Humira 20 Mg/0.2 Ml Enjeksiyonluk Çözelti İçeren Kullanıma Hazır Enjektör Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIHUMIRA 20 mg/0,2 mL enjeksiyonluk çözelti içeren kullanıma hazır enjektör Steril UYARIDiğer TNF blokerlerinde olduğu gibi, HUMIRA kullanan hastalarda (klinikte sıklıkla yaygın veya akciğer dışı tutulum gösterenler de dahil) tüberküloz vakaları gözlenmiştir. Eğer aktiftüberküloz tanısı konulursa HUMIRA tedavisine başlanmamalıdır (bkz. Bölüm 4.3). Hastalar tüberkülin deri testi yapılarak inaktif (latent) tüberküloz açısından değerlendirilmelidir. İnaktif tüberküloz enfeksiyonu için tüberkülin deri testi yapılırken, hastadaha önce Bacille Calmette-Guerin (BCG) ile aşılanmış olsa dahi, 5 mm veya daha yüksekindurasyon boyutu pozitif olarak kabul edilmelidir. İnaktif tüberküloz teşhis edilen hastalardaHUMIRA tedavisine başlanmadan önce yerel öneriler doğrultusunda uygun bir anti-tüberkülozprofilaksisi yapılmalıdır. 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir kullanıma hazır enjektör, 20 mg/0,2 mL adalimumab içerir. Adalimumab Çin Hamster Yumurtalık hücrelerinde üretilen bir rekombinant insan monoklonal antikorudur. Yardımcı maddeler:Yardımcı maddelerin tam listesi için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti. Berrak, renksiz çözelti 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarJüvenil idiyopatik artritPoliartiküler iüvenil idiyopatik artrit.
1 Bir veya daha fazla hastalık modifiye edici anti-romatizmal ilaca (DMARD) karşı yetersiz yanıt alınan 2-17 yaşları arasındaki çocuklar ve adölesanlarda HUMIRA, metotreksat ilekombinasyon halinde, aktif poliartiküler jüvenil idiyopatik artrit tedavisi için endikedir.HUMIRA metotreksata karşı intolerans durumunda veya metotreksat ile tedaviye devamedilmesinin uygun olmadığı durumlarda monoterapi olarak verilebilir (monoterapideki etkililikiçin bkz. Bölüm 5.1). HUMIRA, 2 yaş altındaki küçük çocuklarda araştırılmamıştır. Entezit ile ilişkili artritHUMIRA, konvansiyonel tedaviye yetersiz yanıt veren veya konvansiyonel tedaviye intoleransı olan 6 yaş ve üstü hastalarda, entezit ile ilişkili artritin tedavisinde endikedir (bkz.Bölüm 5.1) Pediyatrik plak psöriyazisHUMIRA, topikal ve geleneksel sistemik (siklosporin, metotreksat) tedaviler veya fototerapiye yeterli cevap vermeyen ya da uygun aday olmayan 4 yaş ve üstü çocuklarda ve ergenlerdekişiddetli kronik plak psöriyazisin tedavisinde endikedir. Pediyatrik Crohn HastalığıHUMIRA, primer beslenme tedavisi, kortikosteroid ve bir immünomodülatör içeren konvansiyonel tedaviye yetersiz yanıt alınan ya da bu tedaviyi tolere edemeyen veya bu türtedavilerin kontrendike olduğu pediyatrik hastalarda (6 ila 17 yaş) şiddetli aktif Crohn hastalığıtedavisi için endikedir. Pediyatrik ÜveitHUMIRA, 2 yaş ve üstü konvansiyonel tedavilere yetersiz yanıt vermiş olan veya tolere edemeyen ya da konvansiyonel tedavilerin uygun olmadığı pediyatrik hastalarda enfeksiyözolmayan, kronik anterior üveit tedavisinde endikedir. 4.2. Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:HUMIRA tedavisi, HUMIRA'nın endike olduğu hastalıkların tanı ve tedavisinde deneyimli uzman doktorlar tarafından başlatılmalı ve gözlenmelidir. Oftalmologların HUMIRA iletedaviye başlamadan önce uygun bir uzmanla görüşmeleri önerilmektedir (bkz. Bölüm 4.4).HUMIRA ile tedavi olan hastalara özel bir uyarı kartı verilir. Enjeksiyon tekniği konusunda eğitilen hastalar, doktor tarafından uygun bulunursa ve doktor takibi ile HUMIRA enjeksiyonlarını kendileri yapabilirler. HUMIRA ila tedavi sırasında, eş zamanlı diğer tedavilerin (örn., kortikosteroidler ve/veya immünomodülatör ajanlar) optimize edilmesi gerekmektedir. Pediyatrik popülasyon:Jüvenil idiyopatik artrit2 yaş ve üstü poliartiküler jüvenil idiyopatik artrit
2 Poli arti kül er jüvenil idiyopatik artriti olan 2 yaş üzeri hastalar için önerilen HUMIRA dozu, vücut ağırlığına bağlı olarak seçilir (Tablo 1). HUMIRA, subkütan enjeksiyon yoluyla ikihaftada bir uygulanır.
Eldeki veriler, klinik yanıtın genellikle 12 haftalık tedavi içinde sağlandığını göstermektedir. Bu zaman diliminde yanıt vermeyen bir hastada tedaviye devam edilmesi dikkatli bir şekildetekrar değerlendirilmelidir. Bu endikasyonda 2 yaş altındaki çocuklarda HUMIRA'nın ilgili bir kullanımı yoktur. HUMIRA, bireysel tedavi ihtiyaçlarına bağlı olarak diğer yitilikler ve/veya formlarda da mevcut olabilir. Entezit ile ilişkili artritEntezit ile ilişkili artriti olan 6 yaş ve üstü hastalar için önerilen HUMIRA dozu, vücut ağırlığına bağlı olarak seçilir (Tablo 2). HUMIRA, subkütan enjeksiyon yoluyla iki haftada bir uygulanır.
HUMIRA ile entezit ile ilişkili artriti olan 6 yaş altındaki hastalarda çalışma yapılmamıştır. HUMIRA, bireysel tedavi ihtiyaçlarına bağlı olarak diğer yitilikler ve/veya formlarda da mevcut olabilir. Pediyatrik plak psöriyazisPediyatrik plak psöriyazisli 4-17 yaş arasındaki hastalar için önerilen HUMIRA dozu, vücut ağırlığına bağlı olarak seçilir (Tablo 3). HUMIRA, subkütan enjeksiyon yoluyla uygulanır.
16 haftalık tedavi ile yeterli yanıt elde edilemeyen olgularda tedaviye devam kararı dikkatle gözden geçirilmelidir. 3 HUMIRA ile tekrar tedaviye devam edilirse, doz ve tedavi süresi yukarıdaki kılavuz bilgilerine göre uygulanmalıdır. Plak psöriyazisli pediyatrik hastalarda HUMIRA'nın güvenliliği ortalama 13 ay için değerlendirilmiştir. Bu endikasyonda 4 yaş altındaki çocuklarda HUMIRA'nın ilgili bir kullanımı yoktur. HUMIRA, bireysel tedavi ihtiyaçlarına bağlı olarak diğer yitilikler ve/veya formlarda da mevcut olabilir. Pediyatrik Crohn hastalığıCrohn hastalığı olan 6-17 yaş arasındaki hastalar için önerilen HUMIRA dozu, vücut ağırlığına bağlı olarak seçilir (Tablo 4). HUMIRA, subkütan enjeksiyon yoluyla uygulanır. _Tablo 4. Crohn Hastalığı Olan Pediyatrik Hastalar için HUMIRA Dozu_Hasta İndüksiyon Dozu 4. Haftada BaşlayanAğırlığı İdame Dozu< 40 kg 0. haftada 40 mg ve 2. haftada 20 mg İki haftada bir 20 mg Tedaviye daha hızlı bir yanıt gerekli görüldüğünde, daha yüksek indüksiyon dozu kullanımıyla advers olay riskinin daha yüksekolabileceği konusunda dikkatli davranılarak aşağıdaki dozkullanılabilir: _ 0. haftada 80 mg ve 2. haftada 40 mg_> 40 kg 0. haftada 80 mg ve 2. haftada 40 mg Tedaviye daha hızlı bir yanıt gerekli görüldüğünde, daha yüksek indüksiyon dozu kullanımıyla advers olay riskinin daha yüksek İki haftada bir 40 mg olabileceği konusunda dikkatli davranılarak aşağıdaki doz kullanılabilir: _ 0. haftada 160 mg ve 2. haftada 80 mg_Tedaviye yetersiz yanıt veren bazı hastalarda, HUMIRA doz sıklığının haftada bire çıkarılması yarar sağlayabilir: < 40 kg: haftada bir 20 mg > 40 kg: haftada bir 40 mg ya da iki haftada bir 80 mg 12. haftada yanıt vermeyen bir hastada tedaviye devam edilmesi dikkatli bir şekilde tekrar değerlendirilmelidir. Bu endikasyonda 6 yaş altındaki küçük çocuklarda HUMIRA'nın ilgili bir kullanımı yoktur. HUMIRA, bireysel tedavi ihtiyaçlarına bağlı olarak diğer yitilikler ve/veya formlarda da mevcut olabilir.
4 Pediyatrik üveit2 yaş ve üstü üveit hastası çocuklar için önerilen HUMIRA dozu vücut ağırlığınadayanmaktadır (Tablo 5). HUMIRA, subkütan enjeksiyon yoluyla uygulanır.Pediyatrik üveitte, metotreksat ile eş zamanlı tedavi haricinde HUMIRA tedavisi ile ilgili deneyim bulunmamaktadır.
HUMIRA tedavisine başlanırken, idame tedavisinden bir hafta önce 30 kg'ın altındaki hastalar için 40 mg'lık bir yükleme dozu ya da 30 kg'ın üzerindeki hastalar için 80 mg'lık bir yüklemedozu uygulanabilir. 6 yaş altındaki çocuklarda HUMIRA yükleme dozu kullanımı ile ilgiliherhangi bir klinik veri bulunmamaktadır (bkz. Bölüm 5.2) HUMIRA'nın 2 yaş altındaki çocuklarda bu endikasyonda kullanımı mevcut değildir. Uzun süreli aralıksız tedavi ile ilgili yarar ve risklerin yıllık olarak değerlendirilmesi önerilmektedir (bkz. Bölüm 5.1) HUMIRA, bireysel tedavi ihtiyaçlarına bağlı olarak diğer yitilikler ve/veya formlarda da mevcut olabilir. Uygulama şekli:HUMIRA, subkütan enjeksiyon yoluyla uzman bir doktorun rehberliği ve gözetimi altında kullanılır. Detaylı uygulama şekli, ambalajın içindeki kullanma talimatında sunulmaktadır. HUMIRA, farklı yitilik ve formlarda da mevcuttur. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karaciğer yetmezliğiHUMIRA bu popülasyonlarda araştırılmamıştır. Herhangi bir doz önerisi yapılamaz. Geriyatrik popülasyon:Yaşlı hastalarda doz ayarlaması gerekmemektedir. Pediyatrik popülasyon:HUMIRA ile 2 yaş altındaki çocuklarda çalışma yapılmamıştır.
5 4.3. KontrendikasyonlarHUMIRA aşağıdaki durumlarda kontrendikedir: - Adalimumaba veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birinekarşı hipersensitivite gösteren hastalarda; - Aktif tüberküloz veya sepsis gibi şiddetli enfeksiyonlar ve fırsatçı enfeksiyonlarınvarlığında (bkz. Bölüm 4.4). - Orta ila şiddetli derecede kalp yetmezliği olan hastalarda (NYHA [New York HeartAssociation] sınıfı III/IV) (bkz. Bölüm 4.4). 4.4. Özel kullanım uyarıları ve önlemleri65 yaş üstü hastalarda ölümle sonuçlanabilecek ciddi enfeksiyon riski 65 yaş altındakilere göre daha yüksektir.İzlenebilirlikBiyolojik ürünlerin takip edilebilirliğinin sağlanması için, uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. EnfeksiyonlarTNF bloke edici ajan alan hastalar ciddi enfeksiyonlara daha fazla duyarlıdır. Akciğer fonksiyon yetmezliği enfeksiyon gelişme riskini arttırabilir. Hastalar bu nedenle, HUMIRAtedavisinden önce, tedavi sırasında ve tedaviden sonra, tüberküloz dahil olmak üzereenfeksiyonlar açısından yakından izlenmelidir. Adalimumabın eliminasyonu dört aya kadarsürebildiğinden, izlemeye bu dönem boyunca da devam edilmelidir. HUMIRA tedavisi, kronik veya lokalize enfeksiyonlar dahil aktif enfeksiyonları olan hastalarda, enfeksiyon kontrol altına alınana kadar başlatılmamalıdır. Tüberküloza maruz kalanhastalarda ya da tüberküloz veya endemik mikoz (histoplazmoz, koksidiyomikoz veyablastomikoz) riski yüksek bölgelere seyahat etmiş olan hastalarda HUMIRA tedavisibaşlatılmadan önce tedavinin risk ve yararları değerlendirilmelidir (bkz. Diğer fırsatçıenfeksiyonlar). HUMIRA tedavisi sırasında yeni bir enfeksiyon gelişen hastalar yakından izlenmelidir ve tam bir tanısal değerlendirmeye tabi tutulmalıdır. Bir hastada yeni bir ciddi enfeksiyon veya sepsisgeliştiğinde HUMIRA uygulaması kesilmelidir ve enfeksiyon kontrol altına alınan dek uygunantimikrobiyal veya antifungal tedavi başlatılmalıdır. Tekrarlayan enfeksiyon öyküsü olanhastalarda veya hastada enfeksiyona predispozisyon yaratan, eş zamanlı immünosupresifilaçların kullanımı dahil altta yatan nedenler bulunduğunda, doktorlar HUMIRA kullanımıkonusunda dikkatli olmalıdır. Ciddi enfeksiyonlarHUMIRA kullanan hastalarda, sepsis de dahil olmak üzere, bakteriyel, mikobakteriyel, invazif fungal, parazitik, viral ya da listeriyozis, lejyonellozis ve pnömosistis gibi diğer fırsatçıenfeksiyonlara bağlı olarak ciddi enfeksiyonlar bildirilmiştir. 6 Klinik çalışmalarda görülen diğer ciddi enfeksiyonlar arasında; pnömoni, piyelonefrit, septik artrit ve septisemi yer almaktadır. Enfeksiyonlarla bağlantılı olarak hastaneye yatışa sebepolabilen ya da fatal sonuçlar bildirilmiştir. TüberkülozHUMIRA almakta olan hastalarda reaktivasyon ve yeni başlayan tüberküloz dahil olmak üzere, tüberküloz ortaya çıktığı bildirilmiştir. Bu bildirimler pulmoner ve ekstrapulmoner (dissemine)tüberküloz vakalarını içermektedir. HUMIRA tedavisine başlanmadan önce bütün hastalar gerek aktif gerekse inaktif (latent) tüberküloz enfeksiyonu yönünden değerlendirilmelidir. Bu değerlendirme, kişide tüberkülozhikayesi veya daha önce aktif tüberkülozu olan hastalar ile temas öyküsü ve önceki ve/veyahalen sürmekte olan immünosupresif tedaviler dahil ayrıntılı bir tıbbi değerlendirmeiçermelidir. Bütün hastalarda uygun tarama testleri (tüberkülin deri testi, akciğer röntgeni gibi)yapılmalıdır (yerel öneriler uygulanabilir). Bu testlerin yürütülme şekli ve sonuçlarının hastauyarı kartları üzerine kaydedilmesi önerilir. Doktorlara, özellikle ağır hastalığı olan veyaimmünkompromize hastalarda yalancı negatif tüberkülin deri testi sonuçlarının alınma riskihatırlatılmalıdır. Eğer aktif tüberküloz tanısı konulursa HUMIRA tedavisine başlanmamalıdır (bkz. Bölüm 4.3). Aşağıda belirtilen tüm durumlarda tedavinin fayda/risk oranı dikkatli değerlendirilmelidir. Latent tüberküloz kuşkusu olması halinde, tüberküloz tedavisinde uzman olan bir hekime danışılmalıdır. Latent tüberküloz teşhis edildiğinde, HUMIRA başlanmadan önce yerel öneriler doğrultusunda uygun bir anti-tüberküloz profilaksi tedavisine başlanmalıdır. Tüberküloz için test sonucu negatif olan ama tüberküloz enfeksiyonu açısından çeşitli veya anlamlı riski bulunan hastalarda ve yeterli tedavi uygulamasının doğrulanamadığı, öncedengeçirilmiş aktif ya da inaktif tüberküloz öyküsü olan hastalarda da HUMIRA tedavisinebaşlanmadan önce anti-tüberküloz profilaksi tedavisi düşünülmelidir. HUMIRA ile tedavi edilen hastalarda, tüberküloz profilaksisi uygulanmasına rağmen tüberküloz reaktivasyonu ortaya çıkmıştır. Daha önce başarılı bir şekilde aktif tüberküloztedavisi görmüş olan bazı hastalarda, HUMIRA ile yapılan tedavi sırasında yeniden tüberkülozgelişmiştir. Hastalar HUMIRA ile tedavi sırasında ya da tedaviden sonra, tüberküloz enfeksiyonunu düşündüren bulgular/semptomlar (örn., inatçı öksürük, güçten düşme/kilo kaybı, düşük dereceliateş, isteksizlik) ortaya çıktığında doktora başvurmaları konusunda bilgilendirilmelidir. Diğer fırsatçı enfeksiyonlarHUMIRA uygulanan hastalarda invazif fungal enfeksiyonlar dahil fırsatçı enfeksiyonlar gözlemlenmiştir. Bu enfeksiyonlar, TNF bloke edici ajan alan hastalarda her zaman teşhisedilememiştir, bu da uygun tedavinin gecikmesine ve bazen fatal sonuçlara neden olmuştur. 7 Ateş, kırıklık, kilo kaybı, terleme, öksürme, dispne ve/veya pulmoner infiltratlar gibi bulgu ve semptomlar ya da eş zamanlı şok ile birlikte veya şok olmaksızın diğer ciddi bir sistemikhastalık gelişen hastalarda invazif bir fungal enfeksiyon varlığından şüphelenilmeli veHUMIRA uygulaması derhal durdurulmalıdır. Bu hastalarda, teşhis ve ampirik antifungal terapiuygulaması, invazif fungal enfeksiyonu olan hastaların tedavisinde uzman bir doktoradanışılarak gerçekleştirilmelidir. Hepatit B reaktivasyonuHUMIRA dahil, TNF bloke edici ajan kullanan ve hepatit B virüsünün kronik taşıyıcısı olan hastalarda (örn. yüzey antijen pozitif) hepatit B reaktivasyonu ortaya çıkmıştır. Bazı olgularfatal sonuçlanmıştır. HUMIRA tedavisine başlamadan önce hastalar HBV enfeksiyonuaçısından test edilmelidir. Hepatit B enfeksiyonu testleri pozitif bulunan hastalar için hepatit Btedavisinde uzman bir hekime danışılması önerilmektedir. HUMIRA tedavisine ihtiyaç duyan HBV taşıyıcıları, tedavi boyunca ve tedavinin kesilmesinden sonra birkaç ay süreyle aktif HBV enfeksiyonunun bulgu ve semptomlarıbakımından yakından izlenmelidir. HBV taşıyıcısı hastalarda TNF bloke edici ajan tedavisiylebirlikte, HBV reaktivasyonunu önleme amaçlı antiviral tedavi uygulaması konusunda yeterliveri bulunmamaktadır. HBV reaktivasyonu gelişen hastalarda HUMIRA tedavisi durdurulmalıve uygun bir destek tedavisi ile birlikte etkili antiviral tedavisine başlanmalıdır. Nörolojik olaylarHUMIRA dahil TNF bloke edici ajanlar, nadir olgularda multipl skleroz ve optik nörit gibi santral sinir sistemi demiyelinizan hastalıklar ve Guillain Barre sendromu dahil periferikdemiyelinizan hastalıkların ortaya çıkması veya bu hastalıkların klinik semptomlarınınalevlenmesi ve/veya radyografik bulguların ortaya çıkması ile ilişkili bulunmuştur. HastalarınaHUMIRA tedavisi uygulayacak olan doktorlar, önceden var olan ya da yakın zamanda başlamışsantral veya periferik sinir sistemi demiyelinizan hastalıkları bulunan hastalarda HUMIRAkullanma kararını dikkatle gözden geçirilmelidir. Bu bozukluklardan herhangi biri ortayaçıktığı takdirde, HUMIRA'nın kesilmesi düşünülmelidir. Orta üveit ile merkezi demiyelinizanbozukluklar arasında bilinen bir ilişki mevcuttur. Enfeksiyöz olmayan orta üveit hastalarındaönceden mevcut olan veya gelişmekte olan merkezi demiyelinizan bozukluklarındeğerlendirilmesine yönelik olarak HUMIRA tedavisine başlanmadan önce ve tedavi sırasındadüzenli şekilde nörolojik değerlendirme gerçekleştirilmelidir. Alerjik reaksiyonlarKlinik çalışmalar sırasında HUMIRA ile ilişkili ciddi alerjik reaksiyonlar seyrek olarak bildirilmiştir. HUMIRA ile ilişkili ciddi olmayan alerjik reaksiyonların sıklığı, klinik çalışmalaresnasında 'yaygın olmayan' kategorisindedir. HUMIRA uygulamasından sonra, anafilaksidahil olmak üzere ciddi alerjik reaksiyonlar bildirilmiştir. Eğer bir anafilaktik reaksiyon ya dabaşka bir ciddi alerjik reaksiyon gelişirse, HUMIRA uygulaması derhal kesilerek uyguntedaviye başlanmalıdır.
