (koagülasyon Faktör I) içerir.50 ml enjeksiyonluk su ile sulandırılmış ürün yaklaşık 20 mg/ml insan fibrinojeni içerir. Yardımcı maddeler:Sodyum klorür 200-350 mg Sodyum sitrat 50-100 mg Sodyum hidroksit (az miktarda pH ayarlayıcı) Her 1 g fibrinojen 164 mg'a kadar sodyum içerir. Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMI.V. enjeksiyonluk/infüzyonluk çözelti tozu Beyaz toz Hazırlanmış çözelti nötr pH değerine neredeyse renksiz veya sarımsı, berrak ila hafif opalesan bir çözelti. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarKanama diyatezinin tedavisi ve profilaksisinde, aşağıdaki durumlarda kullanılır; Konjenital hipo-, dis- ya da afibrinojenemide Aşağıdaki sebeplerle sonradan edinilmiş hipofibrinojenemide: - Şiddetli karaciğer parankima hasarının sonucu olarak sentez bozuklukları 1 - Artmış damar içi tüketimde (Örnek; dissemine intravasküler koagülasyon vehiperfibrinoliz) - Artmış fibrinojen kayıplarında Bir defibrinasyon sendromu ile ilişkin en önemli klinik durumlar şunlardır: Doğum komplikasyonları, akut lösemiler özellikle promyelositik lösemi, karaciğer sirozu, intoksikasyonlar, ağır yaralanmalar/yanıklar, transfüzyon hataları sonrası hemoliz, cerrahimüdahaleler, enfeksiyonlar, sepsis, tüm şok türleri, akciğer, pankreas, uterus ve prostattümörleridir. 4.2. Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:HAEMOCOMPLETTAN P'nin dozu ve tedavinin süresi hastanın klinik durumuna, kanamanın derecesine ve yerine, hastalığın ciddiyetine göre bu konuda uzman doktorlartarafından belirlenmelidir. Özellikle aşırı doz kullanımını önlemek için, laboratuvarda kontrol yoluyla sübstitüsyon tedavisinin yakından izlenmesi gerekir (Clauss yöntemi gibi fibrinojen aktivitesinin tespitineyönelik uygun yöntemler kullanılarak). Kişiye özel dozu hesaplamak için (fonksiyonel) fibrinojen düzeyi belirlenmelidir; ilacın miktarı ve uygulama sıklığı, kullanılan diğer replasman tedavileri ile hastanın klinikdurumunun sürekli izlenmesi ve plazma fibrinojen düzeyinin düzenli olarak ölçülmesiyle herhasta için özel olarak belirlenmelidir. Kritik plazma fibrinojen düzeyi yaklaşık 0,5-1 g/L olup, bu düzeyin altında hemoraji meydana gelebilir. Normal plazma fibrinojen düzeyi 1,5 - 4,5 g/L aralığındadır. Çocuklarda doz çocuğun vücut ağırlığına ve klinik ihtiyacına göre seçilmelidir. Majör cerrahi girişim durumunda replasman tedavisinin koagülasyon testiyle hassas bir şekilde izlenmesi önemlidir. 