Jakavi 5 Mg Tablet Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİ¡Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır?1. BEŞERİ TIBBİ ÜRÜNÜN ADIJAKAVI 5 mg Tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Ruksolitinib fosfat 6,6 mg (5 mg ruksolitinib serbest bazına karşılık gelir) Yardımcı maddeler:Laktoz monohidrat (sığır kaynaklı) 71,45 mg Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMBir yüzünde NVR ve diğer yüzünde L5 yazan, eğimli, beyaz veya beyaza yakın tabletler 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarMiyelofibrozis (MF): JAKAVI, en az bir seri tedaviye yanıtsız, DIPSS plus skoru orta/yüksek olup, kemik iliği nakline uygun olmayan primer miyelofibrozis, post polistemik miyelofibrozisveya esansiyel trombositemi sonrası ikincil miyelofibrozis tanılı hastalardasplenomegaliye bağlı semptomların tedavisinde endikedir. Polisitemi vera (PV): 1. En az 2 g/gün veya hastanın tolere edebileceği maksimum dozda hidroksiüre kullananpolistemi vera tanılı hastalarda en az 3 aylık tedaviye rağmen a. Hematokriti <%45 tutmak için ayda 1'den fazla flebotomi ihtiyacının devam etmesiveya b. Trombosit sayısının >400000/mm3, beyaz küre sayısının >10000/mm3 olması veya c. Dalak boyutlarında ultrasonografi ile küçülme saptanmaması veya d. Yeni tromboz gelişmesi 2. Tam veya kısmi yanıt için gerekli en düşük hidroksiüre dozunda, mutlak nötrofil sayısı<1000/mm3 , ya da trombosit sayısı <100000/mm3 ya da hemoglobin <10g/dl olması 3. Hidroksiüre ilişkili bacak ülseri, kontrol edilemeyen mukokutanöz belirtiler ortaya çıkmasıhalinde Sayılan durumların en az birini karşılaması halinde kullanımı uygundur. Graft versus Host hastalığı (GvHD): JAKAVI, kortikosteroidler veya diğer sistemik tedavilere yetersiz yanıt veren, akut Graft versus Host hastalığı (aGvHD) veya kronik Graft versus Host hastalığı (kGvHD) olan 12 yaşve üzerindeki hastaların tedavisinde endikedir (bkz. Bölüm 5.1).
14.2 Pozoloji ve uygulama şekliAnti-kanser ilaçların uygulanmasında deneyimli hekimler tarafından tedavi yapılmalıdır. JAKAVI ile tedaviye başlanmadan önce beyaz küre sayısı dahil tam kan sayımı gerçekleştirilmelidir. Beyaz küre sayısı dahil, tam kan sayımları, JAKAVI dozları stabilize olana kadar 2 ila 4 haftada bir, daha sonra klinik durumun gerektirdiği şekilde izlenmelidir (bkz. Bölüm 4.4). Pozoloji:Başlangıç dozu:Ruksolitinibin MF'de önerilen başlangıç dozu, trombosit sayılarına göre Tablo 1'de verilmektedir: Tablo 1 Miyelofibrozis'te başlangıç dozu
Ruksolitinibin PV'de önerilen başlangıç dozu günde iki kez ağız yoluyla 10 mg'dır. Akut ve kronik Graft versus Host hastalığında (GvHD) önerilen JAKAVI başlangıç dozu, günde iki kez oral yolla verilen 10 mg'dır. JAKAVI, kortikosteroidlerin ve/veya kalsinörininhibitörlerinin (CNI'ler) sürekli kullanımına eklenebilir. Doz düzenlemeleri: Dozlarnlilik ve etkililik esas alınarak titre edilebilir. Miyelofibroz ve _polistemi veraEğer etkililik yetersiz görülürse ve kan sayımı değerleri yeterli ise; dozlar, günde iki kere en fazla 5 mg'a kadar arttırılabilir. Maksimum doz günde iki kere 25 mg'ı geçmemelidir. Başlangıç dozu, tedavinin ilk 4 haftasında ve sonrasında 2 haftada birden daha sık aralıklarla arttırılmamalıdır. 50.000/mm3'ten düşük trombosit sayımları ya da 500/mm3'ten düşük mutlak nötrofil sayımları durumlarında tedaviye ara verilmelidir. PV'de, hemoglobin düzeyi 8 g/dL'nin altınadüştüğünde tedaviye ara verilmelidir. Kan değerleri bu düzeylerin üzerine geri döndüğündetedaviye günde iki kez 5 mg ile tekrar başlanabilir ve beyaz küre sayısı dahil, tam kansayımlarının dikkatli izlemi esas alınarak aşamalı olarak artırılabilir. Trombositopeni için doz kesintilerini önlemek amacıyla, Tablo 2'de özetlendiği gibi tedavi sırasında trombosit sayısı azalırsa, doz azaltımı düşünülmelidir.
2
PV'de hemoglobin düzeyi 12 g/dL'nin altına düştüğünde de dozun azaltılması düşünülmelidir, 10 g/dL'nin altına düştüğünde ise dozun azaltılması önerilir. Graft versus Host hastalığıBüyüme faktörleri, anti-enfektif tedaviler ve transfüzyonlar dahil olmak üzere standart destekleyici tedaviden sonra trombositopeni, nötropeni veya total bilirubin yüksekliği olanGvHD hastalarında dozun azaltılması ve tedavinin geçici olarak kesilmesi gerekebilir. Bir dozdüzeyinde azaltma adımı önerilir (günde iki kez 10 mg'dan günde iki kez 5 mg'a veya gündeiki kez 5 mg'dan günde bir kez 5 mg'a). Günde tek doz 5 mg JAKAVI'yi tolere edemeyenhastalarda tedaviye ara verilmelidir. Ayrıntılı doz önerileri Tablo 3'te verilmiştir.
Eşzamanlı güçlü CYP3A4 inhibitörleri veya_ flukonazol ile doz ayarlaması:Ruksolitinib, güçlü CYP3A4 inhibitörleri veya hem CYP2C9 ve hem de CYP3A4 enzimlerinin ikili inhibitörleri (örn; flukonazol) ile birlikte uygulanırken ruksolitinibin toplamgünlük dozu günde iki kez uygulamak üzere yaklaşık %50 azaltılmalıdır (bkz. Bölüm 4.5).Ruksolitinibin günde 200 mg'dan fazla flukonazol dozlarıyla birlikte kullanılmasındankaçınılmalıdır. Güçlü bir CYP3A4 inhibitörüne veya CYP2C9 ve CYP3A4 enzimlerinin ikili inhibitörlerine (örn; flukonazol) başlanırken hematolojik parametrelerin ve ruksolitinib ilişkili adversreaksiyonların klinik işaret ve semptomlarının daha sık izlemi (örn., haftada iki kez) önerilir. Uygulama sıklığı ve süresi:Bir doz atlanırsa, hasta ek bir doz almamalı, reçetede belirtilen bir sonraki dozunu almalıdır. Uygulama şekli:JAKAVI, oral yolla uygulanır, aç veya tok karnına alınabilir. Tedavinin bırakılmasıMF ve PV tedavisi, fayda-risk pozitif kaldığı sürece sürdürülebilir. Bununla birlikte 6 aydan sonra dalak boyutunda bir azalma yoksa veya semptomlarda tedavinin başlangıcından beri biriyileşme görülmemişse tedavi bırakılmalıdır. Bir derece klinik iyileşme göstermiş hastalar için, dalak boyutunda başlangıç boyutuna göre %40 artış korunmuşsa (dalak hacminde kabaca %25 artışa eşdeğer) ve hastalıkla ilişkilisemptomlarda dikkat çekici bir iyileşme görülmüyorsa ruksolitinib tedavisinin bırakılmasıönerilmektedir. GvHD'de, yanıt veren hastalarda ve kortikosteroid tedavisi kesildikten sonra JAKAVI'nin aşamalı olarak azaltılması düşünülebilir. JAKAVI dozunun iki ayda bir %50 azaltılmasıönerilir. JAKAVI'nin dozunun azaltılması sırasında veya sonrasında GvHD'nin belirti veyasemptomları tekrar ortaya çıkarsa, tedavi dozunun yeniden yükseltilmesi düşünülmelidir.
4Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Hafif ila orta derecedeki böbrek yetmezliği olan hastalarda spesifik doz ayarlaması gerekli değildir. Şiddetli böbrek yetmezliği olan hastalarda (kreatinin klerensi (KLkr) < 30mL/dk), trombosit sayısına bağlı olarak önerilen başlangıç dozu MF hastaları için günde iki kez uygulamaküzere yaklaşık %50 azaltılmalıdır. Şiddetli böbrek yetmezliği olan PV ve GvHD hastalarındaönerilen başlangıç dozu günde iki kez 5 mg'dır. Ruksolitinib tedavisi sırasında hastalaretkililik ve güvenlilik açısından dikkatle izlenmelidir. Diyalize giren son dönem böbrek yetmezliği (SDBY) olan hastalar için en iyi doz seçeneğine karar verebilmek açısından sınırlı veri mevcuttur. Bu popülasyonda mevcut verilere dayalıfarmakokinetik/farmakodinamik simülasyonlar, hemodiyalize giren SDBY'li MF hastalarındabaşlangıç dozunun, diyalizden sonra ve sadece hemodiyaliz gününde uygulanmak üzere 15-20mg'lık tek doz ya da 12 saat arayla verilen 10 mg'lık iki doz olduğunu düşündürmektedir.Trombosit sayıları 100.000/mm3 ile 200.000/mm3 arasında olan MF hastalarında 15 mg'lıktek doz önerilmektedir. Trombosit sayıları >200.000/mm3 olan MF hastalarında 20 mg'lık tekdoz ya da 12 saat arayla verilen 10 mg'lık iki doz önerilmektedir. Sonraki dozlar (tekuygulama ya da 12 saat arayla verilen 10 mg'lık iki doz) hemodiyaliz günlerinde her diyalizseansından sonra uygulanmalıdır. Diyalize giren SDBY'li PV hastalarında önerilen başlangıç dozu 10 mg'lık tek doz ya da 12 saat arayla verilen 5 mg'lık iki doz olup diyaliz sonrasında ve sadece hemodiyaliz günündeuygulanmalıdır. Bu doz önerileri simülasyonlara dayanmaktadır ve SDBY'de yapılacakherhangi bir doz düzenlenmesi her hastada güvenlilik ve etkililik açısından dikkatleizlenmelidir. Peritoneal diyalize ya da sürekli venovenöz hemofiltrasyona giren hastalardadoz uygulamasına ilişkin veri bulunmamaktadır (bkz. Bölüm 5.2). Son dönem böbrek yetmezliği (SDBY) olan GvHD hastaları için veri bulunmamaktadır. Karaciğer yetmezliği:Herhangi bir düzeyde karaciğer yetmezliği olan MF hastalarında, trombosit sayımına dayalı olan önerilen başlangıç dozu günde iki kez uygulamak üzere yaklaşık %50 azaltılmalıdır.Ardışık dozlar dikkatli güvenlilik ve etkililik takibi temelinde ayarlanmalıdır. PV hastalarıiçin önerilen başlangıç dozu günde iki kez 5 mg'dır. Ruksolitinib kullanırken karaciğerbozukluğu tanısı alan hastalarda, diferansiyel beyaz kan hücresi sayımı dahil tam kansayımları JAKAVI ile tedavi başlatıldıktan sonra ilk 6 hafta boyunca en az 1-2 haftada bir veardından karaciğer fonksiyon testleri ve kan sayımları stabil seyrettiğinde klinik durumungerektirdiği şekilde takip edilmelidir. Ruksolitinib dozu sitopeni riskini azaltmak amacıylatitre edilebilir. GvHD ile ilişkili olmayan hafif, orta veya şiddetli karaciğer yetmezliği olan hastalarda, ruksolitinibin başlangıç dozu %50 oranında azaltılmalıdır (bkz. Bölüm 5.2). GvHD karaciğer tutulumu olan ve toplam bilirubinin normalin üst sınırının (ULN) >3 katına yükseldiği hastalarda, toksisite açısından kan sayımları daha sık izlenmelidir ve dozun bir dozdüzeyinde azaltılması önerilir. Pediyatrik popülasyon:JAKAVI'nin MF ve PV'si olan çocuklarda ve 18 yaşa kadar ergenlerde güvenliliği ve etkililiği saptanmamıştır. Veri bulunmamaktadır (bkz. Bölüm 5.1)
