Alunbrig 30 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA URUN BILGISI¡ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'a bildirmeleribeklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERI TIBBİ ÜRÜNÜN ADIALUNBRİG 30 mg film kaplı tablet Sitotoksik. 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Brigatinib 30 mg Yardımcı maddeler:Laktoz monohidrat (inek sütü) 56,06 mg Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tabletBir yüzünde U3 baskılı diğer yüzü düz, yaklaşık 7 mm çapında, beyaz-beyazımsı, yuvarlak film kaplı tabletler. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarÖnceden ALK-Hedefli Tedavi Almamış İleri Evre ALK-Pozitif Küçük Hücreli Dışı Akciğer Kanseri (KHDAK)ALUNBRİG (brigatinib)'in, anaplastik lenfoma kinaz (ALK)-pozitifliği (standardize FISH veya RT-PCR veya yeni nesil dizileme yöntemleri ile tespit edilen rearanjman/füzyon varlığı; veyaimmünhistokimya ALK pozitifliği) saptanan, ilerlemiş küçük hücreli dışı akciğer kanseri (KHDAK)olan hastaların tedavisinde progresyona kadar kullanımı endikedir. Önceden Krizotinib Tedavisi Almış ALK-Pozitif İleri Evre veya Metastatik Küçük Hücreli Dışı Akciğer Kanseri (KHDAK)ALUNBRİG (brigatinib)'in, krizotinib ile önceden tedavi edilmiş anaplastik lenfoma kinaz (ALK)-pozitifliği (standardize FISH veya RT-PCR veya yeni nesil dizileme yöntemleri ile tespit edilen rearanjman/füzyon varlığı; veya immünhistokimya ALK pozitifliği) saptanan, ileri evre veyametastatik küçük hücreli dışı akciğer kanseri (KHDAK) olan hastaların tedavisinde progresyonakadar kullanımı endikedir. 4.2. Pozoloji ve uygulama şekliALUNBRİG ile tedavi antikanser tıbbi ürünlerinin kullanımı konusunda deneyimli bir uzman hekim tarafından başlatılmalı ve takip edilmelidir. ALUNBRİG tedavisine başlamadan önce ALK-pozitif KHDAK durumu belirlenmelidir. ALK-pozitif KHDAK hastalarının seçimi için, valide edilmiş bir ALK testi gereklidir (bkz. bölüm 5.1.). ALK-pozitif KHDAK değerlendirmesi, kullanılmakta olan spesifik teknoloji konusunda yetkinliğikanıtlanmış laboratuvarlar tarafından gerçekleştirilmelidir. Pozoloji/uygulama sıklığı ve süresi:ALUNBRİG'in tavsiye edilen başlangıç dozu ilk 7 gün için günde bir defa 90 mg, daha sonra günde bir defa 180 mg'dır. ALUNBRİG advers reaksiyonlar dışındaki nedenlerle 14 gün veya daha uzun süre kesilirse; tedaviye, daha önce tolere edilen doza yükseltilmeden önce 7 gün boyunca günde bir kez 90 mg'dadevam edilmelidir. Bir dozun atlanması veya doz alımı sonrasında kusma meydana gelmesi durumunda, ilave bir doz uygulanmamalı ve bir sonraki doz planlanan zamanda alınmalıdır. Tedavi, klinik yarar sağlandığı sürece devam etmelidir. Doz ayarlamasıBireysel güvenlilik ve tolerabiliteye bağlı olarak dozun kesilmesi ve/veya azaltılması gerekebilir. ALUNBRİG doz azaltma seviyeleri Tablo 1'de özetlenmektedir.
Hastanın günde bir defa 60 mg'lık dozu tolere edememesi durumunda, ALUNBRİG kalıcı olarak kesilmelidir. Advers reaksiyonların yönetimi için ALUNBRİG doz modifikasyonlarına ilişkin tavsiyeler Tablo 2'de özetlenmektedir.
durdurulmalıdır. ALUNBRİG, Tablo 1'de açıklandığı üzere, bir sonraki düşük dozda devam ettirilmelidir. ILD/pnömonitin yeniden meydana gelmesi durumunda ALUNBRİG kalıcı olarak kesilmelidir.
3. veya 4. derece
ALUNBRİG kalıcı olarak kesilmelidir.
Hipertansiyon 3. derece hipertansiyon (Sistolikkanbasıncı (SKB) >160 mmHg veya Diastolikkanbasıncı (DKB) >100 mmHg, tıbbi müdahalegerektirir, birden fazla antihipertansif tıbbi ürün veyadahaönce kullanılandan daha yoğun tedavi gerektirir)_
4. derece hipertansiyon (yaşamı tehdit edicisonuçları olan, acilmüdahale gerektiren) Hipertansiyon derece <1'e (SKB <140 mmHg ve DKB <90 mmHg) kadar iyileşme gösterene kadar ALUNBBRİG tedavisidurdurulmalı, daha sonra aynı dozda devam ettirilmelidir. 3.derece hipertansiyonun tekrarlaması durumunda; ALUNBRİG hipertansiyon derece < 1'e kadar iyileşme gösterene kadardurdurulmalı, daha sonra Tablo 1'e göre bir sonraki daha düşükdoz seviyesinde devam edilmeli veya kalıcı olarak kesilmelidir.