2 1. Kanama eğilimi olan ve konjenital hipofibrinojenemi, disfibrinojenemi veyaafibrinojenemisi olan hastalarda profilaksi. Cerrahi prosedürler sırasında aşırı kanamayı önlemek amacıyla, fibrinojen düzeylerini 1 g/L'ye artırmak ve hemostaz sağlanıncaya kadar bu düzeyde tutmak ve yaraiyileşmesi tamamlanıncaya kadar 0,5 g/L'nin üzerinde tutmak amacıyla profilaktiktedavi önerilir. Bir kanama epizodunun tedavisinde veya cerrahi prosedür sırasındadoz aşağıdaki şekilde hesaplanmalıdır: Fibrinojen dozu = [Hedef seviye (g/L) - ölçülen seviye (g/L)](mg/kg vücut ağırlığı) 0,017 (g/L her bir mg/kg vücut ağırlığı başına) Daha sonraki pozoloji (dozlar ve enjeksiyon sıklığı) hastanın klinik durumuna ve laboratuvar sonuçlarına göre ayarlanmalıdır. Fibrinojenin biyolojik yarı ömrü 3-4 gündür. Bu sebeple, tüketim yokluğunda, insan fibrinojeni ile tekrarlı tedavi çoğunlukla gerekli görülmemektedir. Bir profilaktikkullanım için tekrarlı uygulama sırasında meydana gelen birikim göz önüne alınarak,doz ve uygulama sıklığı belirli bir hasta için doktorun terapötik amaçlarına görebelirlenmelidir. 2. Kanama tedavisiYetişkinler Perioperatif kanama için, genellikle 2 g (veya 30 mg/kg vücut ağırlığı) uygulanır, daha sonra gereken infüzyonlar yapılır. Şiddetli hemoraji durumunda (örn: obstetrikkullanım/plasentanın prematüre ayrılması) büyük miktarlarda (4-8 g) fibrinojenkullanımı gerekebilir. Çocuklar Doz vücut ağırlığına ve çocuğun klinik ihtiyacına göre belirlenmelidir ancak genellikle 20-30 mg/kg'dir. 3 Uygulama şekli:HAEMOCOMPLETTAN P intravenöz infüzyon veya enjeksiyon yolla uygulanır. HAEMOCOMPLETTAN P kullanılmadan önce çözelti berrak veya hafif opak olmalıdır. Filtre edildikten/çekildikten sonra hazırlanmış ürün uygulanma öncesi çökelti maddesi veyarenk bozulmasına karşı göz ile incelenmelidir. Artık içeren (kalıntı/parçacık) veya berrakolmayan çözeltilerin kullanılmaması gerekir. Hastanın rahat olacağı hızda damar içine enjekte edilir veya infüzyon şeklinde verilir. Enjeksiyon veya infüzyonun verilme hızı veya enjeksiyon hızı dakikada 5 mL'yigeçmemelidir. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karaciğer yetmezliği:HAEMOCOMPLETTAN P'nin böbrek/karaciğer yetmezliği olan hastalarda kullanımı ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle HAEMOCOMPLETTAN P'nin buhastalarda kullanımında tedbirli olunmalı ve hasta açısından yarar/zarar değerlendirmesiyapıldıktan sonra kullanılmalıdır. Pediyatrik popülasyon:HAEMOCOMPLETTAN P'nin çocuklarda doz ayarlaması ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle HAEMOCOMPLETTAN P'nin çocuklarda kullanımında tedbirli olunmalıve çocuk açısından yarar/zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır. Çocuklarda doz vücut ağırlığına ve çocuğun ihtiyacına göre seçilecektir. Geriyatrik popülasyon:HAEMOCOMPLETTAN P'nin yaşlılarda kullanımı ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle HAEMOCOMPLETTAN P'nin bu hastalarda kullanımında tedbirliolunmalı ve hasta açısından yarar/zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır. 4.3. KontrendikasyonlarHAEMOCOMPLETTAN P, içeriğinde bulunan etkin maddeye ya da herhangi bir yardımcı maddeye aşırı duyarlılığı bulunan bireylerde kontrendikedir. 