5GvHD'li pediyatrik hastalarda (12 yaş ve üzeri) JAKAVI'nin güvenliliği ve etkililiği, randomize faz 3 çalışmalar olan REACH2 ve REACH3'ten elde edilen kanıtlarladesteklenmektedir. 12 yaş ve üzeri GvHD'li pediyatrik hastalarda JAKAVI dozu,yetişkinlerdeki ile aynıdır. JAKAVI'nin güvenliliği ve etkililiği 12 yaşından küçük hastalardabelirlenmemiştir. Geriyatrik popülasyon (>65 yaş):Yaşlı hastalar için ek olarak doz ayarlaması gerekmemektedir. 4.3 KontrendikasyonlarEtkin maddeye ya da formülasyondaki bileşenlerden herhangi birine aşırı duyarlılığı olan hastalarda kontrendikedir. Gebelikte ve emziren annelerde kontrendikedir. 4.4 Özel kullanım uyarıları ve önlemleriMiyelosupresyonJAKAVI tedavisi, trombositopeni, anemi ve nötropeni dahil hematolojik advers olaylara neden olabilir. JAKAVI ile tedavi başlatılmadan önce beyaz küre sayısı dahil tam kan sayımıyapılmalıdır . Trombosit sayımı 50.000/mm3'ten düşük olan veya mutlak nötrofil sayımı500/mm3'ten düşük olan MF'li hastalarda tedavi bırakılmalıdır (bkz. Bölüm 4.2). Tedavi başlangıcında trombosit sayısı düşük olan (<200.000/mm3) MF'li hastalarda tedavi sırasında trombositopeni gelişme olasılığının daha yüksek olduğu gözlenmiştir. Trombositopeni çoğu durumda geri dönüşlü olmuş ve genellikle dozun azaltılması ya da JAKAVI tedavisine geçici olarak ara verilmesiyle kontrol edilebilmiştir (bkz. Bölüm 4.2 veBölüm 4.8). Ancak, klinik gerekliliğe göre trombosit transfüzyonları gerekli olabilir. Anemi geliştiren hastalarda kan nakli gerekebilir. Anemi geliştiren hastalarda doz düzenlemeleri ya da tedaviye ara verilmesi de düşünülebilir. Hemoglobin düzeyi tedavinin başlangıcında 10 g/dL'nin altında olan hastalar, daha yüksek başlangıç hemoglobin düzeyine sahip hastalara kıyasla 8 g/ dL'lik bir hemoglobin düzeyigelişmesi açısından daha yüksek risk taşır (%30,1'e karşı %79,3). Başlangıç hemoglobini 10g/ dL olan hastalar da hematoloji parametreleri ve JAKAVI ile ilişkili advers ilaçreaksiyonlarının klinik belirti ve semptomlarına ilişkin daha sık takip önerilmektedir. Nötropeni (Mutlak Nötrofil Sayımı (MNS) <500/mm3) genellikle geri dönüşlü olmuştur ve JAKAVI tedavisine geçici olarak ara verilmesiyle kontrol edilebilmiştir (bkz. Bölüm 4.2 veBölüm 4.8). Klinik duruma göre beyaz küre sayısı dahil tam kan sayımı izlemi ve gerektiği takdirde doz ayarlaması yapılmalıdır (bkz. Bölüm 4.2 ve Bölüm 4.8). EnfeksiyonlarJAKAVI ile tedavi edilen hastalarda ciddi bakteriyel, mikobakteriyel, fungal, viral ve fırsatçı enfeksiyonlaroluşmuştur.Hastalar ciddi enfeksiyon gelişme riski açısından değerlendirilmelidir. Hekimler, JAKAVI almakta olan hastaları, enfeksiyon belirti ve semptomları açısından dikkatle gözlemlemeli ve gerektiğinde tedaviyi hızla başlatmalıdır.Aktif ciddi enfeksiyonlar giderilene kadar JAKAVI tedavisi başlanmamalıdır. JAKAVI alan hastalarda tüberküloz bildirilmiştir. Tedaviye başlamadan önce hastalar aktif ve inaktif (latent) tüberküloz açısından yerel gerekliliklere göre değerlendirilmelidir. Bu tıbbiöykü, tüberküloz ile olası önceki temas ve/veya uygun şekilde röntgen, tüberkülin testi 6ve/veya interferon-gama-salıverilme analizi gibi uygun bir taramayı içerebilir. Reçete yazan hekimlere özellikle ağır hasta olan veya immünitesi zayıflamış hastalarda yalancı negatiftüberkülin deri testi bulguları olabileceği hatırlatılmalıdır. JAKAVI alan kronik HBV enfeksiyonu olan hastalarda alanin aminotransferaz ve aspartat aminotransferaz değerlerinde artış ile ilişkili olsun veya olmasın Hepatit B viral yükünde(HBV-DNA titresi), artışlar bildirilmiştir. JAKAVI ile tedaviye başlamadan önce HBVtaraması yapılması önerilir. Kronik HBV enfeksiyonu olan hastalar klinik kılavuzlara göretedavi edilmeli ve izlenmelidir. Herpes ZosterHekimler, herpes zoster işaret ve semptomları hakkında hastaları eğitmeli, mümkün olduğunca kısa sürede tedavi için başvurmalarını tavsiye etmelidir. Progresif multifokal lökoensefalopatiJAKAVI tedavisinde progresif multifokal lökoensefalopati (PML) bildirilmiştir. Hekimler, hastaların fark edemeyebileceği, PML'ye işaret eden semptomlar (örn., kognitif, nörolojik yada psikiyatrik semptomlar veya belirtiler) konusunda özellikle dikkatli olmalıdır. Hastalar buyeni ya da ağırlaşan semptomlar ya da belirtiler için takip edilmeli ve bu tipsemptomlar/belirtiler meydana gelirse, bir nöroloji uzmanına sevk edilmeli ve PML içinuygun tanısal değerlendirmeler düşünülmelidir. PML'den şüphelenilirse, tanı dışlanıncayakadar ilaç verilmemelidir. Melanom dışı deri kanseriRuksolitinib ile tedavi edilen hastalarda bazal hücre, skuamöz hücre ve Merkel hücreli karsinom dahil olmak üzere melanom dışı deri kanserleri (MDDK) bildirilmiştir. Bu MF vePV'li hastaların çoğunun öyküsünde hidroksiüre ile uzun süreli tedavi ya da önceden MDDKveya pre-malign deri lezyonları bulunmaktadır. Ruksolitinib ile nedensellik ilişkisisaptanmamıştır. Deri kanseri açısından risk altında olan hastalarda periyodik deri muayenesiönerilmektedir. Lipid anormallikleri / yükselmeleriJAKAVI ile tedavi total kolesterol, yüksek yoğunluklu lipoprotein (HDL) kolesterol, düşük yoğunluklu lipoprotein (LDL) kolesterol ve trigliserid gibi lipid parametrelerindeki artışlarlailişkilendirilmiştir. Lipid takibi ve klinik kılavuzlara göre dislipidemi tedavisi önerilmektedir. Özel popülasyonlarBöbrek yetmezliğiŞiddetli böbrek yetmezliği olan hastalarda JAKAVI'nin başlangıç dozu azaltılmalıdır. Hemodiyalize devam eden son dönem böbrek yetmezliği olan hastalarda, MF hastaları içinbaşlangıç dozunda trombosit sayıları esas alınmalıdır, ancak PV hastaları için önerilenbaşlangıç dozu tek doz 10 mg'dır (bkz. Bölüm 4.2). Sonraki dozlar (MF hastalarında 20mg'lık tek doz veya 12 saat arayla verilen 10 mg'lık iki doz; PV hastalarında 10 mg'lık tekdoz veya 12 saat arayla verilen 5 mg'lık iki doz) sadece hemodiyaliz günlerinde her diyalizseansından sonra uygulanmalıdır. Güvenlilik ve etkililik dikkatle izlenerek ek dozdüzenlemeleri yapılmalıdır (bkz. Bölüm 4.2 ve Bölüm 5.2). Karaciğer yetmezliğiKaraciğer yetmezliği olan MF ve PV hastalarında JAKAVI'nin başlangıç dozu yaklaşık %50 azaltılmalıdır. Sonraki doz düzenlemeleri için ilacın güvenliliği ve etkililiği esas alınmalıdır.
7GvHD ile ilişkili olmayan karaciğer yetmezliği olan GvHD hastalarında, JAKAVI'nin başlangıç dozu yaklaşık %50 oranında azaltılmalıdır (bkz. Bölüm 4.2 ve Bölüm 5.2). EtkileşimlerEğer JAKAVI, güçlü CYP3A4 inhibitörleri veya hem CYP2C9 ve hem de CYP3A4 enzimlerinin inhibitörleri (örn; flukonazol) ile bir arada uygulanacaksa, doz günde iki kezuygulanmak üzere yaklaşık %50 azaltılmalıdır (monitorizasyon sıklığı için bkz. Bölüm 4.2 veBölüm 4.5). Sitoredüktif tedavilerin JAKAVI ile eşzamanlı kullanımı yönetilebilir sitopeni ile ilişkilidir. (bkz. Bölüm 4.2). Geri çekme etkileri:JAKAVI'nin kesilmesini veya bırakılmasını takiben, miyelofibrozis semptomları yaklaşık bir haftalık bir periyotta geri dönebilir. Özellikle akut araya giren hastalık varlığında, daha ağırsemptomları olan hastaların JAKAVI'yi kullanmayı bıraktığı bildirilmiştir. JAKAVI'ninaniden bırakılmasının bu olaylara katkıda bulunup bulunmadığı belirlenmemiştir. Anidenbırakma gerekli olmadıkça, her ne kadar azaltmanın faydası kanıtlanmamış olsa da JAKAVIdozunun kademeli olarak azaltılması düşünülmelidir. Yardımcı maddeler: JAKAVI laktoz içerir. Nadir kalıtımsal galaktoz intoleransı, Lapp laktaz eksikliği veya glukoz-galaktoz malabsorpsiyonu olan hastalar bu tıbbi ürünü kullanmamalıdır. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriEtkileşim çalışmaları sadece erişkinlerde yürütülmüştür. Ruksolitinib CYP3A4 ve CYP2C9 ile katalizlenen metabolizma ile elimine olur. Böylece bu enzimleri inhibe eden ilaçlar artmış ruksolitinib maruziyetine yol açabilir. Ruksolitinib dozunun azaltılmasını gerektiren etkileşimlerCYP3A4 inhibitörleri
Ruksolitinib, güçlü CYP3A4 inhibitörleri ile bir arada uygulanırken, ruksolitinibin toplam günlük dozu günde iki kez uygulanmak üzere yaklaşık %50 azaltılmalıdır. Hastalar,sitopeniler açısından yakından izlenmelidir (haftada iki kez) ve doznlilik ve etkililikesas alınarak titre edilmelidir (bkz. Bölüm 4.2). Hem CYP2C9 ve hem de CYP3A4 inhibitörleriSağlıklı gönüllülerde, tek başına ruksolitinib ile karşılaştırıldığında, ruksolitinibin (10 mg'lık tek doz) ikili bir CYP2C9 ve CYP3A4 inhibitörü olan flukonazol ile birlikte uygulanması,ruksolitinibin Cmaks ve EAA değerlerinin sırasıyla % 47 ve % 232 daha yüksek çıkmasıylasonuçlanmıştır.