8 İmmünosupresyonHUMIRA ile tedavi edilen 64 romatoid artrit hastasıyla yapılan bir çalışmada, gecikmiş tipte hipersensitivitenin baskılanması, immünoglobülin düzeylerinin baskılanması veya efektör T-,B-, NK-hücrelerinin, monositlerin/makrofajların ve nötrofillerin sayılarında değişme olduğunailişkin kanıtlar bulunmamıştır. Maligniteler ve lenfoproliferatif hastalıklarTNF bloke edici ajanlarla yürütülen kontrollü klinik çalışmalarda, TNF bloke edici ajan verilen hastalarda kontrol hastalarına kıyasla lenfoma dahil daha fazla sayıda malignite olgusugözlemlenmiştir. Ancak bunlar seyrek olarak ortaya çıkmıştır. Pazarlama sonrası dönemde,TNF bloke edici ajanlarla tedavi edilen hastalarda lösemi olguları bildirilmiştir. Uzun birgeçmişe sahip, yüksek derecede aktif, inflamatuvar hastalığı olan romatoid artrit hastalarında,arka planda lenfoma ve lösemi riski artmaktadır; bu durum ise risk tahminini zorlaştırmaktadır.Mevcut bilgiler ile, bir TNF bloke edici ajan ile tedavi edilen hastalarda olası bir lenfoma,lösemi ya da diğer malignitelerin gelişme riski göz ardı edilemez. Pazarlama sonrası dönemdeki adalimumab dahil, TNF bloke edici ajanlarla tedavi edilen (tedavi başlangıcı < 18 yaş) çocuklar, ergenler ve genç erişkinler (22 yaşına kadar olanlar)arasında, bazıları ölümcül olmak üzere, maligniteler bildirilmiştir. Bu vakaların yaklaşık yarısılenfoma olurken, diğer vakalar genellikle immünosupresyon ile bağlantılı nadir maligniteler dedahil olmak üzere diğer çeşitli malignitelerdir. TNF bloke edici ajanlarla ile tedavi edilençocuklar ve ergenlerde malignite gelişmesi riski göz ardı edilemez. Adalimumab ile tedavi edilen hastalarda, pazarlama sonrası dönemde seyrek olarak hepatosplenik T hücreli lenfoma teşhis edilmiştir. Nadir görülen bu T hücreli lenfoma türü, çokagresif bir seyre sahiptir ve genelde ölümcüldür. HUMIRA ile görülen bu hepatosplenik Thücreli lenfoma vakalarının bazıları, inflamatuvar bağırsak hastalığı için HUMIRA ile eşzamanlı azatiyoprin veya 6-merkaptopürin kullanılan genç erişkinlerde görülmüştür.Azatiyoprin veya 6-merkaptopürin ile HUMIRA kombinasyonunun neden olabileceğipotansiyel risk dikkatlice düşünülmelidir. HUMIRA ile tedavi edilen hastalarda, hepatosplenikT hücreli lenfoma gelişim riski göz ardı edilemez (bkz. Bölüm 4.8). HUMIRA kullanan 60 yaş üzeri hastalarda, periyodik servikal kanser taramasına devam edilmelidir. Malignite öyküsü olan ya da HUMIRA kullanırken malignite gelişen ve tedaviye devam edilen hastaları içeren bir çalışma yapılmamıştır. Bu nedenle böyle hastalarda HUMIRA tedavisinedevam kararı daha dikkatli olunmalıdır (bkz. Bölüm 4.8). HUMIRA tedavisine başlanmadan önce ve tedavi esnasında bütün hastalar, özellikle geniş kapsamlı immünosupresif tedavi geçmişi olan hastalar veya PUVA tedavisi geçmişi olanpsöriyazis hastaları, melanoma-dışı cilt kanseri varlığı açısından değerlendirilmelidir.Adalimumab dahil TNF bloke edici ajanlar ile tedavi edilen hastalarda melanoma ve Merkelhücreli karsinoma da bildirilmiştir (bkz. Bölüm 4.8). Bir başka TNF bloke edici ajan olan infliksimabın kullanımının değerlendirildiği, orta ve ağır dereceli kronik obstrüktif akciğer hastalığı (KOAH) olan hastalarda tespit amaçlı olarakgerçekleştirilen klinik bir çalışmada, kontrol grubu hastalar ile karşılaştırıldığında, infliksimab 9 uygulanan hasta grubunda daha çok akciğer veya baş ve boyunda olmak üzere maligniteler bildirilmiştir. Bütün hastalarda yoğun sigara içme hikayesi vardır. Bu nedenle, KOAHhastalarında ve yoğun sigara içilmesine bağlı malignite riski artmış olan hastalarda herhangi birTNF bloke edici ajan kullanılırken dikkatli olunmalıdır. Mevcut veriler ile, adalimumab tedavisinin displazi gelişimi veya kolon kanseri geliştirme riskini etkileyip etkilemediği bilinmemektedir. Yüksek displazi veya kolon karsinoma riski olan(örn., uzun süreli ülseratif kolit veya primer sklerozan kolanjit hastaları) veya önceden displaziya da kolon karsinoma geçmişi olan tüm ülseratif kolit hastalarında, tedaviden önce ve hastalıksüresince düzenli aralıklarla displaziye yönelik tarama yapılmalıdır. Bu değerlendirme, yerelönerilere uygun bir şekilde gerçekleştirilecek kolonoskopi ve biyopsileri kapsamalıdır. TNF bloke edici ajanın kullanımına bağlı olarak lösemi-kan kanseri (akut myeloid lösemi, kronik lenfositik lösemi ve kronik myeloid lösemi) geliştiği bildirilmiştir.Hematolojik reaksiyonlarTNF bloke edici ajanlar ile aplastik anemi dahil pansitopeni olguları seyrek olarak bildirilmiştir. HUMIRA ile, tıbbi açıdan önemli, sitopeni de (örn. trombositopeni, lökopeni) dahil olmaküzere, hematolojik sisteme ait advers olaylar bildirilmiştir. Bütün hastalara, HUMIRAkullanmakta iken kan diskrazilerini belirten bulgu ve semptomların (örn. inatçı ateş, morarma,kanama, solukluk) gelişmesi halinde hemen doktora başvurmaları tavsiye edilmelidir. Varlığıdoğrulanmış önemli hematolojik anormalliklerin bulunduğu hastalarda, HUMIRA tedavisineson verilmesi değerlendirilmelidir. AşılamalarAdalimumab veya plasebo ile tedavi edilen romatoid artritli 226 erişkin üzerinde yürütülen bir çalışmada, standart 23 valanlı pnömokok aşısına ve trivalan influenza virüs aşısına karşı benzerantikor yanıtları gözlemlenmiştir. HUMIRA almakta olan hastalarda enfeksiyonun canlı aşılaryoluyla ikincil iletimi konusunda veri bulunmamaktadır. Pediyatrik hastalara, eğer mümkünse, HUMIRA tedavisine başlamadan önce güncel bağışıklama kılavuzlarına uygun olarak tüm bağışıklık aşılarını tamamlamaları önerilmektedir. HUMIRA tedavisi almakta olan hastalara, canlı aşılar hariç, eş zamanlı aşılar uygulanabilir. Anne karnında adalimumaba maruz kalan bebeklerde, anneye gebelik sırasında yapılan sonadalimumab enjeksiyonunu takip eden 5 ay içinde canlı aşı (örn. BCG aşısı) uygulamasıönerilmemektedir. Konjestif kalp yetmezliğiBaşka bir TNF bloke edici ajan ile yapılan bir klinik çalışmada konjestif kalp yetmezliğinde kötüleşme ve konjestif kalp yetmezliğine bağlı mortalitede artış gözlemlenmiştir. HUMIRAtedavisi gören hastalarda da konjestif kalp yetmezliğinin ağırlaştığı olgular görülmüştür.HUMIRA, hafif kalp yetmezliği (NYHA sınıfı I/II) olan hastalarda dikkatle kullanılmalıdır.HUMIRA, orta ila şiddetli derecede kalp yetmezliğinde kontrendikedir (bkz. Bölüm 4.3).Konjestif kalp yetmezliği semptomları yeni ortaya çıkan veya kötüleşen hastalarda HUMIRA tedavisi kesilmelidir. Bu belge Belge Do 10 Otoimmün süreçlerHUMIRA ile tedavi, otoimmün antikorların oluşmasına yol açabilir. Uzun dönemli HUMIRA tedavisinin otoimmün hastalık gelişmesi üzerindeki etkisi bilinmemektedir. Eğer bir hastadaHUMIRA tedavisinden sonra, lupus benzeri sendromu düşündüren semptomlar gelişirse vehasta çift-sarmallı DNA'ya karşı antikorlar yönünden pozitif ise, HUMIRA tedavisine dahafazla devam edilmemelidir (bkz. Bölüm 4.8). TNF bloke edici ajan veya biyolojik hastalık modifıye edici anti-romatizmal ilaçların (bDMARD) birlikte uygulanmasıAnakinra ve başka bir TNF bloke edici ajan olan etanerseptin birlikte kullanıldığı klinik çalışmalarda ciddi enfeksiyonlar görülmüş ve tek başına etanersept kullanımına göre ek biryarar sağlanmamıştır. Etanersept ve anakinra kombinasyon tedavisinde görülen adversolayların doğası gereği, benzer toksisiteler anakinra ile diğer TNF bloke edici ajanlarınkombinasyonunda da ortaya çıkabilir. Bu nedenle, adalimumab ve anakinra kombinasyonuönerilmemektedir (bkz. Bölüm 4.5). Adalimumabın diğer biyolojik hastalık modifiye edici anti-romatizmal ilaçlarla (örn. anakinra ve abatasept) ya da diğer TNF bloke edici ajanlar ile ¦ eş zamanlı kullanımı, ciddienfeksiyonlar ve diğer potansiyel farmakolojik etkileşimlerin dahil olduğu olası enfeksiyonriski artışı nedeniyle önerilmemektedir (bkz. Bölüm 4.5). CerrahiHUMIRA tedavisindeki hastalarda cerrahi prosedürlerle ilgili güvenlilik deneyimi kısıtlıdır. Eğer bir cerrahi prosedür planlanıyorsa, adalimumab yarı-ömrünün uzun oluşu dikkatealınmalıdır. HUMIRA tedavisinde iken cerrahi işlem gereken bir hasta, enfeksiyonlar açısındanyakından izlenmeli ve uygun önlemler alınmalıdır. HUMIRA almakta iken artroplastiuygulanan hastalara ilişkin güvenlilik deneyimi kısıtlıdır. İnce bağırsak obstrüksiyonuCrohn hastalığı tedavisine yanıt alınamaması, ameliyat gerektirebilecek sabit fibrotik striktür varlığına işaret edebilir. Mevcut veriler, HUMIRA'nın striktürlere neden olmadığını veyakötüleştirmediğini göstermektedir. Geriyatrik popülasyon:HUMIRA ile tedavi edilen 65 yaş üstündeki hastalarda görülen ciddi enfeksiyon sıklığı (%3,7), 65 yaşının altındaki hastalardan daha yüksek (%1,5) bulunmuştur. Bazılarında ölümcülsonuçlar görülmüştür. Yaşlı hastaların tedavisinde enfeksiyon riskine özellikle dikkatedilmelidir. Pediyatrik popülasyon:Yukarıda aşılamalar başlığına bakınız.