4 Yaşamı tehdit eden kanama durumları haricinde, belirgin tromboz ya da miyokard enfarktüsünde kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleriVirüs güvenliliğiİnsan kanından veya plazmasından hazırlanan tıbbi ilaçların kullanımı sonucu oluşan enfeksiyonların önlenmesi için alınan standart önlemler, donörlerin seçimini, bireyselbağışların ve plazma havuzlarının spesifik enfeksiyon göstergeleri için taranmasını vevirüslerin inaktivasyonu/yok edilmesi için etkili üretim basamaklarının eklenmesiniiçerir. Buna rağmen, insan kanından veya plazmasından hazırlanan tıbbi ilaçlaruygulandığı zaman, bulaşıcı ajanların bulaşma olasılığı tamamen engellenemez. Buayrıca bilinmeyen veya yeni görülen virüsler ve patojenler için de geçerlidir.Alınan önlemlerin İnsan immün yetmezlik virüsü (HIV), hepatitis B virüsü (HBV) ve hepatitis C virüsü (HCV) gibi zarflı virüsler ve zarfsız hepatitis A virüsü (HAV) içinetkili olduğu düşünülmektedir.Parvovirus B19 gibi zarflı olmayan virüslere karşı alınan önlemler sınırlı sayıda olabilir.Parvovirus B19 enfeksiyonu, hamile kadınlar (fetal infeksiyon) ve immün yetmezlik ya da kırmızı kan hücre üretiminde artış (hemolitik anemi gibi) olan hastalar için tehlikeliolabilir.Düzenli/tekrarlanan sürelerde insan kanından veya plazmasından hazırlanan tıbbi ilaçları kullanan hastalarda, uygun aşıların (hepatit A ve hepatit B) yaptırılması düşünülmelidir. HAEMOCOMPLETTAN P her uygulandığında, hastayla ürünün seri numarası arasındaki bağlantının korunabilmesi için, ürünün adı ve seri numarası kaydedilmelidir. Alerjik ya da anafilaktik reaksiyonlar oluşursa enjeksiyon/infüzyon hemen durdurulmalıdır. Anafilaktik şok durumunda, şok için standart medikal tedavi uygulanmalıdır. 5 Konjenital eksikliği olan hastalar insan fibrinojeniyle, özellikle yüksek doz veya tekrarlanan dozlarla, tedavi edildiğinde tromboz riski mevcuttur. İnsan fibrinojeni verilen hastalar,tromboz bulgu ve belirtileri açısından yakından takip edilmelidir. Koroner kalp hastalığı veya miyokard infarktüsü öyküsü olan hastalarda, karaciğer hastalığı olan hastalarda, peri- veya post- operatif hastalarda, yenidoğanlarda veya tromboembolikolaylar veya dissemine intravasküler koagülasyon riski taşıyan hastalarda; insan plazmafibrinojeni ile tedavinin potansiyel yararı tromboembolik komplikasyon risklerine karşıtartışılmalıdır. Ayrıca, dikkatli olunmalıdır ve yakın takip gerçekleştirilmelidir. Edinilmiş hipofibrinojenemi, tüm koagülasyon faktörlerinin (sadece fibrinojen değil) ve