8CYP2C9 ve CYP3A4 enzimlerinin inhibitörleri olan tıbbi ürünler (örn., flukonazol) kullanıldığında dozda %50 azaltma düşünülmelidir. Ruksolitinibin günde 200 mg'dan fazlaflukonazol dozlarıyla birlikte kullanılmasından kaçınılmalıdır. Enzim indükleyicileriCYP3A4 indükleyicileri (avasimib, karbamazepin, fenobarbital, fenitoin, rifabutin, rifampin (rifampisin), sarı kantaron (Hypericum perforatum) gibi ancak bunlarla sınırlı olmayan):Hastalar yakından izlenmeli ve doz güvenlilik ve etkililik temelinde titre edilmelidir (bkz.Bölüm 4.2).Potent CYP3A4 indükleyicisi rifampisini (10 gün boyunca günlük 600 mg dozda) takiben 50 mg tek doz olarak ruksolitinib verilen sağlıklı gönüllülerde, ruksolitinib EAA'sı tek başınaruksolitinib uygulamasından sonra olandan %70 daha düşük bulunmuştur. Ruksolitinib aktifmetabolitlerinin maruziyeti değişmemiştir. Genel olarak ruksolitinib farmakodinamikaktivitesi benzer olup, CYP3A4 indüksiyonunun farmakodinamik üzerinde minimumetkisinin olduğunu düşündürmektedir. Bununla birlikte, bu Emaks'a yakın farmakodinamiketkilerle sonuçlanan yüksek ruksolitinib dozu ile ilgili olabilir. Bir hastada güçlü bir enzimindükleyicisi ile tedavi başlatıldığında ruksolitinib dozunun artırılması gerekebilir. Dikkate alınması gereken diğer etkileşimlerHafif veya orta kuvvetli CYP3A4 inhibitörleri (siprofloksasin, eritromisin, amprenavir, atazanavir, diltiazem, simetidin gibi ancak bunlarla sınırlı olmayan):Orta kuvvetli bir CYP3A4 inhibitörü olan eritromisini dört gün süreyle günde iki kez 500 mg dozunda alan sağlıklı gönüllülerde JAKAVI'nin 10 mg'lık tek dozunun tek başınakullanımına kıyasla Cmaks değerinde %8, EAA değerinde %27 artış olmuştur. JAKAVI, hafif veya orta kuvvetli CYP3A4 inhibitörleri (örn, eritromisin) ile bir arada uygulanırken doz ayarlaması gerekmemektedir. Orta kuvvetli CYP3A4 inhibitörleri ile tedavibaşlatılırken hastalar sitopeniler açısından yakından izlenmelidir. Diğer ilaçlar ile etkileşim:P-glikoprotein ve diğer taşıyıcılarla taşınan bileşiklerRuksolitinib bağırsakta meme kanserine direnç proteinini (BCRP) ve P-glikoproteini inhibe edebilir. Bu inhibisyon, debigatran eteksilat, siklosporin, rosuvastatin ve muhtemelendigoksin gibi bu taşıyıcıların substratlarına sistemik maruziyeti artırabilir. Etkilenen maddeiçin terapötik ilaç takibi veya klinik takip tavsiye edilir. Bağırsakta meme kanserine direnç proteini ve P-gp için potansiyel inhibisyon, uygulamalar arasındaki süre mümkün olduğunca uzun tutularak en aza indirilebilir. Sağlıklı gönüllülerle gerçekleştirilen bir çalışma ruksolitinibin oral CYP3A4 substratı midazolamın metabolizmasını inhibe etmediğini göstermiştir. Bu nedenle, ruksolitinib ilekombine edilen CYP3A4 substratlarının maruziyetinde herhangi bir artış beklenmemektedir.Sağlıklı gönüllülerle gerçekleştirilen başka bir çalışma ruksolitinibin etinil estradiol velevonorgestrel içeren bir oral kontraseptifin farmakokinetiğini etkilemediğini göstermiştir.Dolayısıyla, eşzamanlı ruksolitinib uygulanmasıyla bu kombinasyonun kontraseptifetkililiğinin olumsuz yönde etkilenmesi beklenmemektedir. Özel popülasyonlara ilişkin ek bilgilerBöbrek yetmezliği: Böbrek yetmezliği olan hastalarla ilgili bir etkileşim çalışması yapılmamıştır.
9Karaciğer yetmezliği: Karaciğer yetmezliği olan hastalarla ilgili bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon: Pediyatrik hastalarla ilgili bir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi C'dir. Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli bulunan kadınlar, JAKAVI ile tedavi sırasında etkili doğum kontrol yöntemi kullanmalıdır. JAKAVI ile tedavi sırasında gebelik oluşursa, hasta bazında fayda-risk değerlendirmeleri yapılmalı ve fetüs üzerine potansiyel riskler ile ilgili olarak hasta bilgilendirilmelidir (bkz.Bölüm 5.3). Gebelik dönemiGebe kadınlarda JAKAVI ile yeterli çalışma bulunmamaktadır. Hayvanlar üzerinde yapılan çalışmalar ruksolitinibin embriyotoksik ve fetotoksik olduğunu göstermiştir. Sıçanlarda veya tavşanlarda teratojenisite gözlenmemiştir. Bununla birlikte,maruz kalma marjları en yüksek klinik doza kıyasla düşüktür ve dolayısıyla sonuçlarıninsanlarla ilişkisi sınırlıdır (bkz. Bölüm 5.3). İnsanlar için potansiyel risk bilinmemektedir. JAKAVİ gebelikte kontrendikedir. Gebelik süresince JAKAVI kullanılmamalıdır (bkz. Bölüm 4.3). Laktasyon dönemiEmzirme esnasında JAKAVI kullanılmamalıdır (bkz. Bölüm 4.3), bu nedenle tedavi başladığında emzirmeye ara verilmelidir. Ruksolitinib ve/veya metabolitlerinin anne sütü ileatılıp atılmadığı bilinmemektedir. Emzirilen çocuk için risk göz ardı edilemez. Hayvanlardanelde edilen farmakodinamik/toksikolojik veriler ruksolitinib ve/veya metabolitlerinin annesütüne geçtiğini göstermiştir. Üreme yeteneği/FertiliteRuksolitinibin fertilite üzerindeki etkisine dair insanda veri bulunmamaktadır. Hayvan çalışmalarında, fertilite üzerinde herhangi bir etki gözlenmemiştir. 4.7 Araç ve makine kullanımı üzerindeki etkilerJAKAVI'nin sedatif etkisi yoktur, ya da göz ardı edilebilecek kadar azdır. Bununla birlikte, JAKAVI aldıktan sonra baş dönmesi yaşayan hastalar, araç veya makine kullanmaktankaçınmalıdır. 4.8 İstenmeyen etkilerGüvenlilik profilinin özetiMiyelofibrozisEk sık bildirilen advers ilaç reaksiyonları trombositopeni ve anemidir.
10Hematolojik advers reaksiyonlar (tüm Advers Olaylar için Ortak Terminoloji Kriterleri [CTCAE] dereceleri) anemi (% 83,8), trombositopeni (% 80,5) ve nötropeniyi (% 20,8)içermektedir. Anemi, trombositopeni ve nötropeniyi doz ilişkili yan etkilerdir. Hematolojik olmayan en sık üç advers reaksiyon morarma (% 33,3), diğer kanamalar (burun kanaması, prosedür sonrası kanama ve hematüri dahil) (% 24,3) ve baş dönmesidir (% 21,9).Hematolojik olmayan en sık üç laboratuvar anomalisi alanin transaminaz (% 40,7), yüksekaspartat aminotransferaz (% 31,5) ve hipertrigliseridemidir (% 25,2). Faz 3 MF klinikçalışmalarda CTCAE derece 3 veya 4 hipertrigliseridemi veya aspartat aminotransferazyükselmesi ya da CTCAE derece 4 alanin aminotransferaz yükselmesi veyahiperkolesterolemi gözlenmemiştir. Nedensellikten bağımsız olarak advers olaylar nedeniyle tedavinin kesilmesi hastaların % 30,0'unda görülmüştür. Polisitemi veraEn sık bildirilen advers ilaç reaksiyonları anemi ve alanin aminotransferaz artışı olmuştur. Hematolojik advers reaksiyonlar (herhangi bir CTCAE derecesi) anemi (% 61,8) ve trombositopeni (% 25,0) ve nötropeniyi (% 5,3) içermektedir. CTCAE derecesi 3 ve 4 anemive trombositopeni sırasıyla % 2,9 ve % 2,6 oranında bildirilmiştir. Hematolojik olmayan en sık üç advers reaksiyon kilo artışı (% 20,3), baş dönmesi (% 19,4) ve baş ağrısı (% 17,9) olmuştur. Advers reaksiyonlar olarak tanımlanan en sık görülen üç hematoloj i dışı laboratuvar anormalliği (herhangi bir CTCAE derecesi) alanin aminotransferaz düzeyinde yükselme (%45,3), aspartat aminotransferaz düzeyinde yükselme (% 42,6) ve hiperkolesterolemi (% 34,7)olmuştur. CTCAE derece 4 alanin aminotransferaz yükselmesi veya hiperkolesterolemigözlenmemiş ve bir adet CTCAE derece 4 artmış aspartat aminotransferaz gözlenmiştir. Hastaların % 19,4'ünde nedensellikten bağımsız olarak advers reaksiyonlar nedeniyle tedavinin kesilmesi gözlenmiştir. Akut GvHDEn sık bildirilen genel advers ilaç reaksiyonları trombositopeni, anemi ve nötropenidir. Advers ilaç reaksiyonları olarak tanımlanan hematolojik laboratuvar anormallikleri trombositopeni (%85,2), anemi (%75,0) ve nötropeniyi (%65,1) içermiştir. Hastaların%47,7'sinde derece 3 anemi rapor edilmiştir (CTCAE v4.03'e göre derece 4 uygulanamaz).Hastaların sırasıyla %31,3 ve %47,7'sinde derece 3 ve 4 trombositopeni bildirilmiştir. En sık görülen hematolojik olmayan advers ilaç reaksiyonları sitomegalovirüs (CMV) enfeksiyonu (%32,3), sepsis (%25,4) ve idrar yolu enfeksiyonlarıdır (%17,9). Advers ilaç reaksiyonları olarak tanımlanan en sık görülen hematolojik olmayan laboratuvar anormallikleri, alanin aminotransferaz artışı (%54,9), aspartat aminotransferaz artışı (%52,3)ve hiperkolesterolemi (%49,2) olmuştur. Çoğunluğu derece 1 ve 2'dir. Nedenselliğe bakılmaksızın advers olaylar nedeniyle tedavinin kesilmesi hastaların %29,4'ünde gözlemlenmiştir.
11Kronik GvHDEn sık bildirilen genel advers ilaç reaksiyonları anemi, hiperkolesterolemi ve aspartat aminotransferaz artışıdır. Advers ilaç reaksiyonları olarak tanımlanan hematolojik laboratuvar anormallikleri anemi (%68,6), trombositopeni (%34,4) ve nötropeniyi (%36,2) içermiştir. Derece 3 anemihastaların %14,8'inde rapor edilmiştir (CTCAE v4.03'e göre derece 4 uygulanamaz).Hastaların sırasıyla %9,5 ve %6,7'sinde derece 3 ve 4 nötropeni bildirilmiştir. Hematolojik olmayan en sık görülen üç advers ilaç reaksiyonu hipertansiyon (%15,0), baş ağrısı (%10,2) ve idrar yolu enfeksiyonlarıdır (%9,3). Advers ilaç reaksiyonları olarak tanımlanan en sık görülen hematolojik olmayan laboratuvar anormallikleri hiperkolesterolemi (%52,3), aspartat aminotransferaz artışı (%52,2) ve alaninaminotransferaz artışıdır (%43,1). Çoğunluğu derece 1 ve 2'dir. Nedenselliğe bakılmaksızın advers olaylar nedeniyle tedavinin kesilmesi hastaların %18,1'inde gözlemlenmiştir. Klinik çalışmalardan bildirilen advers ilaç reaksiyonlarının tablo halinde listesiMF hastalarında güvenlilik, başlangıçta ruksolitinibe (n=301) randomize edilen ve kontrol tedavilerinden çapraz geçişten sonra ruksolitinib alan hastalardan (n=156) elde edilen verileriiçeren iki faz 3 çalışmadan (COMFORT-I ve COMFORT-II) elde edilen uzun vadeli takipverileri kullanılarak değerlendirilmiştir. MF hastaları için advers ilaç reaksiyonu sıklıkkategorilerinin dayandığı medyan maruziyet 30.5 aydır (aralık 0,3 ila 68,1 ay). PV hastalarında güvenlilik, başlangıçta ruksolitinibe (n=184) randomize edilmiş hastalardan ve kontrol tedavilerinden çapraz geçişten sonra ruksolitinib alan hastalardan elde edilenverileri içeren iki faz 3 çalışmadan (RESPONSE, RESPONSE 