Advate 250 Iu/5 Ml Iv Enjeksiyon İçin Liyofilize Toz İçeren Flakon Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİV Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIADVATE 250 IU/5 mL IV enjeksiyon için liyofilize toz içeren flakon Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Oktokog alfa [İnsan koagülasyon faktör VIII (rDNA)]: Her bir flakon 250 IU Oktokog alfa içermektedir. ADVATE rekonstitüsyon sonrası yaklaşık 50 IU/mL oktokog alfa içerir. Preparatın gücü [Uluslararası Ünite (IU)] Avrupa Farmakopesi kromojenik ölçüm yöntemiyle belirlenmiştir. ADVATE'in spesifik etkinliği yaklaşık 4.000-10.000 IU/mg protein'dir. Oktokog alfa [İnsan koagülasyon faktör VIII (rDNA)] 2332 amino asitli saf bir proteindir. Kobay over [Chinese hamster ovary (CHO)] hücrelerinde rekombinan DNA teknolojisiyleüretilmiştir. Hücre kültürleme, saflaştırmada ya da son formülasyon aşamasında herhangi bir(ekzojen) insan -veya hayvan- kaynaklı protein eklenmeksizin hazırlanmıştır. Yardımcı madde(ler):Bu tıbbi ürün her bir flakonda 0,45 mmol (ya da 10 mg) sodyum içerir. Yardımcı maddeler için 6.1'e bakınız. 3. FARMASOTİK FORMEnjeksiyon çözeltisi için toz ve çözücü. Beyazdan, kirli beyaza değişen renkte gevrek toz. Sulandırıldıktan sonra, çözelti berrak, renksiz, yabancı partiküller bulundurmaz ve pH'sı 6,77,3 arasındadır. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarHemofili A (konjenital faktör VIII eksikliği) hastalarında, kanamanın tedavisi ve profilaksisinde endikedir. ADVATE, farmakolojik olarak etkili miktarlarda von WillebrandFaktörü içermez ve bu nedenle von Willebrand hastalığında endike değildir. 1 4.2. Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:Uygulama hemofili tedavisinde deneyimli doktor gözetimi altında ve anafilaksi durumunda derhal temin edilmesi mümkün resüsitasyon olanaklarıyla başlatılmalıdır.Pozoloji ve yerine koyma tedavi süresi faktör VIII eksikliğinin ciddiyetine, kanama yerine, büyüklüğüne ve hastanın klinik durumuna bağlıdır. Faktör VIII (F VIII) dozu, faktör VIII ürünleri için geçerli WHO standardına uygun olarak Uluslararası Birimlerde (IU) ifade edilmektedir. Plazmada Faktör VIII dozu yüzde (normalinsan plazmasına göre) veya IU (plazmada faktör VIII için uluslararası standart) olarak ifadeedilmektedir. Bir IU faktör VIII aktivitesi bir ml normal insan plazmasındaki faktör VIII miktarına karşılık gelmektedir. Kanadıkça tedaviGerekli faktör VIII dozunun hesaplaması bir kilo vücut ağırlığı başına 1 IU faktör VIII'ün plazma faktör VIII aktivitesini 2 IU/dL arttırdığı yönündeki gözlemsel bulguya dayanmaktadır.Doz aşağıdaki formüle göre belirlenir: Gerekli doz (IU) = vücut ağırlığı (kg) x istenen faktör VIII artışı (%) x 0.5Aşağıdaki hemorajik olayların gerçekleşmesi durumunda, faktör VIII aktivitesi ilgili dönemde belirtilen plazma aktivitesi seviyesinin (normal seviyenin %'si veya IU/dl) altına düşmemelidir.Aşağıda sunulan Tablo 1, kanama episodları ve cerrahide dozaj belirlenmesinde kullanılabilir:
2