11 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriHUMIRA monoterapi olarak ve metotreksat ile kombine olarak romatoid artrit, poliartiküler jüvenil idiyopatik artrit ve psöriyatik artrit hastalarında incelenmiştir. HUMIRA antikoroluşumu, metotreksat ile birlikte verildiğinde monoterapi şeklinde kullanımına kıyasla dahadüşüktür. HUMIRA'nın metotreksat olmaksızın uygulanması, antikor oluşumunda artış, ayrıcaadalimumabın klirensinde artma ve etkililiğinde azalma ile sonuçlanmıştır (bkz. Bölüm 5.1). HUMIRA ve anakinra kombinasyonu önerilmemektedir (bkz. Bölüm 4.4). HUMIRA ve abatasept kombinasyonu önerilmemektedir (bkz. Bölüm 4.4). Özel popülasyonlara ilişkin ek bilgilerBöbrek/Karaciğer yetmezliği:Böbrek ya da karaciğer yetmezliği olan hastalarda klinik etkileşim çalışması gerçekleştirilmemiştir. Pediyatrik popülasyon:Pediyatrik popülasyonda klinik etkileşim çalışması gerçekleştirilmemiştir. Geriyatrik popülasyon:Geriyatrik popülasyonda klinik etkileşim çalışması gerçekleştirilmemiştir. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik Kategorisi: B Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli olan kadınların gebeliği önlemek için etkili kontrasepsiyon önlemleri almaları ve bu önlemlere son HUMIRA enjeksiyonundan sonra en az beş ay süreyledevam etmeleri önerilmektedir. Gebelik dönemi1500'den fazlası ilk trimesterde olmak üzere adalimumab maruziyeti olan, canlı doğumla sonuçlanan ve çıktıları bilinen, geniş sayıdaki (yaklaşık 2100) gebelikten prospektif olaraktoplanmış veriler, adalimumabın yenidoğanlarda malformasyon oranlarını arttırdığına işaretetmemektedir. Bir prospektif kohort çalışmasında, en az ilk trimester sırasında adalimumab ile tedavi edilen romatoid artrit (RA) veya Crohn hastalığı (CH) olan 257 kadın ve adalimumab ile tedaviedilmemiş RA veya CH olan 120 kadın kayıt altına alınmıştır. Çalışmanın primer sonlanımBelge Dcnokasıpmajardoğumdkusurlaımnzdoğum prevalansdı^piMjörtdoğöm-tkuroruofenen&zfeir 12 canlı doğum ile sonuçlanan gebelik oranı adalimumab ile tedavi edilen RA hastası kadınlarda 6/69 (%8,7) iken tedavi edilmeyenlerde 5/74 (%6,8) (düzeltilmemiş risk oranı 1,31, %95 güvenaralığı 0,38-4,52); adalimumab ile tedavi edilen CH tanılı kadınlarda 16/152 (%10,5) ikentedavi edilmeyenlerde 3/32 (%9,4) (düzeltilmemiş risk oranı 1,14, %95 güven aralığı 0,31-4,16)olarak bulunmuştur. RA ve CH için kombine (temel farklılıklara göre hesaplanan) düzeltilmişrisk oranı 1,10'dur (%95 güven aralığı 0,45-2,73). İkincil sonlanım noktaları olan kendiliğindendüşük, minör doğum kusurları, erken doğum, doğum ölçüleri ve ciddi veya fırsatçıenfeksiyonlar için adalimumab ile tedavi edilen ve tedavi almamış kadınlar arasında belirginfarklılıklar bulunmamakla birlikte ölü doğum veya malignite vakası bildirilmemiştir. Çalışmaküçük örnek boyutlarını ve randomize olmayan çalışma dizaynını içeren metodolojik limitleresahip olduğu için verilerin yorumlanması değişiklik gösterebilir. Maymunlarda yürütülen bir gelişimsel toksisite çalışmasında herhangi bir maternal toksisite, embriyotoksisite ya da teratojenite göstergesi bulunmamıştır. Adalimumabın postnatal toksisiteüzerindeki etkisine ilişkin klinik öncesi veri bulunmamaktadır (bkz. Bölüm 5.3). TNF-alfa'yı inhibe etmesi nedeniyle, gebelik sırasında verilen adalimumab yenidoğanda normal immün yanıtları etkileyebilir. Adalimumab gebelik sırasında sadece ihtiyaç duyulduğutakdirde kullanılmalıdır. Bununla birlikte, adalimumab gebelik sırasında alındığında plasentadan bebeğin serumuna geçebilir. Sonuç olarak da bu bebeklerde enfeksiyon riskinde artışa neden olabilir. Annekarnında adalimumab maruziyeti bulunan bebeklerde, anneye gebelik sırasında yapılan sonadalimumab enjeksiyonunu takip eden 5 ay içinde canlı aşı (örn. BCG aşısı) uygulamasıönerilmez. Laktasyon dönemiYayımlanan literatürden elde edilen kısıtlı bilgiye göre; insan sütünde anne serum seviyesinin %0,1 ila %1 konsantrasyonlarındaki adalimumab varlığı, adalimumabın anne sütüne çok düşükkonsantrasyonlarda geçtiğini göstermektedir. Oral yolla verilip sindirilen immünoglobülin Gproteinleri, intestinal proteolize uğrar ve düşük biyoyararlanıma sahiptir. Anne sütü ile beslenenyeni doğanlara/bebeklere etkisi beklenmemektedir. Sonuç olarak, HUMIRA emzirme dönemiboyunca kullanılabilir. Üreme yeteneği/FertiliteAdalimumabın fertilite üzerindeki etkilerine ilişkin klinik öncesi veri bulunmamaktadır. (bkz. Bölüm 5.3) 4.7. Araç ve makine kullanımı üzerindeki etkilerHUMIRA'nın taşıt ve makine kullanma yeteneği üzerine minör etkisi bulunabilir. HUMIRA uygulanmasını takiben vertigo ve görme bozukluğu görülebilir (bkz. Bölüm 4.8) 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti
13 HUMIRA, pivotal kontrollü ve açık etiketli çalışmalarda 60 ay veya daha uzun süreyle 9506 hasta üzerinde incelenmiştir. Bu çalışmalar hastalık süresi kısa ya da uzun olan romatoid artrit,jüvenil idiyopatik artrit (poliartiküler jüvenil idiyopatik artrit ve entezit ile ilişkili artrit)hastalarının yanı sıra aksiyal spondiloartrit (ankilozan spondilit, radyografik olarak AS kanıtıolmayan aksiyal spondiloartrit), psöriyatik artrit, Crohn hastalığı, ülseratif kolit, psöriyazis,hidradenitis suppurativa ve üveit hastalarını kapsamaktadır. Pivotal kontrollü çalışmalardaHUMIRA uygulanan 6089 hasta ve kontrollü dönem sırasında plasebo ya da aktif karşılaştırmaajanı uygulanan 3801 hasta dahil edilmiştir. Pivotal çalışmaların çift-kör, kontrollü dönemlerinde advers olaylar nedeniyle tedaviyi bırakan hastaların oranı, HUMIRA alan hastalar için %5,9 ve kontrol tedavisi alan hastalar için %5,4olmuştur. En yaygın olarak bildirilen advers reaksiyonlar; enfeksiyonlar (örn. nazofaranjit, üst solunum yolu enfeksiyonu ve sinüzit), enjeksiyon yeri reaksiyonları (eritem, kaşıntı, kanama, ağrı ya daşişme), baş ağrısı ve kas-iskelet ağrısıdır. HUMIRA için ciddi advers reaksiyonlar bildirilmiştir. HUMIRA gibi TNF bloke edici ajanlar immün sistemi etkilemekte ve bu ajanların kullanımı, vücudun enfeksiyona ve kansere karşıolan savunmasını etkileyebilmektedir. HUMIRA kullanımı ile fatal ve yaşamı tehdit edenenfeksiyonlar (sepsis, fırsatçı enfeksiyonlar ve TB dahil), HBV reaktivasyonu ve çeşitlimaligniteler de (lösemi, lenfoma ve HSTCL dahil) bildirilmiştir. Ciddi hematolojik, nörolojik ve otoimmün reaksiyonlar da bildirilmiştir. Bu reaksiyonlar seyrek pansitopeni, aplastik anemi, santral ve periferik demiyelinizan olay bildirimlerini ve lupus,lupusla ilişkili durumlar ve Stevens-Johnson sendromu bildirimlerini içermektedir. Pediyatrik popülasyonGenel olarak, pediyatrik hastalardaki advers reaksiyonlar sıklık ve tip olarak erişkin hastalarda görülenlere benzerlik göstermiştir. İstenmeyen etkilerAşağıda yer alan advers reaksiyonlar, klinik çalışmalar ve pazarlama sonrası deneyimlere dayanmaktadır ve sistem-organ sınıfı ve sıklık derecesine göre aşağıda gösterilmektedir: çokyaygın (> 1/10); yaygın (> 1/100 ila < 1/10); yaygın olmayan (> 1/1000 ila< 1/100), seyrek (>1/10000 ila <1/1000), çok seyrek (< 1/10000) ve bilinmiyor (mevcut veriler ilehesaplanamamaktadır). Her sıklık derecesi grubundaki istenmeyen olaylar, olayın şiddetdüzeyinde azalma sırasına göre listelenmiştir. Aşağıdaki advers reaksiyon listesi, çeşitliendikasyonlar arasında en sık görülme frekansını göstermektedir. İlave bilgilerin Bölüm 4.3,4.4 ve 4.8. içerisinde yer alması durumunda, sistem organ sınıfı sütununda yıldız (*) işaretibulunmaktadır. Enfeksiyon ve enfestasyonlar*Çok yaygın: Solunum yolu enfeksiyonları, (alt ve üst solunum yolu enfeksiyonları, pnömoni, sinüzit, farenjit, nazofarenjit ve herpes virüsüne bağlı pnömoni dahil) Yaygın: Sistemik enfeksiy°nlar (sepsis, kandidiyazis ve mfluenza dahilX intestinal enfeksiyonlar (viral gastroenterit dahil), deri ve yumuşak 14
Bağışıklık sistemi hastalıkları*Yaygın: Aşırı duyarlılık, alerjiler(mevsimsel alerji dahil) Yaygın olmayan: Sarkoidosis1), vaskülit Seyrek: Anafilaksi1)
Psikiyatrik hastalıklarYaygın: Ruh hali değişikliği (depresyon dahil), anksiyete, insomnia
15 Kulak ve iç kulak hastalıklarıYaygın: Vertigo Yaygın olmayan: Sağırlık, kulak çınlaması Kardiyak hastalıklar*Yaygın: Taşikardi Yaygın olmayan: Miyokard infarktüsü1),aritmi, konjestif kalp yetmezliği Seyrek: Kardiyak arest Vasküler hastalıklarYaygın: Hipertansiyon, ciltte kızarıklık,hematom Yaygın olmayan: Vasküler arteriyel oklüzyon,tromboflebit, aortik anevrizma Solunum, göğüs bozuklukları ve mediyastinal hastalıklar*Yaygın: Astım, dispne, öksürük Yaygın olmayan: Pulmoner embolizm1^ interstisyel akciğer hastalığı, kronik obstrüktif akciğer hastalığı, pnömonit, plevral efüzyon1) Seyrek: Pulmoner fibrosis1) Gastrointestinal hastalıklarÇok yaygın: Yaygın: Yaygın olmayan: Seyrek: Abdominal ağrı, bulantı ve kusma Gİ hemoraji, dispepsi, gastroözofajeal reflü hastalığı, sicca sendromu Pankreatit, disfaji, yüz ödemi İntestinal perforasyon1) Hepatobiliyer hastalıklar*Çok yaygın: Karaciğer enzimlerinin artması Yaygın olmayan: Seyrek: Kolesistit ve kolelitiyazis, hepatik steatoz, yükselmiş bilirubin değerleri Hepatit, hepatit B enfeksiyonunun tekrarlaması (reaktivasyonu)1^otoimmün hepatit1^
Bilinmiyor:
Karaciğer yetmezliği1} Deri ve derialtı doku hastalıklarıÇok yaygın:Yaygın: Döküntü (cilt döküntüsü) (eksfoliyatif döküntü dahil) Yaygın olmayan: Seyrek: Bilinmiyor: Eritema multiforme1), Stevens-Johnson sendromu1), anjioödem1), kütanöz vaskulit1), likenoid cilt reaksiyonu1-* Dermatomiyosit semptomlarının kötüleşmesi1) Kas-iskelet bozuklukları, bağ dokusu ve kemik hastalıklarıÇok yaygın: Kas-iskelet ağrısı Yaygın: Kas spazmları (kan kreatin fosfokinaz değerlerinin yükselmesi dahil) Yaygın olmayan: Rabdomiyoliz, sistemik lupus eritematozus Seyrek: Lupns benzeri sendrom
16 Böbrek ve idrar yolu hastalıklarıYaygın: Renal yetmezlik, hematüri Yaygın olmayan: Noktüri Üreme sistemi ve meme hastalıklarıYaygın olmayan: Erektil disfonksiyon Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar*
Çok yaygın: Yaygın: Yaygın olmayan: AraştırmalarYaygın:
Bilinmiyor:
Enjeksiyon yerinde reaksiyon (enjeksiyon yeri eritemi dahil) Göğüs ağrısı, ödem, pireksi1) (ateş) İnflamasyon Koagülasyon ve kanama bozuklukları (uzamış aktive kısmi tromboplastin süresi dahil), pozitif oto antikor testi (çift sarmal DNAantikor dahil), yükselmiş kan laktat dehidrojenaz düzeyleriKilo artışı2) Yaralanma ve zehirlenmeYaygın: Yara yerinde iyileşmede gecikme * Daha fazla bilgi, Bölüm 4.3, 4.4 ve 4.8'de bulunmaktadır. ** Açık etiketli uzatma çalışmalarını içermektedir. 1) Spontan bildirim verileri dahildir. 2) Erişkin endikasyonlarında, 4-6 aylık tedavi dönemi boyunca plasebo için belirlenen (eksi) -0,4 kg ila 0,4 kg aralığıyla karşılaştırıldığında, adalimumab için vücut ağırlığında başlangıcagöre kaydedilen ortalama değişiklik 0,3 kg ile 1 kg arasında değişmiştir. Kontrol grubuolmaksızın ortalama maruziyetin yaklaşık 1-2 yıl olduğu uzun süreli uzatma çalışmalarında,özellikle Crohn hastalığı ve ülseratif koliti olan hastalarda 5-6 kg'lık vücut ağırlığı artışı dagözlenmiştir. Bu etkinin altında yatan mekanizma bilinmemesine karşın, adalimumabın anti-enflamatuvar etkisiyle bağlantılı olabilmektedir. t Pazarlama sonrası dönemde TNF alfa inhibitörleri kullanan hastalarda seyrek otoimmün hepatit vakaları raporlanmıştır. Üveit:HUMIRA ile iki haftada bir tedavi uygulanan üveit hastalarının güvenlilik profili, HUMIRA'nın bilinen güvenlilik profiliyle tutarlılık göstermiştir. Seçilen advers reaksiyonların tanımı:Enjeksiyon yeri reaksiyonlarıErişkin ve çocuklarda yürütülen pivotal kontrollü klinik çalışmalarda HUMIRA ile tedavi edilen hastaların %12,9'unda enjeksiyon yeri reaksiyonları (eritem ve/veya kaşıntı, hemoraji,ağrı ya da şişme) görülmesine karşın plasebo ya da aktif kontrol alanların %7,2'sinde de bureaksiyonlar görülmüştür. Enjeksiyon yeri reaksiyonları genellikle ilacın kesilmesinigerektirmemiştir. Belge 56aklUaklUaklUQ3NRSHY3ZmxXS3kOBelge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys17 Erişkin ve çocuklarda yürütülen pivotal kontrollü klinik çalışmalarda enfeksiyon oranı, HUMIRA ile tedavi edilen hastalarda hasta yılı başına 1,51, plasebo ve aktif kontrol ajanı iletedavi edilen hastalarda hasta yılı başına 1,46 olmuştur. Enfeksiyonlar primer olaraknazofarenjit, üst solunum yolu enfeksiyonu ve sinüzit olmuştur. Hastaların çoğu enfeksiyoniyileştikten sonra HUMIRA tedavisine devam etmiştir. Ciddi enfeksiyonların insidansı, HUMIRA ile tedavi edilen hastalarda hasta yılı başına 0,04, plasebo ve aktif kontrol ajanı ile tedavi edilen hastalarda hasta yılı başına 0,03 olmuştur. HUMIRA ile erişkin ve çocuklarda yürütülen kontrollü ve açık etiketli çalışmalarda bildirilen ciddi enfeksiyonlar (nadiren ortaya çıkan fatal enfeksiyonlar dahil) arasında tüberküloz (miliyerve ekstrapulmoner yerleşimler dahil) ve invazif fırsatçı enfeksiyonlar (örn. dissemine veyaekstrapulmoner histoplazmozis, blastomikoz, koksidiyoidomikoz, pnömosistis, kandidiyazis,aspergillozis ve listeriyozis) bulunmaktadır. Tüberküloz olgularının çoğu tedavi başlatıldıktansonraki ilk sekiz ay içerisinde görülmüştür ve latent hastalığın yeniden ortaya çıkışını yansıtıyorolabilir. Maligniteler ve lenfoproliferatif bozukluklarJüvenil idiyopatik artrit (poliartiküler jüvenil idiyopatik artrit ve entezit ile ilişkili artrit) hastalarındaki HUMIRA çalışmaları sırasında 655,6 hasta yılı maruziyeti olan 249 pediyatrikhastada malignite gözlemlenmemiştir. Buna ilave olarak, Crohn hastalığı olan pediyatrikhastalarda HUMIRA çalışmaları sırasında 498,1 hasta yılı maruziyeti olan 192 pediyatrikhastada malignite gözlemlenmemiştir. Kronik plak psöriyazisi olan pediyatrik hastalardaki birçalışmasında 80 hasta yılı maruziyeti olan 77 pediyatrik plak psöriyaz hastasında malignitegözlemlenmemiştir. Pediyatrik üveit hastalarındaki bir HUMIRA çalışmasında 58,4 hasta yılımaruziyeti olan 60 pediyatrik hastada hiçbir malignite gözlenmemiştir. Orta ila şiddetli derecede aktif romatoid artrit, psöriyatik artrit, ankilozan spondilit, radyografik olarak AS kanıtı olmayan aksiyal spondiloartrit, Crohn hastalığı, ülseratif kolit, psöriyazis,hidradenitis suppurativa ve üveiti olan erişkin hastalarda yürütülen en az 12 haftalık pivotalçalışmalarının kontrollü dönemleri sırasında lenfoma ve melanoma-dışı cilt kanseri dışındakimaligniteler, tedavisindeki 5291 hastada 1000 hasta yılı başına 6,8 (4,4; 10,5) oranında (%95GA) iken; buna karşın 3444 kontrol hastasında 1000 hasta yılı başına 6,3 (3,4; 11,8) oranındagözlemlenmiştir (medyan tedavi süresi için 4 ay, kontrol tedavisi hastaları için ise 3,8 ayolmuştur). Melanoma-dışı cilt kanserlerinin oranı (%95 GA), tedavisindeki hastalarda 1000hasta yılı başına 8,8 (6; 13), kontrol hastalarında ise 1000 hasta yılı başına 3,2 (1,3; 7,6)olmuştur. Bu cilt kanserleri arasında skuamöz hücreli karsinomlar, tedavisindeki hastalarda1000 hasta yılı başına 2,7 (1,4; 5,4), kontrol hastalarında 1000 hasta yılı başına 0,6 (0,1; 4,5)oranında olmuştur. Lenfomaların oranı (%95 GA), tedavisindeki hastalarda 1000 hasta yılıbaşına 0,7 (0,2; 2,7), kontrol hastalarında da 1000 hasta yılı başına 0,6 (0,1; 4,5) olmuştur. 6427 hastayı kapsayan, medyan süresi yaklaşık 3,3 yıl olan ve 26,439 hasta yılından fazla tedavi süresini temsil eden klinik çalışmaların kontrollü dönemlerinde ve halen sürmekte olan vetamamlanmış açık etiketli çalışmalarda, lenfoma ve melanoma-dışı cilt kanserleri haricigözlemlenen malignite oranı 1000 hasta yılı başına yaklaşık 8,5'tir. Melanoma-dışı ciltkanserlerinin gözlemlenen oranı 1000 hasta yılı başına yaklaşık 9,6'dır ve lenfoma içingözlemlenen oranı 1000 hasta yılı başına yaklaşık 1,3'tür