2) uzun vadeli takip verilerikullanılarak değerlendirilmiştir. (n=156). PV hastaları için ADR sıklık kategorilerinindayandığı ortanca maruziyet 41,7 aydır (aralık 0,03 ila 59,7 ay). JAKAVI'nin akut GvHD hastalarında güvenliliği, faz 3 REACH2 çalışmasında değerlendirilmişolupbudeğerlendirmede başlangıçtaJAKAVI'ye randomizeedilen hastalardan (n=152) ve mevcut en iyi tedaviden geçiş yaptıktan sonra JAKAVI alan hastalardan (n=49) elde edilen verileri içermiştir. Advers ilaç reaksiyonu sıklık kategorilerinindayandığı medyan maruziyet 8,9 haftadır (aralık 0,3 ila 66,1 hafta). JAKAVI'nin kronik GvHD hastalarında güvenliliği, faz 3 REACH3 çalışmasında değerlendirilmişolupbudeğerlendirmede başlangıçtaJAKAVI'ye randomizeedilen hastalardan (n=165) ve mevcut en iyi tedaviden geçiş yaptıktan sonra JAKAVI alan hastalardan (n=61) elde edilen verileri içermiştir. Advers ilaç reaksiyonu sıklık kategorilerinindayandığı medyan maruziyet 41,4 haftadır (aralık 0,7 ila 127,3 hafta). Klinik çalışma programında advers ilaç reaksiyonlarının şiddeti, derece 1=hafif, derece 2=orta, derece 3=şiddetli, derece 4=hayatı tehdit eden veya sakat bırakan, derece 5=ölümolarak tanımlanan CTCAE'ye dayalı olarak değerlendirilmiştir. MF ve PV'de (Tablo 4) ve akut ve kronik GvHD'deki (Tablo 5) klinik çalışmalardan elde edilen advers ilaç reaksiyonları MedDRA sistem organ sınıfına göre listelenmiştir. Klinikçalışmalar programında advers ilaç reaksiyonlarının şiddeti, CTCAE esas alınarak Bdge Dodeğeniftıöiriiffiişti¥:<^erece3f=Fh^ftfs nerece6 2= oı^fdefece^fa'detn- wece4=-ıiyı-famı12tehdit edici ya da maluliyete neden olan, derece 5= ölüm MF ve PV'de (Tablo 4) ve akut ve kronik GvHD'deki (Tablo 5) klinik çalışmalardan elde edilen advers ilaç reaksiyonları MedDRA sistem organ sınıfına göre listelenmektedir. Her birorgan sınıfı içinde advers ilaç reaksiyonları sıklığa göre sıralanmakta, en sık reaksiyonlar ilksırada gösterilmektedir. Ayrıca, her advers ilaç reaksiyonu için karşılık gelen sıklık kategorisi,aşağıdaki sisteme göredir (CIOMS III): çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygınolmayan (>1/1,000 ila <1/100); seyrek (>1/10,000 ila <1/1,000); çok seyrek (<1/10,000);sıklığı bilinmiyor. Tablo 4 MF ve PV'deki Faz 3 çalışmalarda bildirilen advers ilaç reaksiyonlarının sıklık kategorisi
13
14Tedavi kesildikten sonra MF hastalarında yorgunluk, kemik ağrısı, yüksek ateş, prurit, gece terlemeleri, semptomatik splenomegali ve kilo kaybı gibi MF semptomlarında dönüş olabilir.Klinik çalışmalarda MF semptomları için toplam semptom skoru, doz uygulaması kesildiktensonraki 7 gün içinde aşamalı olarak başlangıç değerlerine dönmüştür (bkz. Bölüm 4.4). Tablo 5 GvHD'deki faz 3 çalışmalarında bildirilen advers ilaç reaksiyonlarınınsıklık kategorisi_Akut GvHD(REACH2)Kronik GvHD(REACH3)Advers ilaç reaksiyonuEnfeksiyonlar ve enfestasyonlar
CMV enfeksiyonları
CTCAE3 derece > 3
Sepsis
CTCAE derece > 3 İdrar yolu enfeksiyonları
CTCAE derece > 3 BK virüsü enfeksiyonları
CTCAE derece > 3 Kan ve lenf sistemi hastalıkları
Trombositopeni1
CTCAE derece 3
CTCAE derece 4
Anemi1
CTCAE derece 3 Nötropeni1
CTCAE derece 3
CTCAE derece 4
Pansitopeni 1,2Metabolizma ve beslenme hastalıklarıHiperkolesterolemi1
CTCAE derece 3
CTCAE derece 4
Kilo alma
CTCAE derece >3 Sinir sistemi hastalıkları
Baş ağrısı
CTCAE derece >3 Vasküler hastalıklar
Hipertansiyon
CTCAE derece >3 Gastrointestinal hastalıklarLipaz artışı1
CTCAE derece 3
CTCAE derece 4
Amilaz artışı1
CTCAE derece 3 Sıklık kategorisiSıklık kategorisi
Çok yaygın Çok yaygın
Çok yaygın Çok yaygınÇok yaygınYaygın
Çok yaygın
Çok yaygın Çok yaygınÇok yaygınÇok yaygınÇok yaygınÇok yaygınÇok yaygın
Çok yaygın Çok yaygınYaygınYaygın
Yaygın Yaygın olmayanÇok yaygınYaygınCTCAE derece 4 BelgeÖo Kodu: lZW56aklUQ3NRSHY3M0FyYnUySHY3Z]W?i6W?i615
Yaygın YaygınYaygınYaygınYaygın
Yaygın olmayan
Çok yaygın
Yaygın Çok yaygınÇok yaygınÇok yaygınÇok yaygınYaygınYaygınÇok yaygınYaygınYaygın olmayanYaygınUygulanamaz 5
Çok yaygın YaygınÇok yaygınYaygınÇok yaygın
Yaygın Yaygın olmayanÇok yaygınYaygın
Yayg t'e.gov.Tr
ın Belge Takip Adresi:https: www.turkiye.gov.ir saglik-titck-ebys
AnemiMF'teki Faz 3 klinik çalışmalarda, ilk CTCAE derece 2 veya daha yüksek dereceli aneminin başlamasına kadar geçen medyan süre 1,5 ay olmuştur. Bir hasta (%0,3) anemi nedeniyletedaviden ayrılmıştır. Ruksolitinib alan hastalarda ortalama hemoglobin düşüşleri, tedavinin 8 ila 12 haftasından sonra başlangıcın yaklaşık 10 g/L altında en düşük değerlere ulaşmıştır ve ardından aşamalıolarak düzelerek, başlangıcın yaklaşık 5 g/L altında yeni bir kararlı duruma ulaşmıştır.Hastalarda bu patern, tedavi sırasında transfüzyon almış olup olmamalarından bağımsız birşekilde gözlenmiştir. Randomize, plasebo kontrollü çalışmada (COMFORT-I), JAKAVI tedavisindeki hastaların %60,6'sı ve plasebo uygulanan hastaların %37,7'si randomize tedavi sırasında eritrosittransfüzyonu almıştır. COMFORT-II çalışmasında eritrosit transfüzyonu oranı JAKAVIkolunda %53,4 iken en iyi mevcut tedavi (BAT) kolunda %41,1 olmuştur. Pivot çalışmaların randomize periyodunda anemi, PV hastalarında MF hastaları ile karşılaştırıldığında daha düşük sıklıkta görülmüştür (%40,8 karşısında % 82,4). PVpopülasyonunda, CTCAE derecesi 3 ve 4 olaylar %2,7 oranında bildirilirken MF hastalarındasıklık %42,56 olmuştur.
16Faz 3 akut ve kronik GvHD çalışmalarında, hastaların sırasıyla %47,7'sinde ve %14,8'inde CTCAE derece 3 anemi bildirilmiştir. TrombositopeniMF'teki Faz 3 klinik çalışmalarda, derece 3 veya 4 trombositopeni gelişen hastalarda, trombositopeninin başlangıcına kadar geçen medyan süre 8 hafta olmuştur. Trombositopenigenellikle dozun azaltılması ya da ara verilmesiyle geri dönüşümlü olmuştur. 50.000/mm3üzerindeki trombosit sayılarına dönüş için medyan süre 14 gün olmuştur. Randomize periyodboyunca trombosit transfüzyonları, ruksolitinib alan hastaların %4,7'sine ve kontrol rejimlerialan hastaların %4,0'üne uygulanmıştır. Ruksolitinib tedavisindeki hastaların %0,7'si vekontrol rejimleri alan hastaların %0,9'u trombositopeni nedeniyle çalışmadan ayrılmıştır.Çalışmaların randomize periyodu boyunca ruksolitinibe başlamadan önce trombosit sayısı100.000/mm3 ila 200.000/mm3 olan hastalarda derece 3 ya da 4 trombositopeni sıklığı,trombosit sayısı >200.000/mm3 olan hastalar ile karşılaştırıldığında daha yüksek olmuştur(%64,2 karşısında %38,5). Pivot çalışmaların randomize periyodunda, trombositopeni yaşayan hastaların oranı MF (%69,8) hastaları ile karşılaştırıldığında PV (%16,8) hastalarında daha düşük olmuştur.Şiddetli (CTCAE derecesi 3 ve 4) trombositopeni sıklığı PV hastalarında (%2,7), MFhastalarına (%11,6) kıyasla daha düşük bulunmuştur. Faz 3 akut GvHD çalışmasında, hastaların sırasıyla %31,3 ve %47,7'sinde derece 3 ve 4 trombositopeni gözlemlenmiştir. Faz 3 kronik GvHD çalışmasında, derece 3 ve 4trombositopeni, akut GvHD'dekine kıyasla daha düşük olmuştur (%5,9 ve %10,7). NötropeniMF'teki Faz 3 klinik çalışmalarda, derece 3 veya 4 nötropeni geliştiren hastalarda, nötropenin başlangıcına kadar geçen medyan süre 12 hafta olmuştur. Nötropeni nedeniyle dozuygulamalarına ara verilmesi ya da dozun azaltılması, hastaların %1.0'ında söz konusuolmuştur ve hastaların %0,3'ü nötropeni nedeniyle çalışmadan ayrılmıştır. PV hastalarında yapılan Faz 3 çalışmalarının randomize periyodunda, nötropeni referans tedavilerde %7'ye kıyasla ruksolitinibe maruz kalan hastaların % 1,6'sında bildirilmiştir.Ruksolitinib kolunda bir hastada CTCAE derece 4 nötropeni gelişmiştir. Ruksolitinib iletedavi edilen hastaların uzun süreli takibi, CTCAE derece 4 nötropeni bildiren 2 hastagöstermiştir. Faz 3 akut GvHD çalışmasında, hastaların sırasıyla %17,9'unda ve %20,6'sında derece 3 ve 4 nötropeni gözlemlenmiştir. Faz 3 kronik GvHD çalışmasında derece 3 ve 4 nötropeni, akutGvHD'dekine kıyasla daha düşük bulunmuştur (%9,5 ve %6,7). KanamaFaz 3 pivotal çalışmalarda MF kanama olayları (intrakranial ve gastrointestinal, morarma ve diğer kanama olayları dahil) ruksolitinibe maruz kalan hastaların %32,6'sı ve referanstedavileri (plasebo veya en iyi mevcut tedavi) kullananların %23,2'sinde bildirilmiştir. Derece3-4 olayların sıklığı ruksolitinib veya referans tedavilerle tedavi edilen hastalar için benzerdir(%3,1'e karşı %4,7). Tedavi sırasında kanama olayları görülen hastaların çoğu morarmabildirmiştir (%65,3). Morarma olayları referans tedavilere kıyasla ruksolitinib kullananhastalarda daha sık bildirilmiştir (%11,6'ya karşı %21,3). İntrakranial kanama ruksolitinibemaruz kalan hastaların %1'inde ve referans tedavilere maruz kalanların %0,9'undabildirilmiştir. Gastrointestinal kanama referans tedavilere maruz kalanların %3,1'ine kıyaslaruksolitinibe maruz kalan hastaların %5'inde bildirilmiştir. Diğer kanama olayları (burun Buoelge °J\