Hipertansiyon derece < 1'e (SKB <140 mmHg ve DKB <90 mmHg) kadar iyileşme gösterene kadar ALUNBRİG tedavisidurdurulmalı, daha sonra Tablo 1'e göre bir sonraki daha düşükdoz seviyesinde devam edilmeli veya kalıcı olarak kesilmelidir.4.derece hipertansiyonun yeniden meydana gelmesi durumunda;ALUNBRİG kalıcı olarak kesilmelidir.
Bradikardi (Dakikada 60'dandüşük kalp atımhızı)
Semptomatik bradikardi
Yaşamı tehdit edici sonuçları olan bradikardi,acil müdahale gerektiren
Asemptomatik bradikardi sağlanana kadar veya dakikada 60 kalp atım hızı veya daha yüksek dinlenme kalp hızı sağlanana kadarALUNBRİG durdurulmalıdır. Bradikardiye neden olduğu bilinen eş zamanlı kullanılan bir tıbbi ürünün tanımlanması ve kesilmesi veya dozunun ayarlanmasıdurumunda; ALUNBRİG asemptomatik bradikardi sağlandığındaveya dakikada 60 kalp atım hızı veya daha yüksek dinlenme kalphızı sağlandığında, aynı dozda devam ettirilmelidir. Eğer bradikardiye neden olduğu bilinen eş zamanlı kullanılan herhangi bir ilaç belirlenmemiş veya katkı sağlayan eş zamanlıkullanılan ilaçların kullanımına son verilmemiş veya dozayarlaması yapılmamışsa, ALUNBRİG asemptomatik bradikardiveya dakikada 60 kalp atım hızı veya daha yüksek bir dinlenmekalp hızı sağlandığında, Tablo 1'e göre bir sonraki daha düşük 2 1
CPK yüksekliği
Derece >2 kas ağrısı veya zayıflığı ile 3. veya 4.derece CPK yüksekliği(>5 x normalin üst sınırı(NÜS)) ALUNBRİG derece < 1 (< 2.5 x NÜS) CPK yüksekliğine veya başlangıç seviyesine iyileşme olana kadar durdurulmalı, dahasonra aynı dozda devam ettirilmelidir. 3. veya 4. derece CPK yüksekliğinin Derece > 2 kas ağrısı veya zayıflığı ile tekrarlaması durumunda; ALUNBRİG derece < 1 (<2,5 X NÜS) CPK yüksekliğine veya başlangıç seviyesine iyileşmeolana kadar durdurulmalı, daha sonra Tablo 1'e göre bir sonraki daha düşük doz seviyesinde devam ettirilmelidir._Lipaz veya amilaz yüksekliği 3. derece lipaz veya amilaz yüksekliği (>2 xNÜS) ALUNBRİG derece < 1 (<1,5 x NÜS) veya başlangıç seviyesineiyileşme olana kadar durdurulmalı, daha sonra aynı dozda devamettirilmelidir. 3. derece lipaz ve amilaz yüksekliğinin tekrarlaması durumunda;
Uygulama şekli:ALUNBRİG oral kullanım içindir. Tabletler bütün olarak suyla yutulmalıdır. ALUNBRİG aç ya da tok olarak alınabilir. Greyfurt veya greyfurt suyu brigatinibin plazma konsantrasyonlarını artıracağı için tüketilmesinden kaçınılmalıdır (bkz. bölüm 4.5.). Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Hafif veya orta dereceli böbrek yetmezliği olan hastalar için ALUNBRİG doz ayarlaması gerekli değildir (hesaplanan glomerüler filtrasyon hızı (eGFR) >30 mL/dk). Şiddetli böbrek yetmezliği(eGFR) <30 mL/dk) olan hastalar için; ilk 7 gün boyunca günde bir defa 60 mg'lık azaltılmış başlangıç dozu, daha sonra günde bir defa 90 mg tavsiye edilir (bkz. bölüm 5.2.). Şiddetli böbrek yetmezliği olan hastalar özellikle ilk haftada interstisyel akciğer hastalığı (ILD)/pnömonit belirtisiteşkil edebilecek yeni veya kötüleşen solunum semptomları (örneğin: dispne, öksürük vs.) açısındanyakından takip edilmelidir (bkz. bölüm 4.4.). Karaciğer yetmezliği:Hafif dereceli karaciğer yetmezliği (Child-Pugh sınıf A) veya orta derecede karaciğer yetmezliği (Child-Pugh sınıf B) olan hastalar için ALUNBRİG doz ayarlaması gerekli değildir. Şiddetlikaraciğer yetmezliği (Child-Pugh sınıf C) olan hastalar için; ilk 7 gün boyunca günde bir defa 60mg'lık azaltılmış başlangıç dozu, daha sonra günde bir defa 120 mg tavsiye edilir (bkz. bölüm 5.2.) Pediyatrik popülasyon:18 yaşın altındaki hastalarda ALUNBRİG'in güvenliliği ve etkililiği henüz belirlenmemiştir. Veri mevcut değildir. Geriyatrik popülasyon:65 yaş ve üzeri hastalarda ALUNBRİG'in güvenliliği ve etkililiğine ilişkin sınırlı veriler yaşlı hastalarda doz ayarlamasının gerekli olmadığını öne sürmektedir (bkz. bölüm 4.8.). 