Uygulama dozu ve sıklığı her hastada alınan klinik yanıta göre düzenlenmelidir. Belirli koşullarda (örn. düşük titreli inhibitörü varlığı) formül kullanarak hesaplanandan daha yüksekdozlara ihtiyaç duyulabilir. Tedavi esnasında uygulanacak dozun ve tekrarlanan enjeksiyonların sıklığının belirlenmesi için uygun plazma faktör VIII seviyelerinin tayin edilmesi önerilmektedir. Özellikle majör cerrahimüdahalelerde, plazma faktör VIII aktivite tayini ile yerine koyma tedavisinin tam takibigerekmektedir. Farklı hastaların faktör VIII'e verdikleri yanıt yarılanma ömrü ve in vivorecovery seviyelerinin farklı olması yoluyla değişkenlik gösterebilir.ProfilaksiAğır Hemofili A hastalarında kanamalara karşı uzun süreli profilaksi için olağan dozlar 2-3 günlük aralıklarla vücut ağırlığı (kg) başına 20-40 IU faktör VIII'dir. Pediyatrik _ popülasyonADVATE'in etkililiği ve güvenliliği tüm pediatrik yaş gruplarında kanıtlanmıştır. Gereksinime göre, tedavideki dozlar açısından pediyatrik hastalardaki dozlar, erişkinlerdekinden farklıdeğildir. Profilaksi için 6 yaşından küçük hastalarda, haftada 3-4 defa, vücut ağırlığı (kg) başına20-50 IU faktör VIII önerilmektedir. Uygulama şekli:İntravenöz yoldan uygulanır. Sağlık hizmetleri profesyoneli olmayan bir kişi tarafından uygulanması için uygun eğitimin alınması gerekmektedir. Uygulama hızı dakikada 10 mL'yi geçmeyecek şekilde hastanın rahat edeceği bir hızda olmalıdır. Uygulamadan önce ürünün kullanıma hazır hale getirilmesine (çözülmesi) ilişkin talimatlar için bölüm 6.6.'ya bakınız. Özel popülasyonlara ilişkin ek bilgiler:3 Böbrek/karaciğer yetmezliği:.Pediyatrik popülasyon:ADVATE'in çocuklarda uygulaması (intravenöz yoldan)yetişkinlerdeki uygulamadan farklı değildir. FVIII ürünlerinin sık infüzyonuna olanak vermek için Santral Venöz Kateter (SVK) kullanımı gerekli olabilir. Geriyatrik popülasyon:4.3. KontrendikasyonlarADVATE, ürünün etkin maddesine, yardımcı maddelere ya da fare veya hamster proteinlerine karşı aşırı duyarlılığı olduğu bilinen hastalarda kontrendikedir. 4.4. Özel kullanım uyarıları ve önlemleriTakip edilebilirlikBiyolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. Hastayla tıbbi ürünlerin parti numaraları arasındaki bağlantıyı devam ettirmek açısından hastaya her ADVATE uygulamasında ürünün adının ve parti numarasının kayıt altına alınmasışiddetle tavsiye edilir. Aşırı duyarlılık reaksiyonlarıADVATE ile anafilaksi de dahil olmak üzere alerjik tipte aşırı duyarlılık reaksiyonları rapor edilmiştir. Ürün fare ve hamster proteini izleri içermektedir. Hastalara aşırı duyarlılığa ilişkinsemptomların oluşması halinde ürün kullanımını derhal kesmeleri ve doktora başvurmalarıönerilmelidir. Hastalar kurdeşen, yaygın ürtiker, göğüste sıkışma hissi, hırıltılı solunum,hipotansiyon ve anafilaksi dahil aşırı duyarlılık reaksiyonlarının erken belirtileri hakkındabilgilendirilmelidir. Anafilaktik şok durumunda şok tedavisine yönelik mevcut tıbbi standarttedaviler uygulanmalıdır. İnhibitörlerFaktör VIII'e karşı nötralize edici antikor (inhibitörler) oluşumu, hemofili A hastalarının tedavisinde bilinen bir komplikasyondur. Bu inhibitörler genellikle faktör VIII prokoagülanaktiviteye yönelik olan IgG immünoglobülinleridir ve modifiye tetkik kullanılarak her mlplazmada Bethesda Ünitesi (BU) olarak ölçülür. İnhibitör gelişme riski, faktör VIII'emaruziyetin yanı sıra hastalığın şiddeti ile ilişkilidir ve bu risk ilk 50 maruziyet gününde enyüksek seviyededir; ancak risk yaygın görülmemesine rağmen yaşam boyu devam eder. İnhibitör gelişiminin klinik önemi inhibitör titresine bağlı olacaktır; düşük titrenin teşkil ettiği yetersiz klinik yanıt riski, yüksek titreli inhibitörlere kıyasla daha az olacaktır. Genel olarak, koagülasyon faktörü VIII ürünleri ile tedavi edilen tüm hastalar, uygun klinik gözlem ve laboratuvar testleri ile inhibitörlerin gelişimi açısından dikkatle izlenmelidir. Eğerbeklenen faktör VIII aktivitesinin plazma düzeylerine ulaşılamazsa veya yeterli doz ilekanama kontrol altına alınamazsa faktör VIII inhibitörü varlığı açısından test yapılmalıdır. 