18 Ocak 2003 ve Aralık 2010 tarihleri arasındaki pazarlama sonrası deneyimlere göre ağırlıklı olarak romatoid artrit hastalarında raporlanan malignite oranı, 1000 hasta yılı başına ortalama2,7'dir. Bildirilen melanoma-dışı cilt kanserleri ve lenfomaların oranı, sırasıyla 1000 hastatedavi yılı başına yaklaşık olarak 0,2 ve 0,3'tür (bkz. Bölüm 4.4). Adalimumab ile tedavi edilen hastalarda, pazarlama sonrası dönemde nadir olarak hepatosplenik T hücreli lenfoma vakaları bildirilmiştir (bkz. Bölüm 4.4). OtoantikorlarI-V numaralı romatoid artrit çalışmalarında değişik zaman noktalarında hastaların serum örneklerinde otoantikor testleri yapılmıştır. Bu çalışmalarda HUMIRA ile tedavi edilenhastaların %11,9'unda, plasebo ve aktif kontrol ajanıyla tedavi edilen hastaların %8,1'indebaşlangıç döneminde negatif olan anti-nükleer antikorların 24. haftada pozitif titrelerde olduğubildirilmiştir. Bütün romatoid artrit ve psöriyatik artrit çalışmalarında HUMIRA ile tedaviedilen 3441 hastadan 2'sinde yeni başlayan lupusa benzer sendromu düşündüren klinikbelirtiler gözlemlenmiştir. Tedavinin kesilmesinden sonra hastalarda düzelme olmuştur. Hiçbirhastada lupus nefriti veya santral sinir sistemi semptomları gelişmemiştir. Hepatobiliyer olaylarHUMIRA ile romatoid artrit ve psöriyatik artrit hastalarında yürütülen, 4 ila 104 hafta aralığında kontrol periyodu olan Faz 3 kontrollü klinik çalışmalarda, HUMIRA ile tedavi görenhastaların %3,7'sinde ALT yükselmeleri >3 x NÜS (normalin üst sınırı) iken, kontrol grubundabu oran %1,6 olmuştur. HUMIRA'nın kontrollü Faz 3 çalışmalarında 4 ila 17 yaş arasındaki poliartiküler jüvenil idiyopatik artriti olan hastalar ve 6 ila 17 yaş arasındaki entezit ile ilişkili artriti olan hastalarda,> 3 x NÜS ALT yükselmeleri, HUMIRA ile tedavi edilen hastalarda %6,1 ve kontrol tedavisialan hastalar %1,3 olmuştur. ALT yükselmelerinin çoğu eş zamanlı metotreksat kullanımındagörülmüştür. HUMIRA 'nın poliartiküler jüvenil idiyopatik artriti olan 2 ila 4 yaş arasındakihastalardaki Faz 3 çalışmasında >3 x NÜS olan herhangi bir ALT yükselmesigerçekleşmemiştir. HUMIRA ile Crohn hastalarında ve ülseratif kolit hastalarında yürütülen 4 ila 52 hafta aralığında kontrol periyodu olan Faz 3 kontrollü klinik çalışmalarda, HUMIRA ile tedavi edilenhastaların %0,9'unda ALT yükselmeleri >3 x NÜS olmakla birlikte, kontrol grubunda da buoran %0,9 olmuştur. Pediyatrik Crohn hastalığı olan hastalarda yürütülen, tedavinin 52. haftasına dek vücut ağırlığına göre ayarlanan indüksiyon tedavisinin ardından vücut ağırlığına göre ayarlananidame doz rejimlerinin etkililik ve güvenliliğinin araştırıldığı bir Faz 3 çalışmasında başlangıçta4'ünün eş zamanlı olarak immünosupresan almış olduğu hastaların %2,6'sında (5/192) ALTyükselmeleri > 3 x NÜS olarak görülmüştür. HUMIRA ile plak psöriyazis hastalarında yürütülen 12 ila 24 hafta aralığında kontrol periyodu olan Faz 3 kontrollü klinik çalışmalarda, HUMIRA ile tedavi edilen hastaların %1,8'inde ALTyükselmeleri >3 x NÜS olmakla birlikte, kontrol grubunda bu oran %1,8 olmuştur. Plak psöriyazisli pediyatrik hastalar ile yapılan HUMIRA Faz 3 çalışmasında >3 x NÜS ALT artışı meydana gelmemiştir. Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys19 Erişkin üveit hastaları üzerinde 80 haftaya varan süreyle gerçekleştirilen, HUMIRA tedavisi ve kontrol tedavisinin sırasıyla 166,5 gün ve 105 günlük medyan (ortanca) maruziyetin olduğukontrollü HUMIRA çalışmalarında (başlangıç dozu 0. haftada 80 mg dozunu takiben 1.haftadan başlanarak iki haftada bir 40 mg), HUMIRA tedavisi uygulanan hastaların %2,4'ündeve kontrol tedavisi uygulanan hastaların %2,4'ünde >3 x NÜS düzeyinde ALT yükselmeleriortaya çıkmıştır. Tüm endikasyonlar için yapılan klinik çalışmalara dahil olan hastalarda ALT değerlerindeki yükselme asemptomatik olup, vakaların çoğunda geçici olmuştur ve tedavinin devamıesnasında bu durum ortadan kalkmıştır. Bununla beraber, pazarlama sonrası raporları,adalimumab alan hastalarda otoimmün hepatiti de kapsayan hepatit gibi karaciğeryetmezliğinin öncesinde seyredebilen ciddi karaciğer bozuklukları daha az olmakla beraberkaraciğer yetmezliği bildirilmiştir. Azatioprin / 6-merkaptopurin ile eş zamanlı tedavi Erişkin Crohn hastalarında yapılan çalışmalarda, HUMIRA'nın tek başına kullanımı ile kıyaslandığında, HUMIRA ve azatiyoprin/6-merkaptopurin kombinasyonunda daha yüksekmalignite olasılığı ve ciddi enfeksiyona bağlı yan etkiler görülmüştür. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr[email protected]. Doz aşımı ve tedavisiKlinik çalışmalar sırasında doz kısıtlayıcı toksisite gözlemlenmemiştir. Değerlendirilen en yüksek doz düzeyi 10 mg/kg'lık multipl intravenöz dozlar olup; önerilen dozun yaklaşık 15katıdır. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: İmmünosupresanlar, Tümör nekröz faktörü alfa (TNF-alfa) inhibitörleri ATC kodu: L04AB04 Etki mekanizması Adalimumab, TNF'ye spesifik olarak bağlanır ve TNF'nin p55 ve p75 hücre yüzey reseptörleriyle etkileşimini bloke ederek bu faktörün biyolojik fonksiyonunu nötralize eder.
20 Adalimumab, lökosit migrasyonundan sorumlu adezyon moleküllerinin düzeylerindeki değişiklikler de dahil olmak üzere, TNF tarafından indüklenen veya düzenlenen biyolojikyanıtları da modüle eder (ELAM-1, VCAM-1 ve ICAM-1 için IC50 değeri 0,1-0,2 nM). Farmakodinamik etkilerRomatoid artrit hastalarında HUMIRA tedavisinden sonra inflamasyonun akut faz reaktanlarında (C-reaktif protein (CRP) ve eritrosit sedimentasyon hızı (ESR)) ve serumsitokinlerinin (IL-6) düzeylerinde başlangıç değerlerine kıyasla hızlı bir düşüş gözlemlenmiştir.HUMIRA uygulamasından sonra, kıkırdak hasarından sorumlu olan ve dokunun yenidenyapılanması sürecine yol açan matriks metalloproteinazların (MMP-1 ve MMP-3) düzeyleri dedüşmüştür. HUMIRA ile tedavi edilen hastalarda kronik inflamasyonun hematolojikbulgularında çoğunlukla düzelme meydana gelmiştir. Poliartiküler jüvenil idiyopatik artrit, Crohn hastalığı, ülseratif kolit ve hidradenitis suppurativa hastalarında, HUMIRA tedavisinden sonra CRP düzeylerinde de hızlı bir düşüşgözlemlenmiştir. Crohn hastalarında, TNF-alfa ekspresyonundaki anlamlı azalma da dahilolmak üzere, kolondaki inflamasyon göstergelerini eksprese eden hücre sayısında düşüş olduğugözlemlenmiştir. İntestinal mukozada yapılan endoskopik çalışmalarda, adalimumab ile tedaviedilen hastalarda mukoza iyileşmesine ilişkin kanıtlar olduğu gösterilmiştir. Klinik etkililik ve güvenlilikRomatoid artriti olan erişkinlerHUMIRA, bütün romatoid artrit klinik çalışmalarında 3000'i aşkın hasta üzerinde değerlendirilmiştir. HUMIRA'nın etkililiği ve güvenliliği beş adet randomize, çift kör ve iyikontrollü çalışmada belirlenmiştir. Bazı hastalar 120 aya varan sürelerle tedavi edilmiştir. RA I çalışmasında, en az bir tane hastalık modifiye edici anti-romatizmal ilaç tedavisiyle başarısız kalınan ve haftada 12,5-25 mg (metotreksat intoleransı durumunda 10 mg) metotreksattedavisiyle yeterli etki görülmeyen ve metotreksat dozu haftada 10-25 mg arasında sabitseyreden, 18 yaş ve üstü 271 orta ila şiddetli derecede aktif romatoid artrit hastasıdeğerlendirilmiştir. Hastalara 24 hafta süreyle 2 haftada bir, 20, 40 veya 80 mg HUMIRA veyaplasebo dozları verilmiştir. RA II çalışmasında en az bir adet hastalık modifiye edici anti-romatizmal ilaç tedavisiyle başarısız kalınan, 18 yaş ve üstü 544 orta ila şiddetli aktif romatoid artrit hastasıdeğerlendirilmiştir. 26 hafta süreyle, 20 veya 40 mg dozunda HUMIRA subkütan enjeksiyonyoluyla (alternatif haftalarda plasebo verilmek üzere) iki haftada bir, ya da her hafta uygulanmışve aynı süreyle her hafta plasebo verilmiştir. Başka hiçbir hastalık modifiye edici ilacınkullanılmasına izin verilmemiştir. RA III çalışmasında haftada 12,5-25 mg metotreksat tedavisiyle yeterli etki görülmeyen veya 10 mg/hafta metotreksata intoleransı olan 18 yaş ve üstü 619 orta ila şiddetli derecede aktifromatoid artrit hastası değerlendirilmiştir. Bu çalışmada üç grup vardır. İlk gruba 52 haftasüreyle her hafta plasebo enjeksiyonu uygulanmıştır. İkinci gruba 52 hafta süreyle her hafta 20mg HUMIRA verilmiştir. Üçüncü gruba dönüşümlü olarak alternatif haftalarda plaseboenjeksiyonları yapılmak üzere iki haftada bir 40 mg HUMIRA uygulanmıştır. İlk 52 haftatamamlandıktan sonra, 457 hasta. 10 yıla varan süreyle iki haftada bir 40 mg HUMIRA/MTXuygulanan açık etiketli bir uzatma fazına kaydedilmiştir. 21 RA IV çalışmasında, 18 yaş ve üstü 636 orta ila şiddetli derecede aktif romatoid artrit hastasında primer olarak güvenlilik değerlendirilmiştir. Hastaların ya hiç hastalık modifiye edici anti-romatizmal ilaç kullanmamış olmalarına ya da tedavinin en az 28 gün stabil olması şartıyla,mevcut romatolojik tedavilerini sürdürmelerine izin verilmiştir. Bu tedavi rejimleri metotreksat,leflunomid, hidroksiklorokin, sulfasalazin ve/veya altın tuzlarını içermektedir. Hastalar 24hafta süreyle iki haftada bir 40 mg HUMIRA veya plasebo alacak şekilde randomize edilmiştir. RA V çalışmasında, daha önce hiç metotreksat kullanmamış, erken dönem (ortalama hastalık süresi 9 aydan daha az), orta ila şiddetli derecede aktif romatoid artriti olan 799 erişkin hastadeğerlendirilmiştir. Bu çalışmada 104 hafta süreyle iki haftada bir 40 mg HUMIRA vemetotreksat kombinasyon tedavisi ile iki haftada bir 40 mg HUMIRA monoterapisi vemetotreksat monoterapisinin, romatoid artrit bulgu ve semptomları ile eklem hasarındakiprogresyon hızını azaltmadaki etkililikleri incelenmiştir. İlk 104 haftanın tamamlanmasındansonra, 497 hasta 10 yıla kadar iki haftada bir 40 mg HUMIRA'nın uygulandığı açık etiketli fazçalışmasına dahil edilmiştir. RA I, II ve III çalışmalarının primer sonlanım noktası ve RA IV çalışmasının sekonder sonlanım noktası 24. veya 26. haftalarda ACR 20 yanıtına ulaşan hastaların yüzdesidir. RA Vçalışmasında primer sonlanım noktası, 52. haftada ACR 50 yanıtı elde edilen hastalarınyüzdesidir. RA III ve V çalışmalarında, 52. haftada hastalık progresyonunun (röntgensonuçlarıyla saptanan) gerilemesi, ilave bir primer sonlanım noktası olmuştur. RA IIIçalışmasında aynı zamanda yaşam kalitesindeki değişimler de primer sonlanım noktasıolmuştur. ACR yanıtıHUMIRA ile tedavi edilen ve ACR 20, 50 ve 70 yanıtlarına ulaşan hastaların yüzdesi RA I, II ve III çalışmaları arasında tutarlı olmuştur. İki haftada bir 40 mg dozu ile alınan sonuçlar, Tablo6'da özetlenmektedir. Tablo 6. Plasebo - Kontrollü Çalışmalardaki ACR Yanıtları (Hasta Yüzdesi)
22 a RA I çalışmasında 24. haftada, RA II çalışmasında 26. haftada ve RA III çalışmasında 24 ve 52. haftalarda b İki haftada bir uygulanan 40 mg HUMIRA c MTX = metotreksat**p< 0,01, plaseboya karşı HUMIRAGD= Geçerli değil RA I-IV çalışmalarında ACR yanıtı kriterlerinin bütün bireysel bileşenleri (duyarlı ve şiş eklemlerin sayısı, doktor ve hasta tarafından yapılan hastalık aktivitesi ve ağrıdeğerlendirmeleri, Sağlık Değerlendirme Anketi (HAQ) skorları ve CRP (mg/dL) değerleri),24. veya 26. haftalarda plaseboya göre düzelme göstermiştir. RA III çalışmasında budüzelmeler 52 hafta boyunca sürdürülmüştür. RA III çalışmasının açık etiketli uzatma fazında 10 yıla kadar izlenen hastaların çoğunluğunda ACR yanıt oranları devam etmiştir. İki haftada bir 40 mg HUMIRA dozuna randomize edilmişolan 207 hastadan 114'ü, 5 yıl boyunca iki haftada bir 40 mg HUMIRA almaya devam etmiştir.Bu hastalar arasında 86 hastada (%75,4) ACR 20 yanıtları, 72 hastada (%63,2) ACR 50 yanıtlarıve 41 hastada (%36) ACR 70 yanıtları elde edilmiştir. 207 hastadan 81'i iki haftada bir 40 mgHUMIRA tedavisine 10 sene boyunca devam etmişlerdir. Bunlar arasında, 64 hastada (%79)ACR 20 yanıtları, 56 hastada (%69,1) ACR 50 yanıtları ve 43 hastada (%53,1), ACR 70yanıtları elde edilmiştir. RA IV çalışmasında HUMIRA artı standart tedavi gören hastaların ACR 20 yanıtı, plasebo artı standart tedavi ile tedavi edilen hastalardaki yanıta göre istatistiksel anlamlılık taşıyacak şekildedaha iyi olmuştur (p< 0,001). RA I-IV çalışmalarında, HUMIRA ile tedavi gören hastalarda tedaviye başlandıktan sonra bir-iki hafta gibi erken bir sürede, plasebo alan hastalar ile karşılaştırıldığında istatistiksel olarak anlamlı ACR 20 ve 50 yanıtlarına ulaşılmıştır. Erken dönemdeki ve daha önce metotreksat kullanmamış romatoid artrit hastalarının yer aldığı RA V çalışmasında, HUMIRA ve metotreksat ile yapılan kombinasyon tedavisi 52. haftada,metotreksat monoterapisi ve HUMIRA monoterapisinden daha hızlı ve anlamlı olarak dahayüksek ACR yanıtları sağlamış ve bu yanıtlar 104. haftaya kadar sürdürülebilmiştir. (bkz. Tablo 7).