17kanaması, prosedür sonrası hemoraji ve hematüri gibi olaylar dahil) ruksolitinib ile tedavi edilen hastaların %13,3 ve referans tedavilerle tedavi edilenlerin %10,3'ünde bildirilmiştir. MF'de Faz 3 klinik çalışmaların uzun süreli takibi sırasında, kanama olaylarının kümülatif sıklığı, takip süresindeki artışla orantılı olarak artmıştır. Morarma olayları en sık bildirilenkanama olayları olmuştur (% 33,3). İntrakraniyal ve gastrointestinal kanama olayları sırasıylahastaların % 1,3 ve % 10,1'inde bildirilmiştir. PV hastalarındaki faz 3 çalışmaların karşılaştırmalı periyodunda kanama olayları (intrakranial ve gastrointestinal kanama, morarma ve diğer kanama olaylarını içerir) ruksolitinib ile tedaviedilen hastaların %16,8'inde, RESPONSE çalışmasında en iyi mevcut tedavi alan hastaların%15,3'ünde ve RESPONSE 2 çalışmasında en iyi mevcut tedavi alan hastaların %12'sindebildirilmiştir. Morarma, ruksolitinib ile tedavi edilen hastaların %10,3'ünde, RESPONSEçalışmasında en iyi mevcut tedavi alan hastaların %8,1'inde ve RESPONSE 2 çalışmasındaen iyi mevcut tedavi alan hastaların %2,7'sinde bildirilmiştir. Ruksolitinib uygulananhastalarda herhangi bir intrakranial kanama ya da gastrointestinal hemoraji olayıbildirilmemiştir. Ruksolitinib ile tedavi edilen bir hasta bir adet derece 3 kanama olayıyaşamıştır (prosedür sonrası kanama); hiçbir derece 4 kanama bildirilmemiştir. Diğer kanamaolayları (burun kanaması, prosedür sonrası hemoraji, dişeti kanaması dahil), ruksolitinib iletedavi edilen hastaların %8,7'sinde, RESPONSE çalışmasında en iyi mevcut tedavi alanhastaların %6,3'ünde ve RESPONSE 2 çalışmasında en iyi mevcut tedavi alan hastaların%6,7'sinde bildirilmiştir. PV'deki Faz-3 çalışmalarının uzun süreli takibi sırasında, kanama olaylarının kümülatif sıklığı takip süresindeki artışla orantılı olarak artmıştır. Morarma olayları, en çok sık bildirilenkanama olaylarıdır (% 17,4). Kafa içi ve gastrointestinal kanama olayları sırasıyla hastaların% 0,3 ve % 3,5'inde rapor edilmiştir. Faz 3 akut GvHD çalışmasının karşılaştırmalı döneminde, ruksolitinib ve mevcut en iyi tedavi kollarındaki hastaların sırasıyla %25,0 ve %22,0'sinde kanama olayları rapor edilmiştir.Kanama olaylarının alt grupları genellikle tedavi kolları arasında benzer olmuştur: morarmaolayları (ruksolitinibte %5,9'a karşı mevcut en iyi tedavi kolunda %6,7), gastrointestinalolaylar (%9,2'ye karşı %6,7) ve diğer kanama olayları (%13,2'ye karşı %10,7). İntrakraniyalkanama olayları mevcut en iyi tedavi kolundaki hastaların %0.7'sinde rapor edilirken veruksolitinib kolunda hiçbir hastada rapor edilmemiştir. Faz 3 kronik GvHD çalışmasının karşılaştırmalı döneminde, ruksolitinib ve mevcut en iyi tedavi kollarındaki hastaların sırasıyla %11,5 ve %14,6'sında kanama olayları raporedilmiştir. Kanama olaylarının alt grupları genellikle tedavi kolları arasında benzer olmuştur:morarma olayları (ruksolitinibde %4,2'ye karşı mevcut en iyi tedavi kolunda %2,5),gastrointestinal olaylar (%1,2'ye karşı %3,2) ve diğer kanama olayları (%6,7'ye karşı %10,1).Her iki tedavi kolunda da intrakraniyal kanama olayı bildirilmemiştir. EnfeksiyonlarFaz 3 pivotal çalışmalarda hastaların %1'inde derece 3 veya 4 idrar yolu enfeksiyonu, %4,3'ünde herpes zoster ve %1'inde tüberküloz bildirilmiştir. Faz 3 klinik çalışmalarda sepsishastaların %3'ünde bildirilmiştir. Ruksolitinib ile tedavi edilen hastaların uzun süreli takibi,zaman içinde sepsis oranında artış yönünde bir eğilim olmadığını göstermiştir. PV hastalarında yapılan Faz 3 çalışmalarının randomize periyodunda, bir (% 0,5) CTCAE derece 3 idrar yolu enfeksiyonu bildirilmiş, herhangi bir derece 4 idrar yolu enfeksiyonubildirilmemiştir. Herpes zoster oranı PV (% 4,3) hastaları ile MF (% 4,0) hastaları arasındabenzer olmuştur. PV hastaları arasında CTCAE derece 3 post-herpetik nevralji ile ilgili bir 18rapor söz konusu olmuştur. Referans tedavilerde hastaların %1.6'sına kıyasla ruksolitinib ile tedavi edilen hastaların % 0,5'inde pnömoni bildirilmiştir. Ruksolitinib kolundaki hiçbir hastasepsis veya tüberküloz bildirmemiştir. PV'de Faz 3 çalışmalarının uzun süreli takibi sırasında sıklıkla bildirilen enfeksiyonlar idrar yolu enfeksiyonları (% 11,8), herpes zoster (% 14,7) ve pnömoni (% 7,1) olmuştur. Hastaların% 0,6'sında sepsis bildirilmiştir. Uzun süreli takipte hiçbir hasta tüberküloz bildirmemiştir. Faz 3 akut GvHD çalışmasında, karşılaştırmalı dönemde,uzun süreli takiplerisırasında, hastaların %17,9'unda (derece >3, %6,5) idrar yoluenfeksiyonları ve hastaların %32,3'ünde (derece >3, %11,4) CMV enfeksiyonlarıbildirilmiştir. Organ tutulumlu CMV enfeksiyonu çok az hastada görülmüştür; Sırasıyla dört,iki ve bir hastada CMV koliti, CMV enteriti ve herhangi bir derecedeki CMV gastrointestinalenfeksiyonu bildirilmiştir. Herhangi bir derecedeki septik şok dahil sepsis olayları hastaların%25,4'ünde (derece >3, %21,9) rapor edilmiştir.Faz 3 kronik GvHD çalışmasında, karşılaştırmalı dönemde,uzun süreli takiplerisırasında, hastaların sırasıyla %9,3'ünde (derece >3,%1,3) ve %4,9'unda (derece >3, %0,4) idrar yolu enfeksiyonları ve BK virüsü enfeksiyonlarıbildirilmiştir. Hastaların sırasıyla %8,8'inde (derece >3, %1,3) ve %3,5'inde (derece >3,%3,5) CMV enfeksiyonları ve sepsis olayları bildirilmiştir.Lipaz yükselmesiRESPONSE çalışmasının randomize periyodunda, lipaz değerlerinde kötüleşme, kontrol koluna kıyasla ruksolitinib kolunda daha yüksek olup bunun ana nedeni 1. derece yükselmelerarasındaki farklar olmuştur (% 18,2'ye karşı % 8,1). Derece > 2 yükselmeler tedavi kollarıarasında benzer bulunmuştur. RESPONSE 2'de, sıklıklar ruksolitinib ve kontrol kolu arasındakarşılaştırılabilir olmuştur (% 10,8'e karşı % 8). Faz 3 PV çalışmalarının uzun süreli takibisırasında hastaların % 7,4 ve % 0,9'u derece 3 ve derece 4 lipaz değerlerinde yükselmebildirmiştir. Bu hastalarda yüksek lipaz değerleri ile eşzamanlı pankreatit belirti vesemptomları bildirilmemiştir. MF'de yapılan faz 3 çalışmalarda, COMFORT-I ve COMFORT-II çalışmalarında kontrol kollarında sırasıyla % 16,6 ve % 14,0 ile karşılaştırıldığında ruksolitinib kollarındakihastaların % 18,7 ve % 19,3'ünde yüksek lipaz değerleri bildirilmiştir. Lipaz değerleriyükselmiş hastalarda, eş zamanlı pankreatit belirti ve semptomları bildirilmemiştir. Faz 3 akut GvHD çalışmasının karşılaştırmalı dönemindey^iaiiyen}ckv,eya |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
COMFORT-I |
COMFORT-II | ||
|
|
JAKAVI (N=155) |
Plasebo (N=153) |
JAKAVI (N=144) |
Mevcut En İyi Tedavi (N=72) |
|
Zaman noktaları |
24. hafta |
48. hafta | ||
|
Dalak boyutu >%35 küçülen hasta sayısı (%) |
65 (41,9) |
1 (0,7) |
41 (28,5) |
0 |
|
%95 güven aralıkları |
34,1-50,1 |
0-3,6 |
21,3-36,6 |
0-5 |
|
P değeri |
< 0,0001 |
< 0,0001 | ||
JAK2V617F mutasyonunun veya hastalık alt tipinin (primer MF, polisitemi vera sonrası MF, esansiyel trombositemi sonrası MF) varlığına veya yokluğuna bakılmaksızın, JAKAVIgrubundaki hastaların anlamlı derecede daha büyük bir oranı, dalak hacminde başlangıca göre>%35 küçülme elde etmiştir.
T* Bu belge
22TabloJAK mutasyon durumuna göre dalak hacminde başlangıca göre >%35
azalma yaşayan hasta yüzdesi (güvenlilik seti)
|
|
COMFORT-I |
COMFORT-II | ||||||
|
|
JAKAVI |
Plasebo |
JAKAVI |
En iyi mevcut tedavi | ||||
|
JAK mutasyon durumu |
Pozitif (N=113)n (%) |
Negatif (N=40)n (%) |
Pozitif (N=121)n (%) |
Negatif (N=27)n (%) |
Pozitif (N=110)n (%) |
Negatif (N=35)n (%) |
Pozitif (N=49)n (%) |
Negatif (N=20)n (%) |
|
Dalak hacmi >%35 azalmışgönüllü sayısı(%) |
54 (47,8) |
11 (27,5) |
1 (0,8) |
0 |
36 (32,7) |
5 (14.3) |
0 |
0 |
|
Zaman noktası |
24 hafta sonra |
48 hafta sonra | ||||||
JAKAVI tedavisinde en az 24 hafta süreyle dalak yanıtını (>%35 azalma) sürdürme olasılığı COMFORT-I çalışmasında %89 ve COMFORT-II çalışmasında %87 olmuştur; COMFORT-II çalışmasında hastaların %52'si dalak yanıtlarını en az 48 hafta süreyle korumuştur.
COMFORT-I çalışmasında JAKAVI grubundaki hastaların %45,9'unda toplam semptom skorunda (MFSAF günlüğü v2.0 kullanılarak ölçülen) 24. haftada başlangıca göre >%50düzelme elde edilirken bu oran plasebo grubunda %5,3 bulunmuştur (ki-kare testi kullanılarakp<0,0001). 24. haftada Avrupa Kanser Araştırma ve Tedavi Organizasyonu (EORTC) QLQC30 ile ölçülen global sağlık durumundaki ortalama değişiklik JAKAVI için +12,3 ikenplasebo için -3,4 olmuştur (p<0,0001).
COMFORT-I çalışmasında 34,3 aylık medyan takip sonrasında ölüm oranı, ruksolitinib koluna randomize edilen hastalarda %27,1 iken plaseboya randomize edilen hastalarda %35.1bulunmuştur; HR 0,687; %95 GA 0,459-1,029; p=0,0668.
COMFORT-I çalışmasında 61,7 aylık medyan takip sonrasında ölüm oranı, ruksolitinib koluna randomize edilen hastalarda %44,5 (155 hastanın 69'u) iken plaseboya randomizeedilen hastalarda %53,2 (154 hastanın 82'si) bulunmuştur. Ruxolitinib kolunda plaseboyagöre ölüm riskinde % 31'lik bir azalma olmuştur (HR 0,69; % 95 GA 0,50-0,96; p = 0,025).
COMFORT-II çalışmasında 34,7 aylık medyan takip sonrasında ölüm oranı, ruksolitinib koluna randomize edilen hastalarda %19,9 iken mevcut en iyi tedaviye (BAT) randomizeedilen hastalarda %30,1 olmuştur; HR 0,48; %95 GA 0,28-0,85; p=0,009. Her iki çalışmadada ruksolitinib kolunda belirlenen daha düşük ölçüm oranlarının ağırlıklı sebebi, post-polisitemi vera ve post-esansiyel trombositemi alt gruplarında elde edilen sonuçlar olmuştur.
COMFORT-II çalışmasında 55,9 aylık medyan takip sonrasında ölüm oranı, ruksolitinib koluna randomize edilen hastalarda %40,4 (146 hastanın 59'u) iken mevcut en iyi tedaviye(BAT) randomize edilen hastalarda %47,9 (73 hastanın 35'i) olmuştur; Ruksolitinib kolundaplaseboya göre ölüm riskinde % 33'lik bir azalma olmuştur (HR 0,67; % 95 GA 0,44-1,02; p= 0,062).
23
Polisitemi vera
Avrupa LeukemiaNet (ELN) uluslararası çalışma grubunun yayımlamış olduğu kriterlere göre tanımlanan, hidroksiüre tedavisine dirençli veya yanıt vermeyen 222 PV hastası ilerandomize, açık etiketli, aktif kontrollü bir faz 3 çalışma (RESPONSE) gerçekleştirilmiştir.110 hasta ruksolitinib koluna ve 112 hasta BAT koluna randomize edilmiştir. JAKAVI'ninbaşlangıç dozu günde iki kez 10 mg olmuştur. Dozlar daha sonra tolerabilite ve etkililik esasalınarak, maksimum doz günde iki kez 25 mg olacak şekilde hasta bazında düzenlenmiştir.BAT araştırmacı tarafından hasta bazında seçilmiştir ve şunları içermiştir: hidroksiüre(%59,5), interferon/pegile interferon (%11,7), anagrelid (%7,2), pipobroman (%1,8) vegözlem (%15,3).
Başlangıçtaki demografik özellikler ve hastalık karakteristikleri iki tedavi kolu arasında karşılaştırılabilir niteliktedir. Medyan yaş 60'tır (aralık 33-90 yaş). Ruksolitinib kolundakihastalarda medyan 8,2 yıl PV tanısı mevcuttur ve bu hastalar önceden medyan olarak yaklaşık3 yıl süreyle hidroksiüre tedavisi almıştır. Çoğu hastaya (>%80) tarama öncesindeki son 24haftada en az iki flebotomi uygulanmıştır. Uzun süreli sağkalım ve hastalıkkomplikasyonlarının insidansına yönelik karşılaştırmalı veri mevcut değildir.