inhibitörlerin düşük plazma konsantrasyonlarıyla ilişkilidir ve bu nedenle koagülasyonfaktörlerini içeren kan ürünleriyle tedavi (fibrinojen konsantresi uygulanarak veyauygulanmayarak) değerlendirilmelidir. Koagülasyon sisteminin dikkatli takibi gereklidir. Diğer konjenital eksikliklerde koagülasyon faktörleri ile replasman tedavisi durumunda antikor reaksiyonları gözlenmiştir, fakat fibrinojen ile ilgili halihazırda veri mevcut değildir. HAEMOCOMPLETTAN P, her 1 g fibrinojende 164 mg (7,1 mmol)'a kadar sodyum ihtiva eder. Bu durum kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriHAEMOCOMPLETTAN P için hiçbir etkileşim çalışması yapılmamıştır. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin hiçbir klinik etkileşim çalışması yapılmamıştır. Pediyatrik popülasyonPediyatrik popülasyona ilişkin hiçbir klinik etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyon Genel tavsiyeGebelik kategorisi: C 6 Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)HAEMOCOMPLETTAN P'nin çocuk doğurma potansiyeli üzerindeki etkilerine ilişkin herhangi bir veri mevcut değildir. Gebelik dönemiHAEMOCOMPLETTAN P ile hayvan üreme çalışmaları yürütülmemiştir (bkz. bölüm 5.3.). Etkin madde insan kaynaklı olduğu için; hastanın kendi proteiniyle aynı şekilde katabolizeolur. İnsan kanının bu fizyolojik bileşenlerinin üreme veya fetüs üzerinde advers etkilereneden olması beklenmez. İnsan gebeliğinde kullanımına ilişkin HAEMOCOMPLETTAN P'nin güvenliliği kontrollü klinik çalışmalarda kanıtlanmamıştır. Obstetrik komplikasyonların tedavisinde fibrinojen ürünleriyle edinilen klinik deneyim, gebelik süresince veya fetüsün veya yenidoğanın sağlığı üzerinde zararlı etkilerinbeklenmeyeceğini öne sürmektedir. Laktasyon dönemiHAEMOCOMPLETTAN P'nin anne sütüne geçip geçmediği bilinmemektedir. İnsan plazma fibrinojen ürünlerinin laktasyon sırasında kullanımına ilişkin güvenliliği kontrollü klinikçalışmalarda kanıtlanmamıştır. Emzirilen çocuklara yönelik risk göz ardı edilemez. Emzirmenin durdurulup durdurulmayacağına veya HAEMOCOMPLETTAN P tedavisinin durdurulupdurdurulmayacağına, emzirmenin çocuk için yararı ve tedavinin anne için yararı göz önündebulundurularak bir karar verilmelidir. Üreme yeteneği/FertiliteHAEMOCOMPLETTAN P'nin fertilite üzerindeki etkilerine ilişkin herhangi bir veri mevcut değildir. 4.7. Araç ve makine kullanımı üzerindeki etkilerHAEMOCOMPLETTAN P'nin araç ve makine kullanma yeteneği üzerine bir etkisi yoktur. 