4 İnhibitör düzeyleri yüksek olan hastalarda faktör VIII tedavisi etkili olmayabilir ve diğer tedavi seçenekleri dikkate alınmalıdır. Böyle hastaların tedavisi hemofili ve faktör VIIIinhibitörleri tedavisi konusunda deneyimli hekimler tarafından yönlendirilmelidir. Kateter ile ilişkili komplikasyonlarUygulama için Santral Venöz Kateter (SVK) kullanılırsa, lokal enfeksiyonlar, bakteriyemi ve kateter bölgesinde trombozis dahil olmak üzere SVK ile ilişkili komplikasyon riskinin gözönünde bulundurulması gerekmektedir. Yardımcı maddeler ile ilgili konularBu tıbbi ürün her flakonda 10 mg sodyum içerir. Bu miktar, bir yetişkin için Dünya Sağlık Örgütü (WHO) tarafından önerilen maksimum günlük 2 g sodyum alımının %0.5'ineeşdeğerdir. Pediyatrik _ popülasyon:Yukarıda sıralanan uyarı ve önlemler hem erişkinler hem de çocuklar için gereklidir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriADVATE ile diğer tıbbi ürünlerin etkileşimi olup olmadığını araştıran bir çalışma yapılmamıştır. 4.6. Gebelik ve LaktasyonGenel Tavsiye:Gebelik Kategorisi: C. Faktör VIII ile hayvan üreme çalışmaları gerçekleştirilmemiştir. Hemofili A kadınlarda nadir görüldüğünden, gebelik ve emzirme döneminde faktör VIII kullanımına ilişkin deneyim debulunmamaktadır. Bu nedenle faktör VIII preparatları gebelik ve laktasyon döneminde ancakçok gerekliyse kullanılmalıdır. Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon)Bilinen bir etkileşimi yoktur. Gebelik DönemiADVATE'in gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. bölüm5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. ADVATE gerekli olmadıkça (başka bir tedavi seçeneği yoksa) gebelik döneminde kullanılmamalıdır. 5 Laktasyon DönemiBu ilacın anne sütüne geçip geçmediği bilinmemektedir. Birçok ilacın anne sütüne geçtiği bilindiğinden emzirmekte olan annelerde ADVATE ancak çok gerekliyse kullanılmalıdır. Üreme yeteneği/FertiliteADVATE'in insanlarda üreme yeteneği / fertilite üzerindeki etkisini araştıran bir çalışma bulunmamaktadır. 4.7. Araç ve makine kullanımı üzerindeki etkilerAraç ve makine kullanımı üzerine herhangi bir etkisi bulunmamaktadır. 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tabloyaş gruplarına göre farmakokinetik parametrelerinin özeti__ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
ADVATE'in pediyatrik popülasyondaki emniyet ve hemostatik etkinliği erişkin hastalardakiyle aynıdır. Düzeltilmiş recovery ve terminal yarılanma ömrü (f/2) küçük çocuklarda (6 yaşındanküçük), erişkinlere kıyasla yaklaşık %20 daha düşüktür, bu durum küçük yaştaki hastalardavücut ağırlığına göre plazma hacminin daha yüksek olmasından kaynaklanıyor olabilir.
Daha önce tedavi almamış hastalarda ADVATE farmakokinetik verileri günümüzde mevcut değildir.
Emilim:
İntravenöz yoldan enjeksiyon sonrası değişik yaş gruplarında (infantlar, küçük ve büyük çocuklar, adölesanlar ve erişkinler) hastalar için plazmada elde edilen
in vivoDağılım:
11
ADVATE hemofili hastalarına uygulandığında, hastanın dolaşımındaki endojen von Willebrand Faktörüne bağlanır. Faktör VIII / von Willebrand Faktör kompleksi esas olarakintravasküler olarak dağılıma uğrar. İntravenöz yoldan enjeksiyon sonrası değişik yaşgruplarında (infantlar, küçük ve büyük çocuklar, adölesanlar ve erişkinler) hastalar için dağılımhacimlerinin bir özeti yukarıdaki tabloda verilmiştir.