Birincil bileşik sonlanım noktasını hem flebotomiye uygunluğun (HCT kontrolü) olmayışı hem de 32. haftada dalak büyüklüğünde başlangıca göre >%35 azalma durumuna ulaşanhastaların oranı oluşturmuştur. Flebotomiye uygunluk, doğrulanan HCT >%45 şeklinde, yanibaşlangıçta elde edilen HCT'ye kıyasla en az 3 yüzde puanı fazla ya da doğrulanan HCT>%48 şeklinde (hangisinin daha düşük olduğuna bağlı olarak) tanımlanmıştır. Başlıca ikincilsonlanım noktaları arasında birincil sonlanım noktasına ulaşan ve 48. haftada progresyonsuzkalan hastaların yanı sıra 32. haftada tam hematolojik remisyona ulaşan hastaların oranı yeralmıştır.
Çalışma birincil amacını karşılamıştır ve JAKAVI grubundaki hastaların daha yüksek bir oranı birincil bileşik sonlanım noktasına ve bileşenlerinin her birine ulaşmıştır. BAT (%0,9)ile karşılaştırıldığında, JAKAVI ile tedavi edilen hastaların anlamlı derecede daha çoğu (%23)bir birincil yanıta ulaşmıştır (p<0,0001). Hematokrit kontrolü JAKAVI kolundaki hastaların%60'ında elde edilirken bu oran BAT kolunda %18,8 bulunmuştur ve dalak hacminde >%35azalma, JAKAVI kolundaki hastaların %40'ında, BAT kolundaki hastaların ise %0,9'undaelde edilmiştir (Şekil 1).
Ayrıca her iki ikincil sonlanım noktası da karşılanmıştır. Tam hematolojik remisyona ulaşan hastaların oranı JAKAVI kolunda %23,6 iken BAT kolunda %8,0 bulunmuştur (p=0,0013) ve48. haftada kalıcı birincil yanıta ulaşan hastaların oranı JAKAVI ile %20 ve BAT ile %0,9olmuştur (p<0,0001).
2432. haftada birincil sonlanım noktasına ve birincil sonlanım noktasının bileşenlerine ulaşan hastalar
Şekil 1
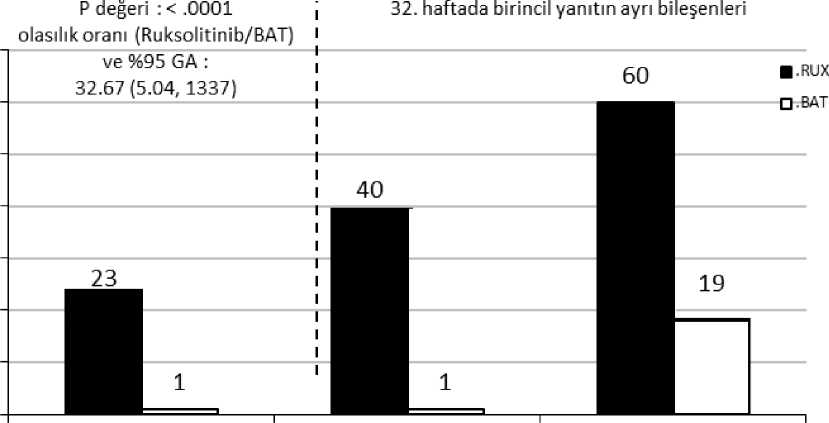 |
32. haftada birincil bileşik sonlanım Dalak Hacminde >%35 azalma Flebotomisiz hematokrit kontrolü noktası |
70
8 60
T3
M
:>- 50
C
¦S 40 ro
41
WJŞ 30
2010
0Semptom yükü, 14 sorudan oluşan MPN-SAF toplam semptom skoru (TSS) elektronik hasta günlüğü kullanılarak değerlendirilmiştir. 32. haftada ruksolitinib ile tedavi edilen hastalarınsırasıyla %49'unda ve %64'ünde TSS-14 ve TSS-5'te >%50 azalma elde edilirken BATtedavisindeki hastalarda bu oranlar sadece %5 ve %11 olmuştur.
Tedavi faydası algısı, Hasta Global Değişiklik İzlenimi (PGIC) anketi kullanılarak ölçülmüştür. BAT ile tedavi edilen hastaların %19'u karşısında ruksolitinib ile tedavi edilenhastaların %66'sı, tedavi başladıktan sonra henüz 4 hafta gibi kısa bir sürede bir düzelmebildirmiştir. Tedavi faydası algısında düzelme 32. haftada da ruksolitinib ile tedavi edilenhastalarda daha yüksek olmuştur (%78 karşısında %33).
RESPONSE çalışmasında, randomizasyonun ardından 80. haftada ve 256. haftada yanıtın kalıcılığına yönelik ek analizler gerçekleştirilmiştir. 32. haftada birincil yanıt elde edilen 25hastadan 3 hasta 80. hafta itibariyle, 6 hasta 256. hafta itibariyle progrese olmuştur. Yanıtın32. haftadan sonra 80. hafta ve 256. haftaya kadar devam etme olasılığı sırasıyla % 92 ve %74 olmuştur (bkz. Tablo 4).
Tablo 8 RESPONSE çalışmasında birincil yanıtın kalıcılığı | ||||||||||||||||||||||||
| ||||||||||||||||||||||||
|
25 |
* Birincil yanıt bileşik sonlanım noktası kriterlerine göre: flebotomi uygunluğunun olmaması (HCT kontrolü) ve dalak hacminde başlangıca göre > % 35 azalma.
n/a : uygulanamaz
Hidroksiüreye dirençli olan ya da hidroksiüreyi tolere edemeyen fakat palpabl splenomegalisi olmayan 149 PV hastasında ikinci bir randomize, açık etiketli, aktif kontrollü, faz 3b çalışma(RESPONSE 2) gerçekleştirilmiştir. 28. haftada HCT kontrolü (flebotomiye uygunluğunolmaması) elde edilen hastaların oranı şeklinde tanımlı birincil sonlanım noktasıkarşılanmıştır (JAKAVI kolunda %62,2 karşısında BAT kolunda %18,7). 28. haftada tamhematolojik remisyona ulaşan hastaların oranı şeklinde tanımlı ana ikincil sonlanım noktasınada ulaşılmıştır (JAKAVI kolunda %23 karşısında BAT kolunda %5,3).
Graft-versus-host hastalığı
İki randomize faz 3, açık etiketli, çok merkezli çalışmada allojenik hematopoietik kök hücre transplantasyonundan (alloSCT) sonra akut GvHD (REACH2) ve kronik GvHD'si(REACH3) olan ve kortikosteroidlere/veya diğer sistemik tedavilere yetersiz yanıt veren 12yaş ve üzerindeki hastalarda JAKAVI incelenmiştir. JAKAVI'nin başlangıç dozu günde ikikez 10 mg olmuştur.
Akut graft-versus-host hastalığı
REACH2'de, kortikosteroidlere derece II ila IV dirençli, akut GvHD'li 309 hasta, JAKAVİ veya mevcut en iyi tedaviye 1:1 oranında randomize edilmiştir. Hastalar, randomizasyonsırasında akut GvHD'nin ciddiyetine göre katmanlandırılmıştır. Kortikosteroid refrakterliği,hastalarda en az 3 gün sonra progresyon olduğunda, 7 gün sonra yanıt alınamadığında veyakortikosteroid azaltımının başarısız olduğu durumlar olarak belirlenmiştir.
Mevcut en iyi tedavi(BAT), araştırmacı tarafından hasta bazında seçilmiştir ve anti-timosit globulin (ATG), ekstrakorporeal fotoferez (ECP), mezenkimal stromal hücreler (MSC), düşükdoz metotreksat (MTX), mikofenolat mofetil (MMF), mTOR inhibitörleri (everolimus veyasirolimus), etanersept veya infliksimabi içermiştir.
JAKAVI veya mevcut en iyi tedaviye ek olarak, hastalar anti-enfektif tıbbi ürünler ve transfüzyon desteği dahil olmak üzere standart allojenik kök hücre nakli destekleyici bakımalmış olabilir. Ruksolitinib, enstitülerin kılavuzlarına göre kortikosteroidlerin ve/veyasiklosporin veya takrolimus gibi kalsinörin inhibitörlerinin (CNI'ler) ve/veya topikal veyainhale kortikosteroid tedavilerinin sürekli kullanımına eklenmiştir.
Akut GvHD için kortikosteroidler ve kalsinörin inhibitörlerinin (CNI) dışında önceden bir sistemik tedavi almış hastalar çalışmaya dahil edilmeye uygun bulunmuştur. Kortikosteroidlerve kalsinörin inhibitörlerine (CNI) ek olarak, akut GvHD için önceki sistemik tıbbi ürünün,yaygın tıbbi uygulamaya göre yalnızca akut GvHD profilaksisi için kullanılması durumunda(yani akut GvHD teşhisinden önce başlamışsa), devamına izin verilmiştir.
Mevcut en iyi tedavi alan hastalar, aşağıdaki kriterleri karşılamaları halinde 28. günden sonra ruksolitinibe geçebilmişlerdir:
28. günde birincil sonlanım noktası yanıt tanımı (tam yanıt [CR] veya kısmi yanıt [PR])karşılanamamışsa; VEYA
Sonrasında yanıtı kaybetmişse ve akut GvHD için yeni ek sistemik immünosupresiftedavi gerektiren progresyon, karışık yanıt veya yanıtsızlık kriterlerini karşılamışsa, VE
BuDelge
Belge DoJbelirtiIeri/§emıploml'arıı\olmamış>sae Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys
26
Tedavi yanıtı olan hastalar için 56. gün vizitinden sonra JAKAVI'nin azaltılmasına izin verilmiştir.
Başlangıçtaki demografik özellikler ve hastalık özellikleri, iki tedavi kolu arasında dengelidir. Medyan yaş 54'tür (12 ila 73 yaş). Çalışmaya %2,9 adölesan, %59,2 erkek ve %68,9 beyazhasta dahil edilmiştir. Kayıtlı hastaların çoğunda altta yatan malign hastalık vardır.
Akut GvHD'nin şiddeti, JAKAVI ve mevcut en iyi tedavi kollarının sırasıyla %34 ve %34'ünde derece II, %46 ve %47'sinde derece III ve %20 ve %19'unda derece IV olmuştur.
JAKAVI ve mevcut en iyi tedavi kollarında hastaların kortikosteroidlere yetersiz yanıt vermesinin nedenleri i) 7 günlük kortikosteroid tedavisinden sonra yanıt elde edilememesi(sırasıyla %46,8 ve %40,6), ii) kortikosteroid azaltımında başarısızlık (sırasıyla %30,5 ve%31,6) veya iii) 3 günlük tedaviden sonra hastalığın ilerlemesidir (sırasıyla %22,7 ve %27,7).
Tüm hastalar arasında akut GvHD'de en sık tutulan organlar deri (%54,0) ve alt gastrointestinal sistem (%68,3) olmuştur. Mevcut en iyi tedavi koluna kıyasla (deri: %47,7 vekaraciğer: %16,1) JAKAVI kolundaki daha fazla hastada deri (%60,4) ve karaciğeri (%23,4)içeren akut GvHD söz konusu olmuştur.
En sık kullanılan önceki sistemik akut GvHD tedavileri kortikosteroidler+kalsinörin inhibitörleridir (CNI) (JAKAVI kolunda %49,4 ve mevcut en iyi tedavi kolunda %49,0).
Birincil sonlanım noktası, daha erken bir progresyon için ek sistemik tedavilere gerek duymadan her bir kolda tam yanıt (CR) veya kısmi yanıt (PR) olan hastaların oranı olaraktanımlanan 28. günde genel yanıt oranı (ORR), karışık yanıt veya Harris ve ark. (2016)kriterlerine göre araştırmacı değerlendirmesine dayalı yanıtsızlıktır.
Kritik ikincil sonlanım noktası, 28. günde bir tam yanıt (CR) veya kısmi yanıt (PR) elde eden ve 56. günde bir tam yanıt (CR) veya kısmi yanıt (PR) sürdüren hastaların oranıdır.
REACH2 birincil hedefine ulaşmıştır. Tedavinin 28. gününde genel yanıt oranı (ORR), mevcut en iyi tedavi koluna (%39,4) kıyasla JAKAVI kolunda (%62,3) daha yüksektir.Tedavi kolları arasında istatistiksel olarak anlamlı bir fark söz konusu olmuştur (katmanlıCochrane-Mantel-Haenszel testi p<0.0001, iki taraflı, OR: 2,64; %95 GA: 1,65, 4,22).
Ayrıca mevcut en iyi tedavi koluna (%19,4) kıyasla JAKAVI kolunda (%34,4) tam yanıt verenlerin oranı daha yüksek bulunmuştur.
28. günde genel yanıt oranı (ORR), JAKAVI kolunda derece II GvHD için %76, derece III GvHD için %56 ve derece IV GvHD için %53 ve mevcut en iyi tedavi kolunda derece IIGvHD için %51, derece III GvHD için %38 ve derece IV GvHD için %23 olmuştur.