7 4.8. İstenmeyen etkilerAdvers ilaç reaksiyonlarının (ADR) tablo halinde listesi Bu tablo, klinik çalışmalar ve pazarlama sonrası deneyimlerden elde edilen advers reaksiyonların birleşimidir. Tabloda sunulan sıklıklar, diğer cerrahi prosedürlerle diğer cerrahiprosedürler olmaksızın aortik cerrahide gerçekleştirilen iki firmanın sponsor olduğu klinikçalışmalarda (BI3023-2002 (N=61) ve BI3023_3002 (N=152)) aşağıdaki sıralamaya görehavuzlanmış analizlere dayanmaktadır: (Çok yaygın >1/10, yaygın (>1/100 ila <1/10, yaygınolmayan >1/1.000 ila <1/100, seyrek >1/10.000 ila <1/1.000, çok seyrek (<1/10.000),bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Pazarlama sonrası advers ilaç reaksiyonları için, raporlama kriteri bilinmeyen olarak kategorilenmiştir. Bu çalışmaların aort ameliyatları olan sınırlı bir popülasyonda yürütüldüğügöz önüne alındığında; bu çalışmalarda gözlemlenen advers ilaç reaksiyon oranları, klinikuygulamada gözlenen oranları yansıtmayabilir ve çalışılan endikasyon dışındaki klinikortamlar için bilinmemektedir.
Bulaşıcı ajanların güvenliliği konusunda 4.4. Özel kullanım uyarıları ve önlemleri bölümüne bakınız. 8 Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr;e- posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99) 4.9. Doz aşımı ve tedavisiHAEMOCOMPLETTAN P'nin doz aşımından sakınmak için tedavi süresince hastanın plazma fibrinojen seviyesinin düzenli izlemi gereklidir (bkz. Bölüm 4.2). Doz aşımı durumunda, tromboembolik komplikasyonların gelişme riski artabilir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: Antihemorajikler, insan fibrinojeni ATC kodu: B02B B01 Etki mekanizmasıİnsan fibrinojeni (koagülasyon faktör I); trombin, kalsiyum iyonları ve aktive edilmiş koagülasyon faktör XIII (FXIIIa) varlığında üç boyutlu, stabil ve elastik yapılı fibrinhemostatik pıhtısına dönüşür. İnsan fibrinojeni uygulaması plazma fibrinojen düzeyini artırır ve fibrinojen eksikliği bulunan hastalardaki koagülasyon bozukluğunu geçici olarak düzeltir. Pivot Faz II çalışmada tek dozluk farmakokinetik özellikleri incelenmiş (bkz. Bölüm 5.2 Farmakokinetik özellikler) ve alternatif sonlanma noktası maksimum pıhtı sıkılığı (MCF) ilegüvenlilik verileri sağlanmıştır. Her hasta için, 70 mg/kg v.a. HAEMOCOMPLETTAN P dozu uygulanmadan önce (başlangıç) ve sonra MCF belirlenmiştir. HAEMOCOMPLETTAN P'nin, tromboelastometriile ölçüldüğü üzere konjenital fibrinojen eksikliğinde (afibrinojenemi) pıhtı sıkılığınıartırmada etkili olduğu belirlenmiştir. Akut kanama epizodlarında hemostatik etkililiği veMCF ile korelasyonu, pazarlama sonrası çalışmada doğrulanmaktadır. 