23
RA V çalışması için yapılan açık etiketli uzatma çalışmasında, 10 yıla varan bir süre boyunca takip edildiğinde ACR yanıt oranları korunmuştur. İki haftada bir HUMIRA 40 mg grubunarandomize edilmiş olan 542 hastanın 170'i, iki haftada bir HUMIRA 40 mg tedavisine 10 yılboyunca devam etmiştir. Bunlar arasında yer alan 154 hastada (%90,6) ACR 20, 127 hastada(%74,7) ACR 50 ve 102 hastada (%60) ACR 70 yanıtları elde edilmiştir. 52. haftada HUMIRA/metotreksat kombinasyon tedavisi gören hastaların %42,9'unda klinik remisyon (DAS28 (CRP) < 2,6) sağlanırken, bu oran metotreksat monoterapisi alan hastalarda%20,6 ve HUMIRA monoterapisi alan hastalarda %23,4 olmuştur. HUMIRA/metotreksatkombinasyon tedavisi, yakın dönemde tanı konulmuş orta ila şiddetli derecede romatoid artritiolan hastalarda düşük hastalık durumunun sağlanmasında metotreksat ve HUMIRAmonoterapilerinden klinik ve istatistik olarak daha üstündür (sırasıyla p < 0,001 ve p < 0,001).İki monoterapi kolundaki yanıtlar benzerdir (p=0,447). Açık etiketli uzatma çalışmasına katılanve ilk olarak HUMIRA monoterapisi ya da HUMIRA/metotreksat kombinasyon tedavisigrubuna randomize edilmiş olan 342 hastanın 171'i 10 yıllık HUMIRA tedavisinitamamlamıştır. Bunlar arasında yer alan 109 hastanın (%63,7) 10 yıl sonunda remisyondaolduğu bildirilmiştir. Radyografik yanıtHUMIRA ile tedavi edilen hastalardaki ortalama romatoid artrit süresinin yaklaşık 11 yıl olduğu RA III çalışmasında, yapısal eklem hasarı radyografik olarak değerlendirilmiş veModifiye Total Sharp Skoru (mTSS) ve bileşenlerinde, erozyon skorunda ve eklem aralığıdaralma skorunda değişim şeklinde ifade edilmiştir. HUMIRA/metotreksat hastaları, 6. ve 12.aylarda tek başına metotreksat alan hastalara kıyasla anlamlı olarak daha az radyografikprogresyon göstermişlerdir (bkz. Tablo 8). RA III çalışmasının açık etiketli uzatma fazında, yapısal harabiyetin progresyon hızındaki azalma bir grup hastada 8 ve 10 yd süreyle devam diştin ^angjçta iki haftada bir 40 mgBelge doHUMIRA: iletedavikedüen/ 207 rhastadaakSUi 8. yıldaraady©graf})k//ol^akkdeğ©Blendirilmiş$ir. 24 Bu hastaların 48'inde, 0,5 veya daha az mTSS değişimi olarak tanımlanan, yapısal hasarda ilerleme olmaması durumu tespit edilmiştir. Başlangıçta iki haftada bir 40 mg HUMIRA iletedavi edilen 207 hastadan 79'u 10. yılda radyografik olarak değerlendirilmiştir. Bu hastaların40'ında, 0,5 veya daha az mTSS değişimi olarak tanımlanan, yapısal hasarda ilerleme olmamasıdurumu tespit edilmiştir.
aralıkları c Sıralı analize (rank analysis) dayalı olarak d Eklem Aralığında DaralmaGA: Güven Aralığı RA V çalışmasında, yapısal eklem hasarı radyografik olarak değerlendirilmiş olup modifiye Total Sharp Skorunda değişim şeklinde ifade edilmiştir (bkz. Tablo 9).
a p değeri, metotreksat monoterapisi ve HUMIRA/metotreksat kombinasyon tedavisi arasında Mann-Whitney U testi kullanılarak yapılan çiftli kıyaslamadan elde edilmiştir.b p değeri, HUMIRA monoterapisi ve HUMIRA/metotreksat kombinasyon tedavisi arasındaMann-Whitney U testi kullanılarak yapılan çiftli kıyaslamadan elde edilmiştir.c p değeri, HUMIRA monoterapisi ve metotreksat monoterapisi arasında Mann-Whitney Utesti kullanılarak yapılan ikili kıyaslamadan elde edilmiştir. GA: Güven Aralığı 52 haftalık ve 104 haftalık tedavilerden sonra progresyon göstermeyen (modifiye Total SharpSkoru'nda başlangıç dönemine göre değişim < 0,5) hastaların yüzdesi, HUMIRA/metotreksatkombinasyon tedavisi ile (sırasıyla %63,8 ve %61,2), metotreksat monoterapisi(sırasıyla %37,4 ve %33,5, p< 0,001) ve HUMIRA monoterapisine (sırasıyla %50,7, p< 0,002ve %44,5, p< 0,001) kıyasla anlamlı derecede daha yüksektir. RA V çalışması için yapılan açık etiketli uzatma çalışmasında; başlangıçta metotreksat monoterapisi, HUMIRA monoterapisi ve HUMIRA/metotreksat kombinasyon tedavisi
25 gruplarına randomize edilen hastaların, 10. yılda modifiye Total Sharp Skoru'nda başlangıca göre elde edilen ortalama değişiklik sırasıyla 10,8, 9,2 ve 3,9 olarak belirlenmiştir. Radyografikprogresyon saptanmayan hastaların oranları ise sırasıyla %31,3, %23,7 ve %36,7 olmuştur. Yaşam kalitesi ve fiziksel fonksiyonSağlık Değerlendirme Anketi'nin (HAQ) iş göremezlik indeksi kullanılarak sağlığa ilişkin yaşam kalitesi ve fiziksel fonksiyon, orijinal uygunlukta ve iyi-kontrollü dört çalışmadadeğerlendirilmiştir; bu değerlendirmeler RA III çalışmasının 52. haftasında öncedentanımlanmış bir primer sonlanım noktasıdır. Dört çalışmanın tamamında, HUMIRA'nın bütündozları/doz şemaları plasebo ile karşılaştırıldığında 6. ayda HAQ anketinin iş göremezlikindeksinde başlangıç dönemine göre istatistiksel olarak anlamlı bir iyileşme olduğu görülmüşve aynı durum RA III çalışmasında 52. haftada da gözlemlenmiştir. Bu dört çalışmanınhepsinde Kısa Sağlık Çalışma Formu (SF 36) sonuçları, Fiziksel Bileşen Özeti (PCS) skorlarıistatistiksel anlamlılık taşıyacak şekilde, HUMIRA'nın bütün dozları/doz şemaları için bubulguları desteklemektedir, aynı zamanda ağrı ve vitalite skorları ise, iki haftada bir 40 mg dozuiçin istatistiksel olarak anlamlıdır. Kronik Hastalık Tedavisi Fonksiyonel Değerlendirme(FACIT) skorlarının değerlendirildiği üç çalışmanın hepsinde de (RA I, III, IV çalışmaları), buskorlar ile ölçülen yorgunlukta istatistiksel olarak anlamlı bir azalma görülmüştür. RA III çalışmasında fiziksel fonksiyonda iyileşme elde eden ve tedaviye devam eden hastaların çoğunda iyileşme, açık etiketli tedavinin 520. haftası boyunca (120 ay) devam etmiştir. Yaşamkalitesindeki iyileşmeler 156. haftaya (36 ay) kadar ölçülmüş ve iyileşmeler bu dönem boyuncakorunmuştur. RA V çalışmasında HAQ iş göremezlik indeksi ve SF 36'nın fiziksel bileşenindeki iyileşmeler, HUMIRA/metotreksat kombinasyon tedavisi ile, metotreksat monoterapisi ve HUMIRAmonoterapisine kıyasla 52. haftada daha yüksektir ve bu yükseklik 104. haftada da devametmektedir (p< 0,001). Açık etiketli uzatma çalışmasını tamamlayan 250 hastada, 10 yıllıktedavi boyunca fiziksel fonksiyonlardaki iyileşme sürdürülmüştür. Erişkin plak psöriyazisHUMIRA'nın güvenliliği ve etkililiği randomize, çift-kör çalışmalarda sistemik tedavi veya fototerapi adayı olan erişkin kronik plak psöriyazis (Ps) (> %10 Vücut Yüzey Alanı (VYA)tutulumu ve Psöriyazis Alan ve Şiddet İndeksi (PASI) > 12 veya > 10) hastalarındadeğerlendirilmiştir. Psöriyazis I ve II çalışmasına kayıt edilen hastaların %73'ü öncedensistemik tedavi veya fototerapi almışlardır. HUMIRA'nın güvenliliği ve etkililiği ayrıca,randomize çift-kör bir çalışmada (Psöriyazis III çalışması), sistemik tedavi adayı olan ve elve/veya ayak psöriyazisi ile birlikte orta ila şiddetli derecede kronik plak psöriyazisi bulunanerişkin hastalarda değerlendirilmiştir. Psöriyazis I çalışmasında (REVEAL) üç tedavi dönemi içinde 1212 hasta değerlendirilmiştir. A döneminde, hastalar plasebo veya başlangıç 80 mg HUMIRA dozunu takiben, başlangıçdozundan bir hafta sonra başlanarak iki haftada bir 40 mg HUMIRA almışlardır. 16 haftalıktedaviden sonra en az PASI 75 yanıtına ulaşan hastalar (PASI skorunda başlangıca göre en az%75 düzelme olması) B dönemine girmişler ve iki haftada bir açık etiketli olarak 40 mgHUMIRA almışlardır. 33. haftada > PASI 75 yanıtını koruyan ve A döneminde orijinal olarakaktif tedaviye randomize edilmiş olan hastalar, tekrar randomize edilerek C döneminde ilave19 hafta boyunca, iki haftada bir 40 mg HUMIRA tedavisi veya plasebo almışlardır. Tüm tedaviBelge DtğupiaMda z^rtalHMâutaşianiğışYPASIss&koru 1 $,9dfe!pp vee&&ş^bgçtakiyeHeklmiin-tit@lobal 26 Değerlendirme (PGA) skoru orta (dahil edilen hastaların %53'ü), şiddetli (%41) ila çok şiddetli (%6) oranlarında değişmiştir. Psöriyazis II çalışmasında (CHAMPION), 271 hastada metotreksat ve plasebo karşısında HUMIRA'nın etkililiği ve güvenliliği karşılaştırmıştır. Hastalar, başlangıç dozu olarak 7,5 mgMTX almışlar ve daha sonra doz, 12. haftaya kadar arttırılarak maksimum 25 mg'a ulaşacakşekilde plasebo veya 16 hafta boyunca 80 mg HUMIRA başlangıç dozunu takiben iki haftadabir 40 mg HUMIRA (başlangıç dozundan bir hafta sonra başlayarak) verilmiştir. 16 haftalıktedavinin ötesinde HUMIRA ve MTX'ı karşılaştıran veri mevcut değildir. 8. hafta ve/veya 12.haftada > PASI 50 yanıtına ulaşan MTX alan hastalarda, doz daha fazla arttırılmamıştır. Tümtedavi gruplarında ortalama başlangıç PASI skoru 19,7'dur ve başlangıçtaki PGA skoru hafif'(< %1) orta (%48) şiddetli (%46) çok şiddetli (%6) oranlarında değişmiştir. Tüm Faz II ve Faz III psöriyazis çalışmalarına katılan hastalar en az 108 hafta daha HUMIRA verilen açık etiketli uzatma çalışmasına katılmaya uygundu. Psöriyazis I ve II çalışmasında, primer sonlanım noktası başlangıca göre 16. haftada PASI 75 yanıtına ulaşan hastaların oranıdır (Tablo 10 ve 11).