JAKAVI ve mevcut en iyi tedavi kollarında 28. günde yanıt vermeyenlerin sırasıyla %,2,6 ve %8,4'ü hastalık progresyonuna sahiptir.
Genel sonuçlar Tablo 9'da sunulmuştur.
27
Tablo 9 REAC |
H2'de 28. günde genel yanıt oranı | |||
|
|
JAKAVIN=154 |
Mevcut En Iyi Tedavi N=155 | ||
|
|
n (%) |
%95 GA |
n (%) |
%95 GA |
|
Genel yanıt |
96 (62,3) |
54,2; 70,0 |
61 (39,4) |
31,6; 47,5 |
|
OR (%95 GA) |
2,64 (1,65; 4,22)_ | |||
|
p-değeri (2-taraflı) |
p <0.0001 | |||
|
Tam yanıt |
53 (34,4) |
30 (19,4) | ||
|
Kısmi yanıt |
43 (27,9) |
31 (20,0) | ||
Çalışma, birincil veri analizine dayalı olarak kilit ikincil sonlanım noktasına ulaşmıştır (veri kesim tarihi: 25-Temmuz-2019). 56. günde kalıcı genel yanıt oranı, JAKAVI kolunda %39,6(%95 GA: 31,8, 47,8) ve mevcut en iyi tedavi kolunda %21,9 (%95 GA: 15,7, 29,3) olmuştur.İki tedavi kolu arasında istatistiksel olarak anlamlı bir fark söz konusu olmuştur (OR: 2,38;%95 GA: 1,43, 3,94; p=0.0007). Kısmi yanıtı olan hastaların oranı, mevcut en iyi tedavikolunda %16,1'e karşılık JAKAVI kolunda %26,6 olmuştur. Genel olarak, başlangıçtamevcut en iyi tedavi koluna randomize edilen 49 hasta (%31,6) JAKAVI koluna geçmiştir.
Kronik graft-versus-host hastalığı
REACH3'te, kortikosteroide orta veya şiddetli dirençli, kronik GvHD'si olan 329 hasta, JAKAVI veya mevcut en iyi tedaviye 1:1 oranında randomize edilmiştir. Hastalar,randomizasyonsırasında kronik GvHD'nin ciddiyetine göre katmanlandırılmıştır.
Kortikosteroid refrakterliği, hastalarda 7 gün sonra yanıtsızlık veya hastalık progresyonu olduğunda veya hastalık 4 hafta boyunca sebat ettiğinde veya kortikosteroid azaltımının ikikez başarısız olduğu durumlar olarak belirlenmiştir.
Mevcut en iyi tedavi, araştırmacı tarafından hasta bazında seçilmiştir ve ekstrakorporeal fotoferez (ECP), düşük doz metotreksat (MTX), mikofenolat mofetil (MMF), mTORinhibitörleri (everolimus veya sirolimus), infliksimab, rituksimab, pentostatin, imatinib veyaibrutinibi içermiştir.
JAKAVI veya mevcut en iyi tedaviye ek olarak, hastalar anti-enfektif tıbbi ürünler ve transfüzyon desteği dahil olmak üzere standart allojenik kök hücre nakli destekleyici bakımalabilmişlerdir. Siklosporin veya takrolimus gibi kortikosteroidlerin ve kalsinörininhibitörlerinin (CNI) sürekli kullanımına ve topikal veya inhale kortikosteroid tedavilerineenstitülerin kılavuzlarına göre izin verilmiştir.
Kronik GvHD için kortikosteroidler ve/veya kalsinörin inhibitörleri dışında önceden bir sistemik tedavi almış hastalar çalışmayadahil edilmeye uygun bulunmuştur.
Kortikosteroidlere ve kalsinörin inhibitörlerine ek olarak, kronik GvHD için önceki sistemik tıbbi ürünün, yaygın tıbbi uygulamaya göre yalnızca kronik GvHD profilaksisi içinkullanılması durumunda (yani kronik GvHD teşhisinden başlamışsa), devamına izinverilmiştir.
Mevcut en iyi tedavi alan hastalar, 7. döngü 1. günde ve sonrasında hastalık progresyonu, karışık yanıt veya değişmeyen yanıt, mevcut en iyi tedaviye toksisite veya kronik GvHDalevlenmesi nedeniyle ruksolitinibe geçebilmişlerdir.
Kortikosteroidleri ve herhangi bir sistemik tedaviyi azaltmadan aktif akut GvHD'den kronik
Bırbelge, ehvenli ejekti'omk mı/a ne imzalanmıştır. . .
Bdge DtGvHDyâ : geçışaıyspa»Rfeastal(aEdafevsetkiiilık bilmmemrastedrs i DoöörYteftfosıio^flfügsonttnıdan
28
(DLI) sonra akut veya kronik GvHD'de ve steroid tedavisini tolere edemeyen hastalardaki etkililik bilinmemektedir.
7. döngü 1. gün vizitinden sonra JAKAVI'nin azaltılmasına izin verilmiştir.
Başlangıçtaki demografik özellikler ve hastalık özellikleri, iki tedavi kolu arasında dengelidir. Medyan yaş 49'dur (12 ila 76 yaş). Çalışmaya %3,6 adölesan, %61,1 erkek ve %75,4 beyazhasta dahil edilmiştir. Kayıtlı hastaların çoğunda altta yatan malign hastalık söz konusuolmuştur.
Kortikosteroide dirençli kronik GvHD'nin tanı anındaki ciddiyeti, JAKAVI ve mevcut en iyi tedavi kollarında sırasıyla %41 ve %45 orta ve %59 ve %55 şiddetli olmak üzere iki tedavikolu arasında dengelidir.
JAKAVI ve mevcut en iyi tedavi kolundaki hastaların kortikosteroidlere yetersiz yanıtı, i) 1 mg/kg/gün prednizon eşdeğerlerinde kortikosteroid tedavisinden en az 7 gün sonra yanıtsızlık(sırasıyla %37,6 ve %44,5) veya hastalık progresyonu ii) 0,5 mg/kg/gün dozunda 4 haftasonra hastalığın kalıcılığı (%35,2 ve %25,6) veya iii) kortikosteroid bağımlılığı (sırasıyla%27,3 ve %29,9) ile karakterize edilmiştir.
Tüm hastalar arasında, mevcut en iyi tedavi kolunda %69 ve %41'e kıyasla, JAKAVI kolunda %73 ve %45'inde deri ve akciğer tutulumu söz konusu olmuştur.
En sık kullanılan önceki sistemik kronik GvHD tedavileri sadece kortikosteroidler (JAKAVI kolunda %43 ve mevcut en iyi tedavi kolunda %49) ve kortikosteroidler+kalsinörininhibitörleridir(CNI) (JAKAVI kolunda %41 hasta ve mevcut en iyi tedavi kolunda %42).
Birincil sonlanım noktası, daha erken bir progresyon için ek sistemik tedavilere gerek duymadan her bir kolda tam yanıt(CR) veya kısmi yanıtı(PR) olan hastaların oranı olaraktanımlanan 7. döngünün 1. gününde genel yanıt oranı (ORR), karışık yanıt veya Ulusal SağlıkEnstitüleri (NIH) kriterlerine göre yanıtsızlık olmuştur.
Kritik bir ikincil sonlanım noktası, aşağıdaki olayların en erken görülenini içeren, bileşik bir olaya kadar geçen zaman sonlanım noktası olan başarısızlık olmadan sağkalımdır (FFS): i)altta yatan hastalığın relapsı veya nüksetmesi veya altta yatan hastalığa bağlı ölüm, ii) relapsdışı mortalite veya iii) kronik GvHD için başka bir sistemik tedavinin eklenmesi veyabaşlatılması.
REACH3 birincil hedefine ulaşmıştır. Birincil analiz sırasında (veri kesim tarihi: 08 Mayıs 2020), 24. haftadaki genel yanıt oranı (ORR), mevcut en iyi tedavi koluna (%25,6) kıyaslaJAKAVI kolunda (%49,7) daha yüksektir. Tedavi kolları arasında istatistiksel olarak anlamlıbir fark söz konusu olmuştur (katmanlı Cochrane-Mantel- Haenszel testi p<0.0001, iki taraflı,OR: 2,99; %95 GA: 1,86, 4,80). Sonuçlar Tablo 10'da sunulmuştur.
JAKAVI ve mevcut en iyi tedavi kollarında 7. döngü 1. günde yanıt vermeyenlerin sırasıyla %2.4 ve %12.8'i hastalık progresyonuna sahiptir.
Tablo 10 REACH3'te 7. döngü 1. günde genel yanıt oranı
JAKAVI
Mevcut En Iyi Tedavi=164
BelgeDo Kodu: lZW56aklUQSNRSHY3M0FyYnUy
Belge Taküp Adresi:https://ww\\Cturlayc.gov.tr/saglik-titck-cbys
29
|
|
n (%) |
%95 GA |
n (%) |
%95 GA |
|
Genel yanıt |
82 (49,7) |
41,8; 57,6 |
42 (25,6) |
19,1; 33,0 |
|
OR (%95 GA) |
2,99 (1,86; 4,80) | |||
|
p-değeri (2-taraflı) |
p<0.0001 | |||
|
Tam yanıt |
11 (6,7) |
5 (3,0) | ||
|
Kısmi yanıt |
71 (43,0) |
37 (22,6) | ||
Kritik ikincil sonlanım noktası olan başarısızlık olmadan sağkalım(FFS), mevcut en iyi tedaviye karşı JAKAVI'de istatistiksel olarak anlamlı %63'lük risk azalması göstermiştir(HR: 0,370; %95 GA: 0,268, 0,510. p<0.0001). 6. ayda, başarısızlık olmadan sağkalım(FFS)olaylarının çoğu kronik GvHD için başka bir sistemik tedavinin eklenmesi veyabaşlatılması olmuştur (bu olayın olasılığı sırasıyla JAKAVI ve mevcut en iyi tedavi kollarıiçin %13,4'e karşı %48,5 bulunmuştur). Altta yatan hastalığın nüksetmesi ve relaps dışımortalite sonuçları JAKAVI ve mevcut en iyi tedavi kollarında sırasıyla %2,46'ya karşı%2,57 ve %9,19'a karşı %4,46 olmuştur. Yalnızca relaps dışı mortaliteye odaklanıldığındatedavi kolları arasında kümülatif insidans farkı gözlemlenmemiştir.
Pediyatrik popülasyon
Avrupa İlaç Ajansı (EMA), MF ve PV tedavisi için pediyatrik popülasyonun tüm alt gruplarında JAKAVI ile yapılan çalışmaların sonuçlarını sunma zorunluluğundanvazgeçmiştir. GvHD pediyatrik hastalarda (12 yaş ve üzeri), JAKAVI'nin güvenliliği veetkililiği, randomize faz 3 REACH2 ve REACH3 çalışmalarından elde edilen kanıtlarladesteklenmektedir (pediyatrik kullanım hakkında bilgi için bkz. Bölüm 4.2). REACH2'de, 28.günde, ruksolitinib kolunda akut GvHD'li 4/5 adölesan hastada (3'ünde tam yanıt (CR) ve1'inde kısmi yanıt(PR) söz konusu olmuştur) ve mevcut en iyi tedavi kolunda 3/4 adölesanhastada (3'ünde tam yanıt (CR) söz konusu olmuştur) yanıtlar gözlemlenmiştir. REACH3'te,ruksolitinib kolunda kronik GvHD'li 3/4 adölesan hastada (tümünde kısmi yanıt (PR) sözkonusu olmuştur) ve mevcut en iyi tedavi kolunda 2/8 adölesan hastada (her ikisinde de kısmiyanıt (PR) söz konusu olmuştur) 7. döngü 1. günde yanıtlar gözlemlenmiştir.
5.2 Farmakokinetik özelliklerGenel özellikler
Emilim
Biyofarmasötik Sınıflandırma Sistemine göre ruksolitinib yüksek permeabilite, yüksek çözünürlük ve hızlı dağılım karakteristiklerine sahip bir Sınıf 1 moleküldür. Klinikçalışmalarda, ruksolitinib, oral uygulamadan sonra hızla emilmekte, maksimal plazmakonsantrasyonuna (Cmaks) dozdan yaklaşık 1 saat sonra ulaşılmaktadır. İnsanda ruksolitinibinoral emilimi > %95'tir. Ortalama ruksolitinib Cmaks ve toplam maruziyet (EAA) değerleri, 5ila 200 mg tek doz aralığında orantılı olarak artmıştır. Yüksek oranda yağ içeren bir öğünlebirlikte uygulandığında ruksolitinibin farmakokinetiğinde klinik olarak anlamlı bir değişiklikolmamıştır. Yüksek oranda yağ içeren öğünle verildiğinde Cmaks orta düzeyde azalmagöstermiş (%24), ortalama EAA ise neredeyse hiç değişmemiştir (%4 artış).