9 5.2. Farmakokinetik özelliklerGenel özelliklerİnsan plazma flbrinojeni, insan plazmasının normal bir bileşenidir ve endojen fibrinojen olarak etki gösterir. Plazmada, fibrinojenin biyolojik yarılanma ömrü 3 ila 4 gündür.Bozunmayla ilgili olarak HAEMOCOMPLETTAN P endojen fibrinojen olarak etki gösterir. HAEMOCOMPLETTAN P intravenöz olarak uygulanır ve plazma konsantrasyonu hemen uygulanan doza cevap verir. Bir farmakokinetik çalışmada konjenital afibrinojenemi hastalarında insan fibrinojeni konsantresi uygulaması öncesi ve sonrasındaki tek dozluk farmakokinetik özelliklerincelenmiştir. Bu ileriye dönük, açık etiketli, kontrolsüz, çok merkezli çalışmaya 8 ila 61 yaşaralığında 5 kadın ve 10 erkek katılmıştır (2 çocuk, 3 adolesan, 10 yetişkin). Ortalama doz77,0 mg/kg vücut ağırlığı idi (aralık: 76,6 - 77,4 mg/kg). Başlangıçtaki ve infüzyon tamamlandıktan 14 gün sonraki fibrinojen aktivitesini belirlemek üzere 15 hastadan (14'ü ölçülebilir) kan numunesi alınmıştır. Ayrıca, artan in vivo gerikazanım (IVR) (mg/kg vücut ağırlığı dozuna göre fibrinojen plazma düzeylerindekimaksimum artış olarak tanımlanır), infüzyon sonrasındaki 4 saate kadar elde edilendüzeylerden belirlenmiştir. Ortalama artan IVR, mg/kg v.a. dozunda 1,7 mg/dL (aralık: 1,302,73) idi. Bu çalışmanın farmakokinetik sonuçları aşağıda yer almaktadır.
t1/2 = Terminal eliminasyon yarılanma ömrü h = Saat Cmaks = 4 saat içerisindeki maksimum konsantrasyon EAA = Eğri altı alanıCl = Klerens MRT = Ortalama kalış süresi Vss = Kararlı haldeki dağılım hacmiSD = Standard sapmaIVR = In vivo geri kazanım 10 Emilim:HAEMOCOMPLETTAN P intravenöz olarak uygulanır ve plazma konsantrasyonu hemen uygulanan doza cevap verir. Uygulama yeri açısından (intravenöz) ilaç direkt kana karışır.Cmaks 140 ± 27 (100-210) mg/dL' dir. Kilogram başına 70 mg doz için EAA 124,3 ± 24,16(81,73-156,40) mg*sa/mL' dir. Dağılım:Fibrinojen konsantrelerinin dağılımları vasküler kompartımanlarla sınırlandırılmıştır. Ortalama vücutta alıkonma zamanı (MRT) 92,8 ± 20.11 (66,14-126,44) saat'tir. Kararlıdurumdaki dağılım hacmi 52,7 ± 7,48 (36,22-67,67) mL/kg' dır. Biyotransformasyon:HAEMOCOMPLETTAN P'nin degradasyonu endojen plazma koagülasyon Faktor I gibidir. Eliminasyon:Ortalama yarılanma ömrü 78,7 ± 18,13 (55,73-117,26) saattir. Klerensi 0,59 ± 0,13 (0,450,86) mL/h/kg'dır. Doğrusallık / Doğrusal olmayan durum:Doz cevap ilişkisi doğrusaldır. 5.3. Klinik öncesi güvenlilik verileriKonvensiyonel güvenlilik farmakolojisi ve tek doz toksisite çalışmalarına dayanarak, klinik dışı veriler insanlar için özel bir tehlike ortaya koymamaktadır. Tekrarlı doz uygulamaları ile gerçekleştirilen klinik öncesi çalışmalar (kronik toksisite, kanserojenite ve mutagenisite) heterolog insan proteinlerinin uygulanmasını takiben antikorgelişimi nedeniyle geleneksel hayvan modellerinde gerçekleştirilemez. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiİnsan albumini Sodyum klorürL-arjinin hidroklorürSodyum sitrat Sodyum hidroksit (pH ayarlayıcı) Enjeksiyonluk su 11 6.2. GeçimsizliklerHAEMOCOMPLETTAN P, Bölüm 6.6'da belirtilenler haricinde diğer tıbbi ürünlerle, seyrelticilerle veya çözücülerle karıştırılmamalıdır. Oda sıcaklığındaki sulandırılmışçözeltinin intravenöz uygulaması için standart bir infüzyon seti önerilmektedir. 6.3. Raf ömrü60 ay Sulandırılmış ürünün fizikokimyasal stabilitesi oda sıcaklığında (maksimum 25°C) 8 saat süreyle kanıtlanmıştır. Mikrobiyolojik açıdan, ürün sulandırmayı takiben hemenkullanılmalıdır. Sulandırılmış ürünün hemen kullanılmaması durumunda; saklama odasıcaklığında (maksimum 25°C) 8 saatten uzun olmamalıdır. Sulandırılmış ürün buzdolabındasaklanmamalıdır. 6.4. Saklamaya yönelik özel tedbirlerHAEMOCOMPLAETTAN P'yi çocukların göremeyeceği, erişemeyeceği yerlerde ve ambalajında saklayınız. 2-8 °C arasında buzdolabında saklayınız. Dondurmayınız. Işıktan korumak için, flakonu kapalı dış kartonunda saklayınız. Donmuş ürünü çözüp kullanmayınız! Sulandırılmış ürünün fizikokimyasal stabilitesi oda sıcaklığında (maksimum 25°C) 8 saat süreyle kanıtlanmıştır. Mikrobiyolojik açıdan, ürün sulandırmayı takiben hemenkullanılmalıdır. Sulandırılmış ürünün hemen kullanılmaması durumunda; saklama odasıcaklığında (maksimum 25°C) 8 saatten uzun olmamalıdır. Sulandırılmış ürün buzdolabındasaklanmamalıdır. 6.5. Ambalajın niteliği ve içeriğiLatekssiz bir tıpa (bromobütil kauçuk), ve alüminyum kapak ve plastik disk ile kapatılmış renksiz cam bir flakon (Tip II Avrupa Farmakopesi). Enjeksiyonluk su: Bromobütil kauçuk bir tıpa ve plastik diskli bir alüminyum kapaktan oluşan kombinasyon kapaklarla kapatılmış 50 mL'lik renksiz cam bir flakon (Tip I AvrupaFarmakopesi). Ürünün sunumu: 1 g insan fibrinojeni içeren 1 adet flakon. 1 adet 50 ml enjeksiyonluk su, 1 adet transfer cihazı, 1 adet enjektör filtresi ve 1 adet delici uç. 12 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerGenel talimatlar Ürünün hazırlanması ve sulandırılması aseptik koşullarda gerçekleşmelidir. Sulandırılmış ürün uygulanmadan önce partiküler madde ve renk bozukluğu açısındangörsel olarak incelenmelidir. Çözelti neredeyse renksiz veya sarımsı, berrak ila hafif opelasan ve nötr pH değerindeolmalıdır. Bulanık veya herhangi bir kalıntı partikül (kalıntı/partikül) içeren çözeltilerkullanılmamalıdır. Çözeltinin hazırlanması Aşağıdaki prosedürler, HAEMOCOMPLETTAN P'nin hazırlanması ve sulandırılması için genel kılavuz olarak verilmiştir. HAEMOCOMPLETTAN P'yi hazırlarken ve sulandırırken aseptik teknik kullanın. Ürün uygulanmadan önce oda sıcaklığına veya vücut sıcaklığına (37 °C'den yüksekolmamalı) getirilmelidir. Ürünü sulandırmadan önce ellerinizi yıkayınız veya eldiven kullanınız. 1. Kauçuk tıpanın orta kısmını ortaya çıkarmak için kapağı HAEMOCOMPLETTANP flakonundan çıkarın (Şekil 1). 