Psöriyazis I çalışmasında, PASI 75 yanıtına ulaşan ve 33. haftada plasebo almak üzere yeniden randomize edilen hastaların %28'i, HUMIRA'ya devam edenlerin %5'ine kıyasla (p< 0,001)yeterli yanıtın kaybını yaşamıştır (33. haftadan sonra ve 52. hafta veya öncesinde 33. haftayakıyasla PASI skorunda en az 6 puanlık artış ile başlangıca kıyasla < PASI 50 yanıtı ile tpv* OT1\belge, güvenli elektronik imza ile imzalanmıştır,Bdge Doian^aın ^^SLsfeefiMİuPclasffeşyâJıe^iao randomiezgee e&ı&ta 27 ve ardından açık etiketli uzatma çalışmasına alınan hastaların %38 (25/66) ve %55'i (36/66) sırasıyla 12 ve 24 haftalık yeniden tedavi sonrasında PASI 75 yanıtına ulaşmıştır. 16. hafta ve 33. haftada PASI 75 yanıtına ulaşan toplam 233 hasta, Psöriyazis I çalışmasında 52 hafta devamlı HUMIRA tedavisi almış ve açık etiketli uzatma çalışmasında HUMIRAalmaya devam etmiştir. İlave 108 haftalık açık etiketli tedaviden sonra (toplam 160 hafta) buhastalardaki PASI 75 ve temiz veya minimal PGA yanıt oranları, sırasıyla %74,7 ve %59olmuştur. Advers olaylar veya etkililik görülmemesinden dolayı çalışmadan ayrılan veya dozarttırılan tüm hastaların yanıt vermeyen hasta olarak değerlendirildiği bir analizde, buhastalardaki PASI 75 veya temiz veya minimal PGA yanıt oranları, ilave 108 haftalık açıketiketli tedaviden sonra (toplam 160 hafta) sırasıyla %69,6 ve %55,7 olmuştur. Toplam 347 stabil yanıt veren hasta, açık etiketli bir uzatma çalışmasında tedaviyi kesme ve yeniden tedaviye başlama yönünden değerlendirmeye katılmıştır. Tedaviyi kesme periyodunda,psöriyazis semptomları zaman içerisinde geri dönmüş, medyan nüks (PGA orta veya dahakötü seviyesine düşüş) süresi yaklaşık 5 ay olmuştur. Bu hastaların hiçbirinde tedaviyidurdurma döneminde rebound etkisi görülmemiştir. Yeniden tedavi periyoduna alınanhastaların toplam %76,5'inin (218/285), tedaviyi kesme döneminde nüks yaşayıpyaşamadıklarından bağımsız şekilde, 16 haftalık yeniden tedaviden sonra PGA yanıtı temizveya minimal olmuştur (tedaviyi kesme döneminde nüks yaşayan ve yaşamayan hastalardasırasıyla %69,1 [123/178] ve %88,8 [95/107]). Tekrar tedavi döneminde, tedaviyi kesmedöneminden öncesine benzer bir güvenlilik profili gözlemlenmiştir. DLQI'de (Dermatoloji Yaşam Kalitesi İndeksi) plasebo (Psöriyazis I ve II çalışması) ve MTX (Psöriyazis II çalışması) ile karşılaştırıldığında, başlangıca göre 16. haftada anlamlı düzelmelerolduğu gösterilmiştir. Çalışma I'de, SF 36'nın fiziksel ve mental skorlarındaki iyileşmelerplasebo düzeyleri ile karşılaştırıldığında anlamlıdır. Açık etiketli bir uzatma çalışmasında, PASI skorunun %50'nin altında olması nedeniyle dozu iki haftada bir 40 mg'dan haftada bir 40 mg'a yükseltilen hastalar için, 12. ve 24. haftalardasırasıyla hastaların %26,4 (92/349) ve %37,8'inde (132/349) PASI 75 yanıtı elde edilmiştir. Orta ila şiddetli derecede kronik plak psöriyazisi ve el ve/veya ayak psöriyazisi bulunan 72 hastada gerçekleştirilen Psöriyazis III çalışmasında (REACH), HUMIRA'nın plaseboya karşıetkililiği ve güvenliliği karşılaştırılmıştır. Hastalara 16 hafta boyunca, 80 mg HUMIRAbaşlangıç dozu, bunu takiben her iki haftada bir 40 mg (ilk dozdan bir hafta sonra başlayarak)HUMIRA veya plasebo verilmiştir. 16. haftada, plasebo alan hastalara kıyasla, HUMIRA alanhastaların istatistiksel olarak anlamlı derecede büyük bir çoğunluğunda, el ve/veya ayaklarda'temiz' veya 'neredeyse temiz' PGA sonucu elde edilmiştir (sırasıyla %30,6'ya kıyasla %4,3,[P=0,014]). Psöriyazis IV çalışmasında, orta ila şiddetli tırnak psöriyazis hastalığı olan 217 erişkin hastada HUMIRA'nın plaseboya karşı etkililiği ve güvenliliği karşılaştırılmıştır. Hastalar, ya 80 mgHUMIRA başlangıç dozunun ardından iki haftada bir 40 mg HUMIRA (başlangıç dozundanbir hafta sonra başlayarak) ya da 26 hafta boyunca plasebo ve bunu takiben ilave 26 haftaboyunca açık etiketli HUMIRA tedavisi almıştır. Tırnak psöriyazis hastalığına ilişkindeğerlendirmeler arasında Modifiye Tırnak Psöriyazisi Şiddet İndeksi (mNAPSI), HekiminTırnak Psöriyazisine İlişkin Genel Değerlendirmesi (PGA-F) ve Tırnak Psöriyazis Şiddetİndeksi (NAPSI) yer almaktadır (bkz. Tablo 12). HUMIRA, deri tutulumu derecesi farklı olan
28 (VYA > %10 [hastaların %60'ı] ve VYA < %10 ve > %5 [hastaların %40'ı]) tırnak psöriyazisli hastalarda tedavi yararı göstermiştir.
Erişkin Crohn HastalığıHUMIRA dozlarının güvenliliği ve etkililiği, 1500'ü aşkın orta ila şiddetli derecede aktif Crohn hastasında (Crohn Hastalığı Aktivite İndeksi (CDAI) > 220 ve < 450) randomize, çift-kör,plasebo-kontrollü çalışmalarda değerlendirilmiştir. Aminosalisilatların, kortikosteroidlerinve/veya immünomodülatör ajanların stabil dozlarda eş zamanlı kullanımına izin verilmiş vehastaların %80'i bu ilaçlardan en az birini almaya devam etmiştir. Klinik remisyonun indüksiyonu (CDAI < 150 olarak tanımlanmıştır), CD I (CLASSIC I) ve CD II çalışması (GAIN), isimli iki çalışmada değerlendirilmiştir. CD I çalışmasında daha önceTNF bloke edici ajan kullanmamış olan 299 hasta dört tedavi grubundan birine randomizeedilmiştir: 0. ve 2. haftada plasebo, 0. haftada 160 mg ve 2. haftada 80 mg HUMIRA, 0. haftada80 mg ve 2. haftada 40 mg HUMIRA ve 0. haftada 40 mg ve 2. haftada 20 mg HUMIRA. CDII çalışmasında, infliksimaba yanıtı kaybolmuş veya infliksimabı tolere edemeyen 325 hasta, 0.haftada 160 mg ve 2. haftada 80 mg HUMIRA veya 0. hafta ve 2. haftada plaseboya randomizeedilmişlerdir. Primer yanıt vermeyen hastalar çalışmanın dışında bırakılmış ve bu nedenle buhastalar daha fazla değerlendirilmemiştir. Klinik remisyonun idamesi CD III çalışmasında (CHARM) değerlendirilmiştir. CD III çalışmasında 854 hasta, önce açık etiketli olarak 0. haftada 80 mg HUMIRA ve 2. haftada 40mg HUMIRA almışlardır. Hastalar daha sonra 4. haftada iki haftada bir 40 mg HUMIRA,haftada bir 40 mg HUMIRA veya plaseboya randomize edilmişlerdir. Toplam çalışma süresi56 haftadır. 4. haftada klinik yanıt veren hastalar (CDAI > 70'e düşüş) katmanlara ayrılmış ve4. haftada klinik yanıt vermeyenlerden ayrı olarak analiz edilmişlerdir. 8. haftadan sonrakortikosteroidin azaltılarak kesilmesine izin verilmiştir.
29 CD I ve CD II çalışmasındaki remisyon indüksiyonu ve yanıt oranları Tablo 13'te sunulmaktadır.
8. haftada, 160/80 mg HUMIRA ve 80/40 mg HUMIRA rejimleriyle benzer remisyon oranları gözlemlenmiş ve 160/80 mg HUMIRA grubunda advers olaylar daha sık olarak bildirilmiştir. CD III çalışmasında, 4. haftada hastaların %58'i (499/854) klinik yanıt vermiş ve primer analizde değerlendirilmiştir. 4. haftadaki klinik yanıt alınanların %48'i, daha önce başka TNFbloke edici ajan kullanmış olan hastalardır. Remisyonun idamesi ve yanıt oranları Tablo 14'tesunulmuştur. Klinik remisyon sonuçları, daha önce TNF bloke edici ajan uygulanmasına bağlıolmaksızın rölatif olarak sabit kalmıştır. 56. haftada, plasebo ile karşılaştırıldığında hastalığa bağlı hastaneye yatma ve cerrahi müdahaleler, adalimumab ile istatistiksel olarak anlamlı bir şekilde azalmıştır.
30 4. haftada yanıt vermeyen hastalar arasından, HUMIRA idame grubunda 12. haftada yanıt verenlerin oranı %43 iken plasebo idame hastalarında bu oran %30 olmuştur. Bu sonuçlar 4.haftada yanıt vermeyen bazı hastaların, sürdürülen idame tedavisinden 12. haftada yarargörebileceğini düşündürmektedir. 12. haftadan sonra sürdürülen tedavi, anlamlı ölçüde dahafazla yanıtla sonuçlanmamıştır (bkz. Bölüm 4.2). CD I çalışmasında 117/276 hasta ve CD II ve III çalışmasında ise 272/777 hasta en az 3 yıl boyunca açık etiketli olarak adalimumab tedavisi ile takip edilmiştir. Sırasıyla 88 ve 189 hastaklinik remisyonda kalmaya devam etmiştir. Klinik yanıt (CR-100) sırasıyla 102 ve 233 hastadakorunmuştur. Yaşam kalitesiCD I ve II çalışmalarında plaseboyla karşılaştırıldığında, 80/40 mg HUMIRA ve 160/80 mg HUMIRA tedavisine randomize edilmiş hastalarda 4. haftada hastalığa özgü İnflamatuarBağırsak Hastalığı Anketi (IBDQ) total skorunda istatistiksel olarak anlamlı düzelme eldeedilmiş ve CD III çalışmasının 26. ve 56. haftalarında adalimumab tedavi gruplarında IBDQbaşlangıç skorlarında plasebo grubuna kıyasla istatistiksel olarak anlamlı düzelme görülmüştür. Erişkin üveitHUMIRA'nın güvenliliği ve etkililiği randomize, çift-kör, plasebo kontrollü iki çalışmada (UV I ve II), izole anterior üveit görülen hastalar dahil edilmeyerek, enfeksiyöz olmayan orta,posterior ve panüveit görülen erişkin hastalarda değerlendirilmiştir. Hastalar 80 mg'lıkHUMIRA başlangıç dozu ve onu takiben ilk dozdan bir hafta sonra başlanarak iki haftada biruygulanan 40 mg HUMIRA veya plasebo almışlardır. Tek bir biyolojik olmayanimmünosupresanın eş zamanlı stabil dozlarına izin verilmiştir. UV I çalışmasında kortikosteroid (10 ila 60 mg/gün dozunda oral prednizon) tedavisine rağmen aktif üveiti olan 217 hasta değerlendirilmiştir. Tüm hastalar çalışma başlangıcında 2 haftasüreyle 60 mg/gün standard prednizon dozunun ardından zorunlu doz azaltma programı takipedilerek 15. haftada kortikosteroid tamamen kesilmiştir. UV II çalışmasında çalışma başlangıcında kronik kortikosteroid tedavisi (10-35 mg/gün oral prednizon) gerektiren 226 inaktif üveit hastası değerlendirilmiştir. Hastalar ardından 19.haftada kortikosteroidi tamamen kesecek şekilde zorunlu doz azaltımı dönemine girmişlerdir.Primer etkililik sonlanım noktası her iki çalışmada da 'tedavi başarısızlığına kadar geçen süre'olarak belirlenmiştir. Tedavi başarısızlığı; inflamatuvar korioretinal ve/veya inflamatuvarretinal vasküler lezyonlar, ön kamara (AC) hücre değerlendirmesi, vitröz bulanıklık (VH)derecelendirmesi ve en iyi düzeltilmiş görme keskinliğini (BCVA) esas alan çok bileşenli birsonuç ile tanımlanmıştır. UV I ve UV II çalışmalarını tamamlayan hastalar, süresi başlangıçta 78 hafta olarak planlanan, kontrolsüz, uzun süreli bir uzatma çalışmasına kaydolmaya uygun bulunmuştur. HastalarınHUMIRA'ya erişimleri olana kadar, 78. haftadan sonra çalışma medikasyonuna devametmelerine izin verilmiştir.
31 Klinik yanıtHer iki çalışmadan elde edilen sonuçlar HUMIRA ile tedavi uygulanan hastalarda plasebo alan hastalardakine kıyasla tedavi başarısızlığı riskinde istatistiksel açıdan anlamlı azalma ortayakoymuştur (bkz. Tablo 15). Her iki çalışmada, HUMIRA'nın plaseboya kıyasla tedavibaşarısızlığı oranında erken ve uzun süreli bir etkisi olduğunu göstermiştir. (bkz. Şekil 1).
UV I Çalışmasında 6. Hafta veya Sonrasında Tedavi Başarısızlığına Kadar Geçen SürePrimer Analiz (ITT) Adalimumab 11060(54,5)5.60,5 0,36,0,7< 0,001UV II Çalışmasında 2. Hafta veya Sonrasında Tedavi Başarısızlığına Geçen Süre Primer Analiz (ITT) Adalimumab 11545(39,1)NEc0,57 0,39,0,840,004Not: 6. hafta veya sonrasında (UV I çalışması) ya da 2. hafta veya sonrasında (UV II çalışması) tedavide başarısızlık, olay olarak sayılmıştır. Tedavi başarısızlığı dışındaki nedenlerdenkaynaklanan tedaviyi bırakmalar, bırakma zamanı sırasında dahil edilmemiştir.aTedavinin bir faktör olduğu orantılı riskler regresyonunda plasebo karşısında adalimumabınHR değeri. bLog rank testinden 2 yönlü Pdeğeri.cNE= Hesaplanabilir değil. Risk altındaki hastaların yarısından azında bir olay meydana gelmiştir. Şekil 1: 6. Hafta veya Sonrasında (UV I Çalışması) ya da 2. Hafta veya sonrasında (UV II Çalışması) Tedavi Başarısızlığına Kadar Geçen Süreyi Özetleyen Kaplan-MeierEğrileri
32 z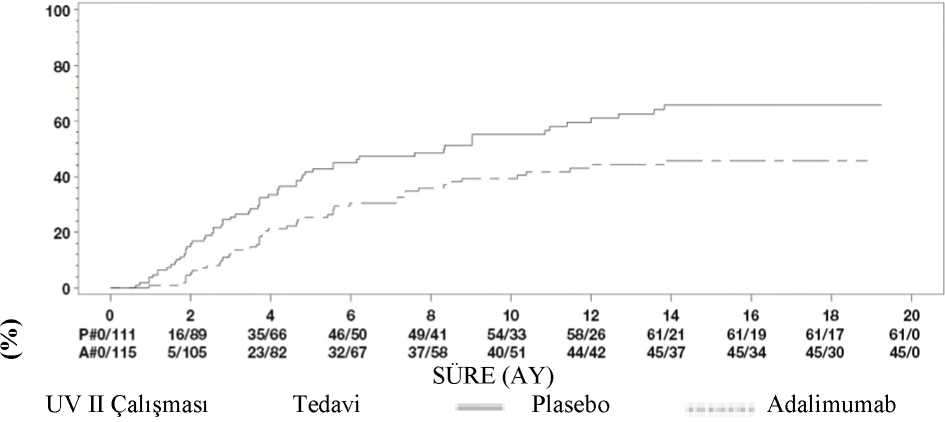
Not: P# = Plasebo (Olay Sayısı/Risk Altındakilerin Sayısı); A# = HUMIRA (Olay Sayısı/Risk Altındakilerin Sayısı ). UV I çalışmasında tedavi başarısızlığı bileşenlerinin her birinde plasebo karşısında adalimumab lehine istatistiksel olarak anlamlı farklılıklar gözlemlenmiştir. UV II çalışmasında görüşkeskinliği konusunda istatistiksel olarak anlamlı farklılıklar elde edilirken, diğer tümbileşenlerin numerik olarak adalimumab lehine olduğu görülmüştür. UV I ve UV II çalışmalarının kontrolsüz uzun süreli uzatmasına dahil edilen 424 hastadan 60'ı uygun görülmemiştir (örn., katarakt ameliyatı veya vitrektomi nedeniyle, sapmalar nedeniyleveya diyabetik retinopatiye bağlı sekonder komplikasyonlar nedeniyle) ve primer etkililikanalizinden çıkarılmıştır. Kalan 364 hastadan, 269 hasta (%74) açık etiketli adalimumabtedavisini 78 hafta boyunca almışlardır. Gözlemlenen veri yaklaşımına göre, eşzamanlı olarakgünde < 7,5 mg steroid alan 216 (%80,3) hasta sakindi (aktif inflamatuvar lezyonbulunmamaktadır, AC hücre derecesi < 0,5+, VH derece < 0,5+) kalmıştır ve 178 (%66,2)hasta steroid almadan hastalık sakin kalmıştır. BCVA, 78. haftada gözlerin %88,6'sıiyileştirilmiştir veya korunmuştur (< 5 harf bozulması). 78. haftadan sonraki veriler busonuçlarla genel olarak tutarlıdır, ancak bu sürenin sonunda kayıtlı hasta sayısı azalmıştır.Genel olarak, hastalar arasında adalimumab tedavisini bırakanların %18'i advers olaylardan ve%8'i yetersiz yanıttan dolayı çalışmayı bırakmıştır. Yaşam kalitesiGörüşle ilişkili işlevsellik konusunda hasta tarafından bildirilen sonuçlar, her iki klinik çalışmada NEI VFQ-25 kullanılarak değerlendirilmiştir. HUMIRA, UV I çalışmasında genelgörme, oküler ağrı, yakın görme, mental sağlık ve toplam skor; UV II çalışmasında ise genelgörme ve mental sağlık olmak üzere alt skorların çoğunda istatistiksel olarak anlamlı ortalamafarklarla sayısal açıdan üstünlük sergilemiştir. Görmeyle ilişkili etkilerin UV I çalışmasındarenk görüşü, UV II çalışmasında ise renk görüşü, periferik görüş ve yakın görüş için numerikolarak açıdan HUMIRA lehine olmadığı görülmüştür.