Biyotransformasyon:
Uygulanabilir değil.
Eliminasyon:
Günümüzde Faktör VIII klerensinin, aralarında düşük dansiteli lipoprotein reseptör-ilişkili protein (LPR) ve heparin sülfat proteoglikanlarının (HSPGs) bulunduğu vasküler reseptörlerce,tam olarak bilinmeyen bazı mekanizmalarca yönetildiğine inanılmaktadır.
Doğrusallık / doğrusal olmayan durum:
Bilinmemektedir.
Hastalardaki karakteristik özellikler:
Pediyatrik hastalar:
Bu popülasyonda ADVATE'in güvenliliği ve hemostatik etkililiği, erişkinlerdekine benzer. Çocuklarda düzeltilmişin vivoelde edilen miktarları ve yarıömürleri, erişkinlerdekinden %20 kadar düşük bulunmuştur.
Farmakokinetik/farmakodinamik ilişki(ler):
Bilinmemektedir.
5.3. Klinik öncesi güvenlilik verileri
Güvenlilik farmakolojisi, akut toksikoloji, tekrarlayan doz toksisitesi, lokal toksisite ve genotoksisite konusunda yapılan çalışmalara dayanarak, klinik dışı veriler insana yönelik özelbir risk olmadığını göstermektedir.
6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesi
Kuru toz
- Mannitol
- Sodyum klorür
- Histidin
- Trehaloz
- Kalsiyum klorür
- Trometamol
- Polisorbat 80
- Glutatyon (indirgenmiş)
Çözücü
12
- Steril enjeksiyonluk su
6.2. Geçimsizlikler
ADVATE ile gerçekleştirilen bir geçimsizlik çalışması bulunmamaktadır. Bu nedenle diğer ilaçlarla ve çözücülerle karıştırılmamalıdır.
6.3. Raf ömrü
Açılmamış kuru toz flakon
24 ay.Sulandırıldıktan sonra
Kimyasal ve fiziksel geçerli stabilitesi 25°C'de 3 saat olduğunu göstermektedir. Mikrobiyolojik açıdan, ürün sulandırıldıktan hemen sonra kullanılmalıdır.
6.4. Saklamaya yönelik özel tedbirlerBuzdolabında saklayınız (2° ila 8 °C arasında). Dondurmayınız.
Raf ömrü süresince, 6 ayı aşmayacak tek bir dönemde ürün oda sıcaklığında (25°C'ye kadar) saklanabilir. Oda sıcaklığına çıkarıldığı tarih, ürün ambalajı üzerine kaydedilmelidir.
Oda sıcaklığında bekletilen ürün yeniden buzdolabına konulmamalıdır.
Kapalı blisteri ışıktan korumak için orijinal ambalajı içinde saklayınız.
Tıbbi ürün sulandırıldıktan sonraki saklama koşulları için Bölüm 6.3'e bakınız.
6.5. Ambalajın niteliği ve içeriği
Toz flakon klorobütil, 5 mL çözücü içeren flakon klorobütil veya bromobütil lastik tıpalar ile kapatılmış Tip- I camdır. Ürün aşağıdaki şekilde sağlanmaktadır:
BAXJECT III sistemi: Her bir ambalaj, kapalı bir blisterde bir kullanıma hazır BAXJECT III sistemi (sulandırma için sistem ile önceden birleştirilmiş toz flakonu ve 5 mL çözücü içerenflakon) uygulama seti kapsamında kelebek iğneli infüzyon seti ve tek kullanımlık enjektöriçerir.
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
ADVATE liyofilize ürünün sulandırılması sonrası intravenöz olarak uygulanır. Sulandırma sonrasında çözelti berrak ve renksiz olmalıdır, yabancı partikül içermemelidir.
- Uygulama için luer enjektörü gereklidir.
- Sulandırdıktan sonra üç saat içinde kullanınız.
- Sulandırma sonrasında preparatı buzdolabına koymayınız.
13
- Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliğive Ambalaj Atıkların Kontrolü Yönetmeliği 'ne uygun olarak imha edilmelidir.
BAXJECT III Sistemiyle Sulandırma:
- Kapak blister üzerinde tamamen kapatılmamışsa kullanmayınız.
1. Ürün hala buzdolabında saklanıyorsa, kapalı blisteri (sulandırmak için sistemle öncedenmonte edilmiş toz ve çözücü flakonları içerir) buzdolabından alınız ve oda sıcaklığına(15°C ile 25°C arasında) ulaşmasını sağlayınız.