Dağılım
MF ve PV hastalarında kararlı durumda görünür dağılım hacmi yaklaşık 75 litredir. Klinik olarak anlamlı ruksolitinib konsantrasyonlarında, in vitro ortamda plazma proteinlerinebağlanma yaklaşık %97 olup ağırlıklı olarak albümine bağlanır. Sıçanlarda tam vücutotoradyografi çalışması, ruksolitinibin kan-beyin bariyerini geçmediğini göstermiştir.
Belge Do Türkiye.gov.tr/laglik-titck-ebys
30
Biyotransformasyon
Ruksolitinib CYP2C9'un ilave katkısı ile başlıca CYP3A4 ile metabolize edilir (>%50). Ana bileşik, insanlarda baskın olup dolaşımdaki ilaçla ilişkili maddelerin yaklaşık %60'ınıoluşturmaktadır. Plazmada ana EAA'nın %25 ve %11'ini temsil eden iki ana ve aktifmetabolit mevcuttur. Bu metabolitler, ana bileşiğin JAK ilişkili farmakolojik aktivitesininyarısı ila beşte birine sahiptir. Tüm aktif metabolitlerin toplamı, ruksolitinibin toplamfarmakodinamik etkisine %18 katkı yapmaktadır. İn vitro çalışmalara göre, ruksolitinib,klinik olarak anlamlı konsantrasyonlarda CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19,CYP2D6 ya da CYP3A4'ü inhibe etmez ve CYP1A2, CYP2B6 ya da hepatik CYP3A4'ünkuvvetli indükleyicisi değildir. In vitro çalışmalara göre ruksolitinib P-gp ve meme kanserinedirenç proteini inhibe edebilir.
Eliminasyon:
Ruksolitinib, büyük oranda metabolizma yoluyla elimine edilir. Ruksolitinibin ortalama eliminasyon yarılanma ömrü yaklaşık 3 saattir. Sağlıklı erişkin gönüllülerde [14C] işaretliruksolitinibin tek oral dozundan sonra eliminasyon başlıca metabolizma yoluyla olmuş,radyoaktivitenin %74'ü idrarda ve %22'si feçeste tespit edilmiştir. Değişmemiş ilaç, atılantoplam radyoaktivitenin %1'inden azını oluşturmuştur.
Doğrusallık/doğrusal olmayan durum:
Tek ve çoklu doz çalışmalarında doz oransallığı gösterilmiştir.
Hastalardaki karakteristik özellikler
Vücut yüzey alanı, yaş, cinsiyet veya ırkın etkileri
Sağlıklı gönüllülerde yapılan çalışmalara göre, cinsiyet ve ırka göre ruksolitinib farmakokinetiğinde anlamlı herhangi bir farklılık gözlenmemiştir. MF hastalarında yapılan birpopülasyon farmakokinetik değerlendirmesinde, oral klirens ile hastanın yaşı veya ırkıarasında herhangi bir ilişki görülmemiştir. Öngörülen oral klirens, MF hastalarında hastalararası %39 değişkenlik ile kadınlarda 17,7 L/saat ve erkeklerde 22,1 L/saat olmuştur. PVhastalarında klirens 12,7 L/saat olup hastalar arası değişkenlik %42 bulunmuş ve PVhastalarında yapılan bir popülasyon farmakokinetik değerlendirmesine göre oral klirens ilecinsiyet, hastanın yaşı veya ırkı arasında herhangi bir ilişki görülmemiştir. Klirens, akutGvHD'li hastalarda 10.4 L/saat ve kronik GvHD'li hastalarda 7,8 L/saat olup, hastalararasında %49 değişkenlik göstermiştir. GvHD hastalarında bir popülasyon farmakokinetikdeğerlendirmesine dayalı olarak, oral klirens ile cinsiyet, hastanın yaşı veya ırkı arasındaherhangi bir ilişki görülmemiştir. Düşük vücut yüzey alanına sahip GvHD hastalarındamaruziyet artmıştır. Vücut yüzey alanı 1 m2, 1,25 m2 ve 1,5 m2 olan hastalarda öngörülenortalama maruziyet (EAA) tipik bir yetişkinden (1,79 m2) sırasıyla %31, %22 ve %12 dahayüksek olmuştur.
Pediyatrik _ popülasyon
MF ve PV'li 18 yaşından küçük pediyatrik hastalarda JAKAVI'nin farmakokinetiği belirlenmemiştir. Akut veya kronik GvHD'li adölesan hastalarda gözlenen farmakokinetikprofil, genel hasta popülasyonu ile benzer olmuştur (bkz. Bölüm 5.1, Pediyatrikpopülasyon). Ruksolitinib, 12 yaşın altındaki akut veya kronik GvHD'li pediyatrikhastalarda henüz değerlendirilmemiştir.
Böbrek yetmezliği:
Böbrek fonksiyonu hem Böbrek Hastalığında Diyet Modifikasyonu (MDRD) hem de üriner kreatinin kullanılarak tespit edilmiştir. Ruksolitinibin 25 mg'lık tek dozundan sonra
31
ruksolitinibe maruziyet çeşitli derecede böbrek yetmezliği olan hastalar ile normal böbrek fonksiyonuna sahip olanlar arasında benzer olmuştur. Ancak ruksolitinib metabolitlerininplazma EAA değerleri, böbrek yetmezliğinin şiddeti arttıkça yükselme eğilimi göstermiş, budurum şiddetli böbrek yetmezliği olan hastalarda belirginlik kazanmıştır. Artmış metabolitmaruziyetinin bir güvenlilik endişesi teşkil edip etmediği bilinmemektedir. Şiddetli böbrekyetmezliği ve son dönem böbrek yetmezliği hastaları için doz planında düzenlemeönerilmektedir (bkz. Bölüm 4.2). Dozların sadece diyaliz günlerinde uygulanması metabolitmaruziyetini azaltır fakat başta diyaliz arası günlerde olmak üzere farmakodinamik etkiyi deazaltmaktadır.
Karaciğer yetmezliği:
Çeşitli derecelerde karaciğer yetmezliği olan hastalarda 25 mg'lık tek bir ruksolitinib dozunun uygulanmasının ardından; ruksolitinibin ortalama EAA değeri,hafif, orta ve şiddetli karaciğeryetmezliği olan hastalarda normal karaciğer fonksiyonu olan hastalara kıyasla sırasıyla %87,%28 ve %65 oranlarda artmıştır.Child-Pugh skorlarına dayalı karaciğer yetmezliği derecesiile açık bir ilişki bulunmamaktadır. Sağlıklı kontrollere kıyasla karaciğer yetmezliği olanhastalarda terminal eliminasyon yarı ömrü uzamıştır (2,8 saate karşılık 4,1-5,0 saat).Karaciğer yetmezliği olan MF ve PV hastaları için dozun %50 oranında azaltılmasıönerilmektedir (bkz. Bölüm 4.2).
GvHD ile ilişkili olmayan karaciğer yetmezliği olan GvHD hastalarında, ruksolitinibin başlangıç dozu %50 oranında azaltılmalıdır.
5.3 Klinik öncesi güvenlilik verileri
Ruksolitinib; güvenlilik farmakolojisi, tekrarlı doz toksisitesi, genotoksisite, üreme toksisitesi çalışmalarında ve bir karsinojenisite çalışmasında değerlendirilmiştir. Tekrarlı dozçalışmalarında ruksolitinibin farmakolojik etkisi ile ilişkili hedef organlar kemik iliğini,periferik kanı ve lenfoid dokuları içermiştir. Köpeklerde, genellikle immün sisteminbaskılanmasına bağlı enfeksiyonlar gözlenmiştir. Köpek telemetri çalışmasında kanbasıncında, kalp hızında artışların eşlik ettiği tersine düşüşler ve sıçanlarda solunumçalışmasında dakika hacminde bir tersine düşüş görülmüştür. Köpek ve sıçan çalışmalarındakiadvers olmayan düzeydeki marjlar (bağlanmamış Cmaks bazında), günde iki kez 25 mgşeklindeki maksimum önerilen insan dozundan sırasıyla 15,7 ve 10,4 kat fazladır.Ruksolitinibin nörofarmakolojik etkilerine yönelik bir değerlendirmede herhangi bir etkigörülmemiştir.
Jüvenil sıçan çalışmalarında ruksolitinib uygulaması büyüme ve kemik ölçümleri üzerinde etkili olmuştur. >5 mg/kg/gün dozlarında tedaviye postnatal 7. günde (insanda yenidoğan ilekarşılaştırılabilir) başlandığında ve > 15 mg/kg/gün dozlarında tedaviye postnatal 14 veya 21.günlerde (insanda 1-3 yaş bebek ile karşılaştırılabilir) başladığında azalmış kemik büyümesigözlenmiştir. >30 mg/kg/gün dozlarında tedaviye postnatal 7. günde başlandığında sıçanlardakırıklar ve erken terminasyon gözlemlenmiştir. Bağlanmamış EAA değerine dayanılarak,postnatal 7. gün gibi erken bir dönemde tedavi edilen jüvenil sıçanlarda NOAEL (adversetkinin gözlenmediği düzey) düzeyinde maruziyet, günde iki kez 25 mg dozunda erişkinhastalarda görülenin 0.3 katı olurken günde iki kez 25 mg dozunda erişkin hastalardagörülenin sırasıyla 1.5 ve 13 katı maruziyetlerde azalmış kemik büyümesi ve kırıklargörülmüştür. Etkiler, tedavi postnatal dönemde daha erken başlandığında genellikle dahaşiddetli olmuştur. Kemik gelişimi dışında jüvenil sıçanlarda ruksolitinibin etkisi yetişkinsıçanlardakine benzer olmuştur. Jüvenil sıçanlar, yetişkin sıçanlara göre ruksolitinibtoksisitesine karşı daha hassastır.
Belge DoiotiunibvhâyıvanNçalışmfllayrındaH^Ptasyoftı|eüPaEapsAdrksayıps veye|etaLi|ğırtlAklearydaotiunibvhâyıvanNçalışmfllayrındaH^Ptasyoftı|eüPaEapsAdrksayıps veye|etaLi|ğırtlAklearyda
32
düşüşler ile ilişkilendirilmiştir. Sıçanlarda ve tavşanlarda teratojenik etki yönünde bir kanıt söz konusu olmamıştır. Diğer yandan, en yüksek klinik doz ile karşılaştırıldığında maruziyetmarjları düşük bulunmuştur ve dolayısıyla sonuçlar, insanlar açısından sınırlı öneme sahiptir.Fertilite üzerinde herhangi bir etki gözlenmemiştir. Prenatal ve postnatal gelişim çalışmasındagestasyon süresinde hafif uzama, implantasyon bölgelerinin sayısında azalma ve doğurulanyavru sayısında düşüş gözlenmiştir. Yavru hayvanlarda ilk beden ağırlıkları ortalamasındaazalma ve kısa süreli azalmış ortalama beden ağırlığı artışı gözlenmiştir. Emziren sıçanlardaruksolitinib ve/veya metabolitleri, maternal plazma konsantrasyonlarının 13 katı daha yüksekbir konsantrasyonla süte atılmıştır. Ruksolitinib, mutajenik ya da klastojenik etkigöstermemiştir. Ruksolitinib, Tg.rasH2 transjenik fare modelinde karsinojenik etkigöstermemiştir.
6. FARMASÖTİK ÖZELLİKLER6.1 Yardımcı maddelerin listesi
Mikrokristalin selüloz Magnezyum stearatKoloidal susuz silikaSodyum nişasta glikolat (Tip A)
Hidroksipropilselüloz (300-600 cps)
Povidon K30
Laktoz monohidrat (sığır kaynaklı)
6.2 Geçimsizlikler6.3 Raf ömrü
36 ay
6.4 Saklamaya yönelik özel tedbirler
25°C'nin altındaki oda sıcaklığında saklayınız.
6.5 Ambalajın niteliği ve içeriği
PVC/PCTFE blister ambalajlar
Ambalaj büyüklüğü: 14, 56 ve 168 tablet içeren blister ambalaj
Blister ambalajlar ısıyla kapatılabilir lakla kaplanmış alüminyum folyoyla kapatılmıştır
6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir.
7. RUHSAT SAHİBİ
Novartis Sağlık, Gıda ve Tarım Ürünleri San. ve Tic. A.Ş.
Kavacık / Beykoz / İstanbul
8. RUHSAT NUMARASI:
2019/238
BelgeşrniLKRüHSAT ITARİHİ(RUfiSA¥ ^EnİleMe TTARİHİİtps ://www. turkiye.gov.tr/saglik-titck-ebysRUfiSA¥ ^EnİleMe TTARİHİİtps ://www. turkiye.gov.tr/saglik-titck-ebys
33
10. KÜB'ÜN YENİLEME TARİHİ:
34
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.