33 İmmünojenisiteAnti-adalimumab antikorlarının oluşumu, adalimumabın klirensinde artış ve etkililiğinde ise azalma ile ilişkilidir. Anti-adalimumab antikorları varlığıyla advers olaylar arasında görünürbir bağlantı bulunmamaktadır. 4-17 yaş arasındaki poliartiküler jüvenil idiyopatik artrit hastalarında, adalimumab ile tedavi edilen hastaların %15,8'inde (27/171) anti-adalimumab antikorları saptanmıştır. Eş zamanlımetotreksat verilmeyen hastalarda insidans %25,6 (22/86) iken, adalimumabın metotreksatailave olarak verildiği hastalarda insidans %5,9 (5/85) olmuştur. 2-4 yaşları arasında ya da 4 yaşve üstü olan, 15 kg'ın altındaki poliartiküler jüvenil idiyopatik artrite sahip hastaların %7'sinde(1/15) anti-adalimumab antikorları belirlenmiş ve bir hasta eş zamanlı metotreksat almıştır. Adalimumab ile tedavi edilen entezit ile ilişkili artrit hastalarında, anti-adalimumab antikorları hastaların %10,9'unda (5/46) tespit edilmiştir. Eş zamanlı metotreksat verilmeyen hastalardakiinsidans, adalimumabın metotreksata ilave olarak verildiği hastalar ile karşılaştırıldığında%13,6'ya (3/22) karşı % 8,3 (2/24) olmuştur. Romatoid artrit I, II ve III çalışmalarındaki hastalar, 6'dan 12. aya kadar olan dönemde anti-adalimumab antikorları bakımından birçok zaman noktasında test edilmiştir. Pivotal çalışmalarda, anti-adalimumab antikorları, adalimumab ile tedavi edilen hastaların %5,5'inde(58/1053) saptanmış, plasebo ile bu oran %0,5 (2/370) olmuştur. Eş zamanlı metotreksatverilmeyen hastalarda insidans %12,4 iken, adalimumabın metotreksata ilave olarak verildiğihastalarda %0,6 olmuştur. Pediyatrik psöriyazisi olan hastalarda, 0,8 mg/kg adalimumab monoterapisi ile tedavi edilen 5/38 hastada (%13) anti-adalimumab antikorları saptanmıştır. Psöriyazisli erişkin hastalarda anti-adalimumab antikorları, adalimumab monoterapisi ile tedavi edilen 920 hastanın 77'sinde (%8,4) saptanmıştır. Uzun dönem adalimumab monoterapi tedavisi alan, tedaviyi bırakma ve tekrar tedavi çalışmalarına dahil olmuş plak psöriyazisli erişkin hastalarda tekrar tedavi sonrası anti-adalimumab antikor oranı (11/482 hasta, %2,3) tedaviyi bırakma öncesi gözlemlenen oran ile(11/590 hasta, %1,9) benzer olmuştur. Orta ila şiddetli derecede aktif pediyatrik Crohn hastalığı olan hastalarda adalimumab alan hastalarda anti-adalimumab antikoru gelişme oranı %3,3 olmuştur. Crohn hastalığı olan hastaların 7/269'sinde (%2,6) anti-adalimumab antikorları saptanmıştır. Enfeksiyöz olmayan erişkin üveit hastaları arasında adalimumab tedavisi uygulanan hastaların %4,8'inde (12/249) anti-adalimumab antikorları tespit edilmiştir. İmmunojenisite analizleri ürün için spesifik olduğundan bu üründeki antikor oranlarının başka ürünlerle karşılaştırılması uygun değildir. Pediyatrik popülasyonJüvenil idiyopatik artrit (JIA)
34 Poliartiküler _ jüvenil idiyopatik artrit (pJIA)HUMIRA'nın güvenlilik ve etkililiği, çeşitli JIA başlangıç tiplerinin (en sık olarak romatoid faktör negatif ya da pozitif poliartrit ve yaygın oligoartrit) görüldüğü aktif poliartiküler ya dapoliartiküler seyirli jüvenil idiyopatik artriti olan çocuklarda yapılan iki çalışmada (pJIA I veII) değerlendirilmiştir. pJIA I HUMIRA'nın güvenliliği ve etkililiği, poliartiküler JIA olan 171 çocukta (4-17 yaş) yapılan çok merkezli, randomize, çift-kör, paralel-gruplu bir çalışmada değerlendirilmiştir. Açık etiketligiriş fazında (OL LI) hastalar, MTX (metotreksat) ile tedavi edilen veya edilmeyen (hiç MTXkullanmayan veya çalışmadan en az 2 hafta önce MTX tedavisi sonlandırılmış hastalar) olmaküzere iki gruba alınmışlardır. Hastalarda stabil NSAİİ ve/veya prednizon (< 0,2 mg/kg/gün veyamaksimum 10 mg/gün) kullanımına devam edilmiştir. OL LI fazında, tüm hastalara 16 haftaboyunca iki haftada bir 24 mg/m2'den maksimum 40 mg'a kadar HUMIRA uygulanmıştırHastaların yaş ve OL LI fazında aldıkları minimum, medyan ve maksimum dozlara göredağılımı Tablo 16'da sunulmaktadır. Tablo 16. Hastaların yaşlarına ve OL LI fazında aldıkları adalimumab dozuna göre
Pediyatrik ACR 30 yanıtına 16. haftada gösteren hastalar, çift-kör fazda randomize edilmeye uygun bulunmuş ve bu hastalara ya maksimum 40 mg'a kadar HUMIRA 24 mg/m2 verilmiş yada bu hastalar ilave 32 hafta boyunca veya hastalık alevlenene kadar iki haftada bir plaseboalmıştır. Hastalık alevlenme kriterleri, 6 Pediyatrik ACR temel kriterinden 3 veya 3'tenfazlasında başlangıca göre > %30 kötüleşme, > 2 aktif eklem ve 6 kriterin en fazla 1'inde >%30 iyileşme olarak tanımlanmıştır. Hastalar 32 hafta sonra veya hastalık alevlenmesinde açıketiketli uzatma fazına kaydedilmek üzere uygun kabul edilmiştir.
35 a 48. haftada plasebo ile tedavi edilen hastalardan anlamlı olarak daha yüksek Ped ACR 30/50/70 yanıtlarıb p = 0,015c p = 0,031 16. haftada yanıt verenler (n=144) arasında, çalışma boyunca HUMIRA alan hastalarda OLE fazında Pediyatrik ACR 30/50/70/90 yanıtları, 6 yıla varan bir süre boyunca sürdürülmüştür.Başlangıçta yaş grubu 4-12 olan 11 hasta ile başlangıçta yaş grubu 13-17 olan 8 hasta olmaküzere toplamda 19 hasta tedaviye 6 yıl veya üzerinde devam etmiştir. Tüm yanıtların genel olarak iyi olduğu, tek başına HUMIRA tedavisine kıyasla HUMIRA ve metotreksat kombinasyonu ile tedavi edilen hastalarda daha az sayıda antikor geliştiğisaptanmıştır. Bu sonuçlara bakarak, HUMIRA'nın, metotreksat ile kombinasyon halindekullanılması ve metotreksat kullanımının uygun olmadığı hastalarda monoterapi olarakkullanılması önerilmektedir (bkz. Bölüm 4.2). pJIA II HUMIRA'nın güvenlilik ve etkililiği, orta ila şiddetli derecede aktif poliartiküler JIA olan 32 çocukta (2 ila 4 yaş ya da 4 yaş üzeri ve vücut ağırlığı < 15 kg) gerçekleştirilen açık etiketli,çok merkezli bir çalışmada değerlendirilmiştir. Hastalara en az 24 hafta boyunca subkütanenjeksiyon yoluyla tek doz olarak iki haftada bir maksimum 20 mg'a kadar olmak üzere 24mg/m2 Vücut Yüzey Alanı (VYA) dozunda HUMIRA uygulanmıştır. Çalışma sırasındahastaların çoğu eş zamanlı MTX kullanmış, daha az sayıda ise kortikosteroid ya da NSAİİkullanımı bildirilmiştir. 12. ve 24. haftalarda, gözlemlenen veri yaklaşımı kullanılarak elde edilen, Pediyatrik ACR 30 yanıtları sırasıyla %93,5 ve %90 olarak saptanmıştır. 12. ve 24. haftalarda Pediyatrik ACR50/70/90 yanıtı veren hasta oranları ise, sırasıyla %90,3 / %61,3 / %38,7 ve %83,3 / %73,3 /%36,7 olarak belirlenmiştir. 24. haftada yanıt veren (Pediyatrik ACR 30) hastalar (n=27/30)arasında, bu dönem boyunca HUMIRA uygulanan hastalarda Pediyatrik ACR 30 yanıtları OLEfazında 60 haftaya varan bir süre boyunca sürdürülmüştür. Toplam olarak 20 hastada tedaviye60 hafta ya da daha uzun bir süre devam edilmiştir. Entezit ile ilişkili artritHUMIRA'nın güvenliliği ve etkililiği; çok merkezli, randomize, çift-kör bir çalışmada orta derecede entezit ile ilişkili artriti olan 46 pediyatrik hastada (6 ila 17 yaş arası)değerlendirilmiştir. Hastalar, 12 hafta boyunca iki haftada bir en fazla 40 mg'a kadar 24 mg/m2Vücut Yüzey Alanı'nda (VYA) HUMIRA veya plasebo alacak şekilde randomize edilmiştir.Çift kör periyodunu takiben açık etiketli periyotta hastalar ilave 192 hafta boyunca her ikihaftada bir 24 mg/m2 vücut yüzey alanında, en fazla 40 mg olacak şekilde subkütan olarakHUMIRA almaya devam etmişlerdir. Primer sonlanım noktası, artritli aktif eklem sayısında(deformasyona veya ağrı ve/veya hassasiyeti olan eklemlerde hareket kaybına bağlı olmayanşişme), başlangıca göre 12. haftadaki değişimdir. Plasebo grubundaki hastalarda ortalama % -11,6 olan (medyan yüzde değişimi % -50) bu azalma, HUMIRA grubundaki hastalarda ortalama% -62,6'dır. (medyan yüzde değişimi % -88,9). 156 hafta boyunca OL döneminde, çalışmayadevam eden HUMIRA grubundaki 31 hastanın 26'sında (%84) aktif artritli eklem sayısındakidüzelme korunmuştur. İstatiksel olarak anlamlı olmamasına rağmen hastaların çoğunluğu;Belge DağnteEifti'foölğe 3§ayı&k iHd3&â$EklemSay3$iQ(HES), ŞjşmişiıEklemS^yısi'{SES)^PediyBtPikkACR 36 50 yanıtı ve Pediyatrik ACR 70 yanıtı gibi sekonder sonlanım noktalarında klinik iyileşme göstermiştir. Pediyatrik plak psöriyazisHUMIRA'nın etkililiği şiddetli kronik plak psöriyazisli (Hekimin Global Değerlendirmesi (PGA) > 4 ya da > %20 Vücut Yüzey Alanı (VYA) tutulumu ya da > %10 VYA tutulumu ileberaber çok kalın lezyonlar ya da Psöriyazis Alan ve Şiddet İndeksi (PASI) > 20 ya da > 10 ilebirlikte klinik olarak ilişkili yüz, genital ya da el/ayak tutulumu ile tanımlandığı üzere), 4 yaşve üzeri olan ve topikal tedavi ve helioterapi ya da fototerapi ile yetersiz şekilde kontrol edilen114 pediyatrik hastanın katıldığı randomize, çift-kör kontrollü bir çalışmadadeğerlendirilmiştir. Hastalar iki haftada bir kez HUMIRA 0,8 mg/kg (40 mg'a kadar), iki haftada bir 0,4 mg/kg (20 mg'a kadar) ya da haftalık metotreksat 0,1-0,4 mg/kg (25 mg'a kadar) almıştır. 16. haftada 0,8mg/kg HUMIRA için randomize edilen hastalar, iki haftada bir 0,4 mg/kg ya da metotreksatarandomize edilen hastalara göre daha fazla pozitif etkililik yanıtı (örn. PASI 75) göstermişlerdir.
PASI 75 ve PGA temiz ya da minimal yanıtını elde eden hastalar, 36 haftaya kadar tedaviden kesilmiş ve hastalık kontrolü kaybı açısından izlenmiştir (yani PGA'nın en az 2 derecekötüleşmesi). Daha sonra hastalar ek 16 hafta süreyle, iki haftada bir 0,8 mg/kg adalimumab iletekrar tedavi edilmiş ve tekrar tedavi süresinde gözlemlenen yanıt oranları önceki çift kördönemle benzer bulunmuştur: %78,9'luk PASI 75 yanıtı (19 hastadan 15'i) ve %52,6'lık PGAtemiz ya da minimal yanıtı (19 hastadan 10'u). Çalışmanın açık etiket döneminde, PASI 75 ve PGA temiz ya da minimal yanıtları yeni güvenlilik bulgusu olmadan ilave 52 haftaya kadar korunmuştur. Pediyatrik Crohn hastalığıHUMIRA, Pediyatrik Crohn Hastalığı Aktivitesi İndeksi (PCDAI) skoru > 30 şeklinde tanımlanan orta ila şiddetli derecede Crohn hastalığı (CD) olan 6-17 (sınırlar dahil) yaşlarıarasındaki 192 pediyatrik hastada, vücut ağırlığına (< 40 kg ya da > 40 kg) dayanan dozlardakiindüksiyon ve idame tedavisinin etkililiği ve güvenliliğini değerlendirmek üzere tasarlanmışçok merkezli, randomize, çift-kör bir klinik çalışmada değerlendirilmiştir. Hastalar, CD içinuygulanan konvansiyonel tedavinin (bir kortikosteroid ve/veya bir immünomodülatör içeren)başarısız olduğu hastalardan oluşmuştur. Hastaların, daha önce uygulanan infliksimabtedavisine verdikleri yanıtın ortadan kalkmış olması ya da infliksimabı tolere edememişolmaları mümkündür.
37 Tüm hastalara, başlangıç vücut ağırlıklarına dayanarak belirlenen bir dozda açık etiketli HUMIRA indüksiyon tedavisi uygulanmıştır: >40 kg olan hastalar için 0. haftada 160 mg ve 2.haftada 80 mg, <40 kg olan hastalar için sırasıyla 80 mg ve 40 mg'dır. Hastalar 4. haftada, Tablo 19'da gösterildiği gibi, o andaki vücut ağırlıklarına bağlı olarak 1:1 oranında düşük doz ya da standart doz idame rejimlerine randomize edilmiştir.
Etkililik sonuçlarıÇalışmanın primer sonlanım noktası olarak, 26. haftada PCDAI skorunun < 10 olması şeklinde tanımlanan klinik remisyon kullanılmıştır. Klinik remisyon ve klinik yanıt (PCDAI skorunun başlangıca göre en az 15 puan düşmesi olarak tanımlanmıştır) oranları Tablo 20'de sunulmuştur. Kortikosteroidler ya daimmünomodülatörlerin tedavi kesilme oranları, Tablo 21'de sunulmaktadır.
38 2 İmmunosüpresan tedavisi ancak hastanın klinik yanıt kriterlerini karşılaması halindeçalışmacının takdirine bağlı olarak, 26. haftada veya sonrasında kesilebilir. 3 Başlangıçtan sonra arka arkaya gerçekleştirilen en az 2 vizitte, başlangıçta akıntılı olantüm fistüllerin kapanması şeklinde tanımlanmaktadır Her iki tedavi grubu için, başlangıçtan 26. ve 52. haftaya kadar Vücut Kütle İndeksi (VKİ) ve uzama hızında istatistiksel olarak anlamlı artışlar (düzelme) gözlemlenmiştir. Her iki tedavi grubunda, yaşam kalitesi parametreleri (IMPACT III dahil) için başlangıca göre istatistiksel ve klinik olarak anlamlı düzelmeler de elde edilmiştir. Pediyatrik CD çalışmasında yer alan 100 hasta (n = 100), açık etiketli uzun süreli uzatma çalışmasına devam etmiştir. 5 yıllık adalimumab tedavisinden sonra, çalışmaya devam eden 50hastanın %74'ü (37/50) klinik remisyonda kalmaya devam ederken %92'si (46/50) PCDAI'yagöre klinik yanıtı sürdürmüştür. Pediyatrik üveitHUMIRA'nın güvenliliği ve etkililiği, en az 12 hafta süren metotreksat tedavisine dirençli aktif JIA ile ilişkili enfeksiyöz olmayan anterior üveiti olan 2 ila 18 yaş arasındaki 90 pediyatrikhastanın dahil edildiği randomize çift-kör, kontrollü bir çalışmada değerlendirilmiştir. Hastalarbaşlangıç metotreksat dozları ile kombine olarak iki haftada bir ya plasebo ya da 20 mgadalimumab (<30 kg olmaları durumunda) veya 40 mg adalimumab (> 30 kg olmalarıdurumunda) almışlardır. Primer sonlanım noktası "tedavi başarısızlığına kadar geçen süre" olarak belirlenmiştir. Tedavi başarısızlığını değerlendirmedeki kriterler; oküler inflamasyonda kötüleşme ya da uzun süreboyunca iyileşme olmaması, uzun süreli oküler komorbidite gelişimi ile birlikte kısmi iyileşmeya da oküler komorbiditelerde kötüleşme, izin verilmeyen eş zamanlı ilaç kullanımı vetedavinin uzun bir süreliğine durdurulmasıdır. Klinik yanıtAdalimumab, plaseboya kıyasla tedavi başarısızlığına kadar geçen süreyi belirgin olarak geciktirmiştir (bkz. Şekil 2, P <0,0001, log rank testi). Tedavi başarısızlığına kadar geçenortalama süre, plasebo ile tedavi edilen hastalar için 24,1 hafta iken, adalimumab ile tedaviedilen hastaların yalnızca yarısından azında tedavi başarısızlığı görülmüş olduğundan ortalamasüre hesaplanamamıştır. Adalimumab, risk oranında görüldüğü üzere plaseboya kıyasla tedavibaşarısızlığı riskini %75 oranında anlamlı olarak azaltmıştır (HR = 0,25 [%95 GA: 0,12, 0,49]).