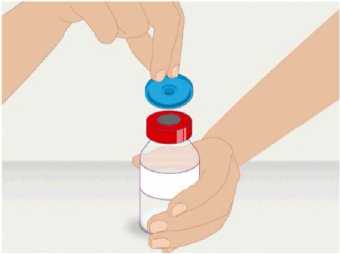 Şekil 1 2. Kauçuk tıpanın yüzeyini antiseptik bir solüsyonla temizleyin ve kurumasınıbekleyin (Şekil 2). 13 Şekil 2 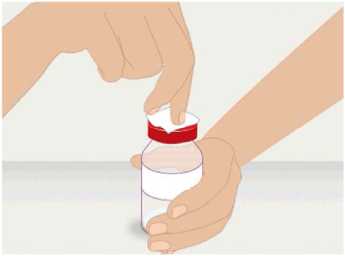 3. HAEMOCOMPLETTAN P 1g ile sunulan transfer setinin bir ucundaki güvenlikkapağını çıkarın ve HAEMOCOMPLETTAN P şişesinin tıpasını delin (Şekil 3). 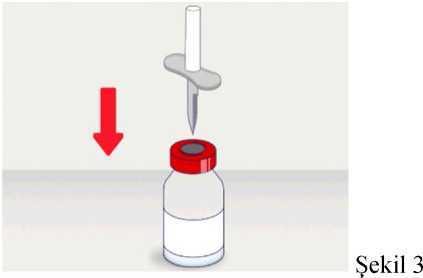 4. Transfersetinindiğer ucundakiemniyet kapağını çıkarın, HAEMOCOMPLETTAN P 1g ile sunulan 50 ml'lik enjeksiyonluk su şişesini ters çevirin, tıpayıdelmekiçinhafif basınç uygulayın ve içindekileri HAEMOCOMPLETTAN P şişesine aktarın (Şekil 4).   Şekil 4 5. Enjeksiyonluk su şişesini atın ve transfer setini HAEMOCOMPLETTAN Pşişesinden çıkarın. 6. Ürünün tamamen çözündüğünden emin olmak için HAEMOCOMPLETTAN Pflakonunu hafifçe döndürün (Şekil 5). 14 Şekil 5 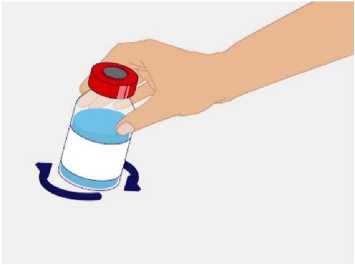 Köpük oluşumuna neden olmamak için sallamaktan kaçının. Toz 15 dakika içinde tamamen sulandırılmış olmalıdır (genellikle 5 ila 10dakika). 7. HAEMOCOMPLETTAN P 1 g ile sunulan delici ucun plastik blisterini açınız(Şekil 6). 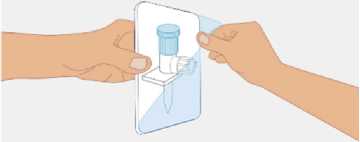 Şekil 6 8. Sunulan delici ucu alınız ve seyreltilmiş ürünün flakon kapağına takınız (Şekil 7). 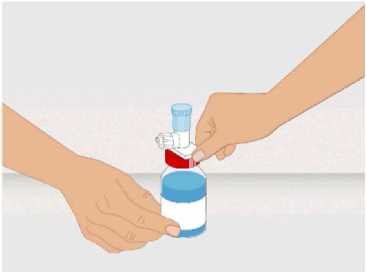 Şekil 7 9. Delici ucu taktıktan sonra, kapağı çıkarınız. Kapak çıkarıldıktan sonra, görünüryüzeye dokunmayınız. 15 10. HAEMOCOMPLETTAN P 1 g ile sunulan filtrenin blisteri açınız (Şekil 8). 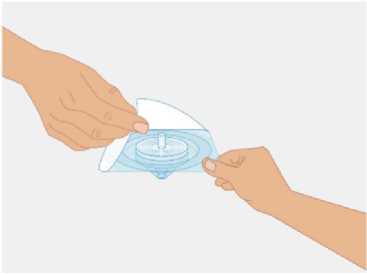 Şekil 8 11. Enjektörü çevirerek filtreye takınız (Şekil 9). 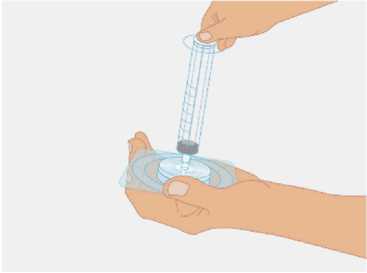 Şekil 9 12. Filtre takılmış enjektörü çevirerek delici uca takınız (Şekil 10). 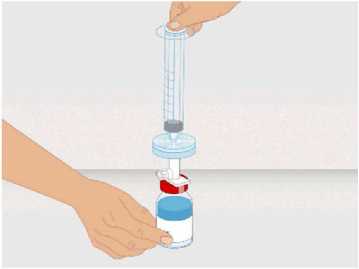 Şekil 10 13. Sulandırılan ürünü enjektöre çekiniz (Şekil 11). 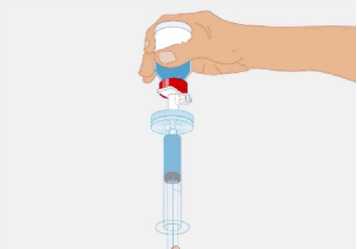 
Şekil 11
16 14.Tamamlandığında, filtreyi, delici ucu ve boş flakonu enjektörden çıkarınız, uygun bir şekilde atınız ve olağan şekilde uygulama işlemine devam ediniz. Bulanık veya herhangi bir kalıntı ya da partikül içeren çözeltiler kullanılmamalıdır. Sulandırılmış ürün hemen farklı bir enjeksiyon/infüzyon yoluyla uygulanmalıdır (bkz.Bölüm 6.3). Uygulama esnasında çözelti ile dolu enjektöre kan girişinin tamamen engellenmesigerekmektedir. Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİCSL Behring Biyoterapi İlaç Dış Ticaret Anonim ŞirketiÜsküdar / İstanbul 8. RUHSAT NUMARASI2015/704 9. İLK RUHSAT TARİHİ/ RUHSAT YENİLEME TARİHİ31/08/2015 10. KÜB' ÜN YENİLENME TARİHİ17 |
İlaç BilgileriHaemocomplettan P 1 G I.v. Enjeksiyonluk/infüzyonluk Çözelti TozuEtken Maddesi: İnsan Fibrinojeni (koagülasyon Faktör I) Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.