2. Ellerinizi sabun ve ılık su kullanarak iyice yıkayınız.
3. Etiketi soyarak ADVATE ambalajını açınız. BAXJECT III sistemini blisterden çıkarınız.
4. ADVATE'i seyreltici flakonu üstte olacak şekilde düz bir yüzeye yerleştiriniz (Şekil 1).Seyreltici flakonunun mavi bir şeridi vardır. Mavi kapağı daha sonraki bir adımda talimatverilinceye kadar çıkarmayınız.
5. Bir eliniz BAXJECT III sistemindeki ADVATE'i tutarken, sistem tamamen çökene veseyreltici ADVATE flakonunun içine akana kadar seyreltici flakonun üzerine diğerelinizle sıkıca bastırınız (Şekil 2). Aktarım tamamlanana kadar sistemi eğmeyiniz.
6. Seyreltici aktarımının tamamlandığını doğrulayınız. Tüm materyal çözünene kadaryavaşça döndürünüz. ADVATE tozunun tamamen çözündüğünden emin olunuz, aksihalde sulandırılan çözeltilerin tümü cihaz filtresinden geçmeyecektir. Ürün hızlı birşekilde çözünür (genellikle 1 dakikadan az). Sulandırıldıktan sonra çözelti berrak, renksizve partikül içermiyor olmalıdır.
Şekil 1Şekil 2Şekil 3 |
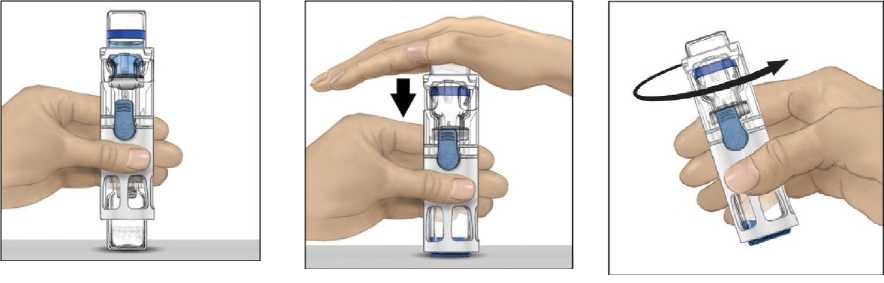 |
Uygulama: |
Aseptik Teknik Kullanınız.
Parenteral tıbbi ürünler, çözelti ve ambalajının uygun olduğu durumlarda, uygulama öncesinde partikül madde açısından incelenmelidir. Yalnız berrak ve renksiz olan çözeltilerkullanılmalıdır.
1. Mavi kapağı /BAXJECT III cihazından ayırınız. ENJEKTÖRE HAVA ÇEKMEYİNİZ. Enjektörü /BAXJECT III cihazına bağlayınız.
14
Sistemi ters çeviriniz (sulandırılmış çözeltinin bulunduğu flakon üstte olmalıdır). Pistonu yavaşça geri çekerek enjektöre sulandırılmış çözeltiyi çekiniz.
3. Enjektörü ayırınız.
4. Enjektöre kelebek iğne takınız. İntravenöz olarak enjekte ediniz. Çözelti hastanın konforseviyesine göre, dakikada 10 mL'lik miktarları geçmeyecek şekilde, yavaşçauygulanmalıdır. ADVATE uygulaması öncesinde ve sonrasında nabız ölçülmelidir.Önemli bir artış meydana gelirse uygulama hızının düşürülmesi veya enjeksiyona geçiciolarak ara verilmesi genellikle semptomların hızlıca giderilmesini sağlamaktadır (bkz.Bölüm 4.4 ve 4.8).
7. RUHSAT SAHİBİ
Takeda İlaç Sağlık Sanayi Ticaret Limited Şirketi Levent-Şişli/İSTANBUL
8. RUHSAT NUMARASI
2016/70
9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİ
İlk ruhsatlandırma tarihi: 01.02.2016 Son ruhsat yenileme tarihi:
10. KÜB'ÜN YENİLENME TARİHİ
15
İlaç Bilgileri
Advate 250 Iu/5 Ml Iv Enjeksiyon İçin Liyofilize Toz İçeren Flakon
Etken Maddesi: Oktokog Alfa
Kullanma talimatı ve kısa ürün bilgileri
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.