39
Pediyatrik popülasyonPediyatrik kullanıma yönelik bilgiler için, Bölüm 4.2'ye bakınız. 5.2. Farmakokinetik özelliklerGenel özelliklerEmilim:4 ila 17 yaşındaki poliartiküler jüvenil idiyopatik artrit (JIA) hastalarına iki haftada bir subkütanolarak 24 mg/m2 (maksimum 40 mg'a kadar) uygulanmasının ardından, ortalama kararlı durum(20-48. haftalar arasında ölçülen değerler) serum adalimumab çukur konsantrasyonu, eşzamanlı metotreksat olmadan adalimumab kullanımında 5,6 ± 5,6 mikrogram/mL (%102 CV)ve metotreksatla eş zamanlı kullanımında 10,9 ± 5,2 mikrogram/mL (%47,7 CV) olmuştur. 24 mg/m2 dozda adalimumab uygulanan 2 ila. 4 yaş arasında veya 4 yaşın üzerinde olup, vücut Belge Dcgğirhğj!o<lı5wkgkoiatfujiAQftâ§tHlai0ın:da,3kortalamSekaS'airfa Aduums ^seromrkadalrmumfebtcişukur 40 konsantrasyonları, eş zamanlı metotreksat olmadan adalimumab kullanımında 6 ± 6,1 mikrogram/mL (% 101 CV) ve eş zamanlı metotreksat kullanımında 7,9 ± 5,6 mikrogram/mL(%71,2 CV) olarak saptanmıştır. 6-17 yaşları arasındaki entezit ile ilişkili artritli hastalarda subkütan enjeksiyon yoluyla iki haftada bir 24 mg/m2 (maksimum 40 mg'a kadar) uygulanması sonrasında ortalama kararlıdurum (24. haftada ölçülen değerler) serum adalimumab çukur konsantrasyonları eş zamanlımetotreksat olmadan adalimumab kullanımında 8,8±6,6 mikrogram/mL ve eş zamanlımetotreksat kullanımında 11,8 ± 4,3 mikrogram/mL olmuştur. Kronik plak psöriyazis hastalığı olan pediyatrik hastalara iki haftada bir subkütan yoldan uygulanan 0,8 mg/kg (maksimum 40 mg'a kadar) sonrasında, ortalama ± SD kararlı durumadalimumab konsantrasyonu yaklaşık olarak 7,4 ± 5,8 mikrogram/mL (%79 CV) olmuştur. Orta ila şiddetli derecede CD olan pediyatrik hastalarda, açık etiketli adalimumab indüksiyon dozu, 40 kg'lık vücut ağırlığı kesme noktasına bağlı olarak 0 ve 2. haftalarda sırasıyla 160/80ya da 80/40 mg olmuştur. 4. haftada hastalar, vücut ağırlıklarına bağlı olarak 1:1 oranındastandart doz (iki haftada bir 40/20 mg) ya da düşük doz (iki haftada bir 20/10 mg) idame tedavisigruplarına randomize edilmiştir. 4. haftada ulaşılan ortalama (±SD) serum adalimumab çukurkonsantrasyonları, > 40 kg olan hastalar için (160/80 mg) 15,7±6,6 mikrogram/mL ve <40 kgolan hastalar için (80/40 mg) 10,6 ± 6,1 mikrogram/mL olarak belirlenmiştir. Randomize edildikleri tedavi grubunda kalan hastalar için, 52. haftadaki ortalama (± SD) serum adalimumab vadi konsantrasyonları, standart doz grubu için 9,5±5,6 mikrogram/mL ve düşükdoz grubu için 3,5 ± 2,2 mikrogram/mL olarak saptanmıştır. 52 hafta boyunca iki haftada biradalimumab tedavisinin sürdürüldüğü hastalarda ortalama çukur konsantrasyonlar devametmiştir. Doz sıklığı arttırılarak iki haftada bir uygulamadan haftada bir uygulama rejiminegeçirilen hastalar için, 52. haftadaki ortalama (±SD) serum adalimumab çukurkonsantrasyonları, 15,3± 11,4 mikrogram/mL (haftada bir 40/20 mg) ve 6,7±3,5 mikrogram/mL(haftada bir 20/10 mg) olarak belirlenmiştir. Pediyatrik üveit hastalarında adalimumab maruziyeti, diğer pediyatrik hastalar üzerinden (pediyatrik psöriyazis, jüvenil idiyopatik artrit, pediyatrik Crohn hastalığı ve entezit ile ilişkiliartrit) popülasyon farmakokinetik modellemesi ve çapraz endikasyon farmakokinetiğine dayalısimülasyon kullanılarak hesaplanmıştır. 6 yaşından küçük çocuklarda yükleme dozu kullanımıile ilgili herhangi bir klinik maruziyet verisi bulunmamaktadır. Öngörülen maruziyethesaplamaları, metotreksat yokluğunda yükleme dozunun başlangıçta sistemik maruziyetiarttırabileceğine işaret etmektedir. Dağılım:40 mg'lık tek bir dozun subkütan uygulanmasından sonra adalimumabın emilim ve dağılımı yavaştır ve doruk serum konsantrasyonlarına uygulamadan yaklaşık 5 gün sonra ulaşılmaktadır.Yapılan üç çalışmada, 40 mg'lık tek bir subkütan dozu takiben, adalimumabın ortalama mutlakbiyoyararlanım %64 olmuştur. 0,25 ile 10 mg/kg arasında değişen tek intravenöz dozdansonraki konsantrasyonlar dozla orantılıdır. 0,5 mg/kg (~40 mg) dozdan sonra, klirens 11 ila 15mL/saat arasında değişmiş, dağılım hacmi (Vss) 5 ila 6 litre arasında değişmiş ve ortalamaterminal faz yarı ömrü yaklaşık iki hafta olmuştur. Çeşitli romatoid artritli hastalardaki sinovyalsıvıdaki adalimumab konsantrasyonları serumdakinin %31-%96 oranında değişmiştir.
41 Pediyatrik popülasyonda maruziyet-yanıt ilişkisi:JIA hastalarında (pJIA ve ERA) yapılan klinik araştırmalara dayanarak plazma konsantrasyonları ile Ped ACR 50 yanıtı arasında bir maruziyet-cevap ilişkisi kurulmuştur. PedACR 50 yanıtının (EC50) olasılığının yarısını oluşturan belirgin adalimumab plazmakonsantrasyonu, 3 mikrogram/ml (%95 GA: 1-6 mikrogram/mL)'dir. Şiddetli kronik plak psöriyazisi olan pediyatrik hastalarda adalimumab konsantrasyonu ve etkililiği arasındaki maruziyet-cevap ilişkisi sırasıyla PASI 75 ve PGA temiz veya minimal içinbelirlenmiştir. PASI 75 ve PGA temiz veya minimal, yaklaşık benzer görünen yaklaşık 4,5mikrogram/mL'lik EC50 ile (sırasıyla %95 GA 0,4-47,6 ve 1,9-10,5) artan adalimumabkonsantrasyonları ile artmıştır. Erişkinler:40 mg'lık tek bir dozun subkütan uygulanmasından sonra adalimumabın emilim ve dağılımı yavaştır ve doruk serum konsantrasyonlarına uygulamadan yaklaşık 5 gün sonra ulaşılmaktadır.Yapılan üç çalışmada, 40 mg'lık tek bir subkütan dozu takiben, adalimumabın ortalama mutlakbiyoyararlanım %64 olmuştur. 0,25 ile 10 mg/kg arasında değişen tek intravenöz dozdansonraki konsantrasyonlar dozla orantılıdır. 0,5 mg/kg (~40 mg) dozdan sonra, klirens 11 ila 15mL/saat arasında değişmiş, dağılım hacmi (Vss) 5 ila 6 litre arasında değişmiş ve ortalamaterminal faz yarı ömrü yaklaşık iki hafta olmuştur. Çeşitli romatoid artritli hastalardaki sinovyalsıvıdaki adalimumab konsantrasyonları serumdakinin %31-%96 arasında değişmiştir. Erişkin romatoid artritli hastaların, 2 haftada bir 40 mg dozunda subkütan HUMIRA uygulamasını takiben ortalama kararlı durum konsantrasyonları eş zamanlı uygulananmetotreksat olmaksızın yaklaşık 5 mikrogram/mL ve eş zamanlı uygulanan metotreksat ile 8-9mikrogram/mL olmuştur. Kararlı durumda serum adalimumabın çukur düzeyleri iki haftada birve haftada bir 20, 40 ve 80 mg'lık subkütan dozu takiben doz ile kabaca orantılı biçimdeyükselmiştir. Erişkin psöriyazisli hastalarda, iki haftada bir adalimumab 40 mg monoterapisiyle 5 mikrogram/mL olan ortalama kararlı durum çukur konsantrasyonuna ulaşılmıştır. Crohn hastalığı olan hastalarda, 0. haftada 80 mg HUMIRA yükleme dozunu takiben 2. haftada 40 mg HUMIRA dozu ile indüksiyon döneminde ortalama 5,5 mikrogram/mL olan serumadalimumab çukur konsantrasyonlarına ulaşılmıştır. 0. haftada 160 mg HUMIRA yüklemedozunu takiben 2. haftada 80 mg HUMIRA ile indüksiyon döneminde ortalama 12mikrogram/mL serum adalimumab çukur konsantrasyonlarına ulaşılmıştır. İki haftada bir 40mg HUMIRA idame dozu alan Crohn hastalarında ortalama 7 mikrogram/mL olan ortalamakararlı durum çukur düzeyleri gözlemlenmiştir. Erişkin üveit hastalarında, 0. haftada 80 mg HUMIRA yükleme dozu ve onu takiben 1. haftadan itibaren iki haftada bir 40 mg adalimumab yaklaşık 8-10 mikrogram/mL ortalama kararlı durumkonsantrasyonuyla sonuçlanmıştır. Adalimumab maruziyeti ve etkililiği, popülasyon farmakokinetik ve farmakokinetik/ farmakodinamik modelleme ve simülasyonu ile 2 haftada bir 80 mg ile tedavi alan hastalar ilehaftada bir 40 mg ile tedavi edilen hastaların (RA, HS, ÜK, CH veya Ps'li erişkin hastalar, Belge Do 42 adolesan HS'li hastalar ve 40 kg ve üzerindeki pediyatrik CH hastaları dahil) karşılaştırılabilir olduğu görülmüştür. Biyotransformasyon:Adalimumabın biyotransformasyonu klinik çalışmalarda incelenmemiştir. Eliminasyon:1300'den fazla romatoid artrit hastasından alınan veriler üzerinde yürütülen popülasyon farmakokinetik analizleri, adalimumabın görünür klirensinin vücut ağırlığında artış ile artmaeğilimi gösterdiğini ortaya çıkarmıştır. Kilo farklılıklarına göre uyarlama yapıldıktan sonra,cinsiyet ve yaşın adalimumab klirensini minimal düzeyde etkiledikleri görülmüştür. Serbestadalimumabın (anti-adalimumab antikorlarına [AAA] bağlı olmayan) serum seviyelerinin,AAA düzeyleri ölçülebilen hastalarda daha düşük olduğu gözlemlenmiştir. Doğrusallık/Doğrusal olmayan durum:0,25 ila 10 mg/kg arasında değişen tek intravenöz dozlardan sonra, konsantrasyonlar dozla orantılıydı. Hastalardaki karakteristik özelliklerKaraciğer/böbrek yetmezliğiKaraciğer ya da böbrek yetmezliği olan hastalarda HUMIRA ile çalışma yapılmamıştır. 5.3. Klinik öncesi güvenlilik verileriKlinik öncesi veriler tek doz toksisitesi, yinelenen doz toksisitesi ve genotoksisite çalışmalarına dayanarak insanlarda özel bir tehlike olmadığını göstermiştir. Sinomolgus maymunlarında 0, 30 ve 100 mg/kg dozlarında bir embriyo-fetal gelişimsel toksisite/perinatal gelişim çalışması yapılmış (9-17 maymun/grup) ve fetüslerde adalimumababağlı hasara ilişkin bulgular görülmemiştir. Kemirgen TNF'sine karşı sınırlı çapraz reaktivitesiolan bir antikor ve kemirgenlerde nötralizan antikor gelişimi için uygun bir modelbulunmadığından adalimumab ile ne karsinojenisite çalışmaları ne de fertilite ve postnataltoksisite için standart değerlendirme çalışmaları gerçekleştirilmemiştir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiMannitol (E421) Polisorbat 80 (E433) Enjeksiyonluk su 6.2. GeçimsizliklerGeçimlilik çalışmaları bulunmadığından bu tıbbi ürün başka tıbbi ürünlerle karıştırılmamalıdır.
43 Raf ömrü
6.3.
24 ay 6.4. Saklamaya yönelik özel tedbirler 2 °C-8°C arasında buzdolabında saklanmalıdır. Ürün dondurulmamalıdır. Kullanıma hazır enjektör ışıktan korunması için kutusunun içerisinde saklanmalıdır. Kullanıma hazır enjektör 25°C'ye kadar varan sıcaklıklarda 14 güne kadar saklanabilir. Enjektör ışıktan korunmalıdır ve 14 gün süre içerisinde kullanılmadığında atılmalıdır. 6.5. Ambalajın niteliği ve içeriği Piston tıpası (bromobütil kauçuk) ve iğne kılıflı (termoplastik elastomer) iğnesi olan HUMIRA 20 mg enjeksiyonluk için kullanıma hazır çözelti içeren enjektör (tip I cam). Her bir ambalaj 2 adet kullanıma hazır enjektör (0,2 mL steril çözelti), her biri için blister içerisinde 1 alkollü ped 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler Kullanılmamış olan ürünler ya da atık materyaller 'Tıbbi Atıkların Kontrolü yönetmeliği ve Ambalaj Atıklarının Kontrolü yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİ Abbvie Tıbbi İlaçlar Sanayi ve Ticaret Limited Şirketi Barbaros Mah. Begonya Sok. Nidakule Ataşehir Batı Blok No: 1 İç Kapı No:33 Ataşehir/İstanbul Tel: 0216 636 06 00 Faks: 0216 425 09 69 8. RUHSAT NUMARASI 2018/106 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİ İlk ruhsat tarihi: 05.03.2018 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENMETARİHİ
44 |
İlaç BilgileriHumira 20 Mg/0.2 Ml Enjeksiyonluk Çözelti İçeren Kullanıma Hazır EnjektörEtken Maddesi: Adalimumab Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
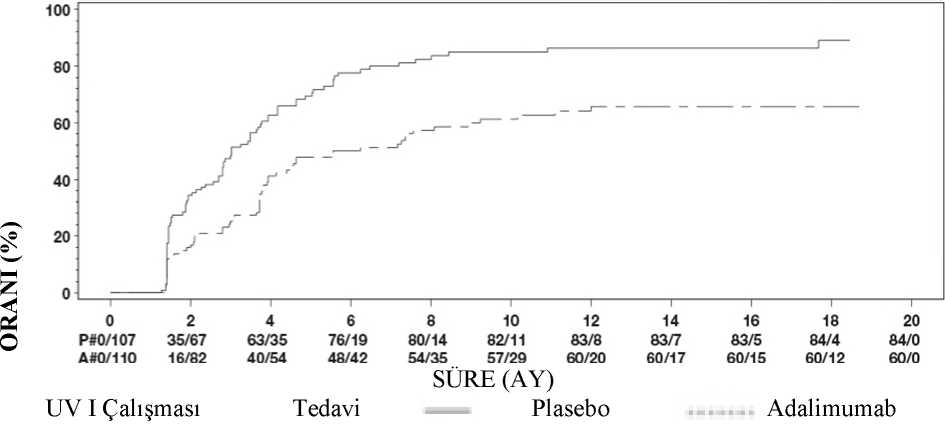
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.