85 yaş üzerihastalara ilişkin veri mevcut değildir. 4.3. KontrendikasyonlarEtkin maddeye veya bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılığı olan hastalarda kullanılmamalıdır. 4.4. Özel kullanım uyarıları ve önlemleriPulmoner advers reaksiyonlarALUNBRİG ile tedavi olan hastalarda ILD/pnömonit ile tutarlı özelliklere sahip olanlar dahil olmak üzere; ciddi, yaşamı tehdit edici ve ölümcül pulmoner advers reaksiyonlar meydana gelebilir (bkz.bölüm 4.8.). Birçok pulmoner advers reaksiyon tedavinin ilk 7 günü içerisinde gözlemlenmiştir. Derece 1-2 pulmoner advers reaksiyonlar, tedavinin kesilmesi veya doz ayarlaması ile giderilmiştir. Yaş artışıve krizotinibin son dozu ve ALUNBRİG'in ilk dozu arasındaki daha kısa zaman aralığı (7 gündenaz) bağımsız olarak bu pulmoner advers reaksiyon oranının artması ile ilişkilendirilmiştir.ALUNBRİG tedavisine başlanırken bu faktörler göz önünde bulundurulmalıdır. ILD veya ilaç-indüklü pnömonit öyküsü olan hastalar pivotal çalışmalardan çıkarılmıştır. Bazı hastalar ALUNBRİG tedavisinden sonra pnömonit geçirmiştir. Hastalar; özellikle tedavinin ilk haftasında, yeni veya kötüleşen solunum semptomları (örn. dispne, öksürük, v.b.) bakımından takip edilmelidir. Kötüleşen solunum semptomlarına sahip herhangi birhastadaki pnömonit bulgusu derhal incelenmelidir. Pnömonitten şüphelenilmesi durumunda;ALUNBRİG tedavisi durdurulmalı ve hasta semptomların diğer nedenleri (örn. pulmonerembolizm, tümör progresyonu ve enfeksiyöz pnömonit) bakımından değerlendirilmeli ve budoğrultuda doz ayarlaması yapılmalıdır (bkz. bölüm 4.2.). HipertansiyonALUNBRİG ile tedavi edilen hastalarda hipertansiyon meydana gelmiştir (bkz. bölüm 4.8.). ALUNBRİG tedavisi sırasında kan basıncı düzenli olarak takip edilmelidir. Hipertansiyon, kan basmcmm kontrol edilmesine yönelik standart kılavuzlara göre tedavi edilmelidir. Kalp hızı; bradikardiye neden olduğu bilinen bir tıbbi ürün ile eş zamanlı kullanımdan kaçınılamadığıdurumda, hastalarda daha sıklıkla takip edilmelidir. Şiddetli hipertansiyon (> derece 3) için;ALUNBRİG, hipertansiyon derece 1 veya başlangıç seviyesine iyileşene kadar durdurulmalıdır. Budoğrultuda doz ayarlaması yapılmalıdır (bkz. bölüm 4.2.) BradikardiALUNBRİG ile tedavi edilen hastalarda bradikardi meydana gelmiştir (bkz. bölüm 4.8.). A^UNBRİG'in bradikardiye neden olduğu bilinen diğer ajanlar ile kombinasyon halindeuygulanması durumunda dikkatli olunmalıdır. Kalp hızı ve kan basıncı düzenli olarak takipedilmelidir. Semptomatik bradikardinin meydana gelmesi durumunda; ALUNBRİG tedavisi durdurulmalı ve bradikardiye neden olduğu bilinen eş zamanlı kullanılan tedaviler değerlendirilmelidir. İyileşmeolduğunda; bu doğrultuda doz ayarlaması yapılmalıdır (bkz. bölüm 4.2.). Yaşamı tehdit edicibradikardi durumunda; katkı sağlayan birlikte kullanılan herhangi bir ilacın tanımlanmaması veyatekrarlama durumunda, ALUNBRİG tedavisi kesilmelidir (bkz. bölüm 4.2.). Görme bozukluğuALUNBRİG ile tedavi edilen hastalarda görme bozukluğu advers reaksiyonları meydana gelmiştir (bkz. bölüm 4.8.). Hastalara, görme semptomlarını raporlaması önerilmelidir. Yeni veya kötüleşenciddi görme semptomların olması durumunda; göz muayenesi ve doz azaltılmasıdeğerlendirilmelidir (bkz. bölüm 4.2.). Kreatin fosfokinaz (CPK) artışıALUNBRİG ile tedavi edilen hastalarda CPK artışı meydana gelmiştir (bkz. bölüm 4.8). Hastalara; açıklanamayan herhangi bir kas ağrısı, hassaslık veya zayıflık durumunu raporlamaları tavsiyeedilmelidir. ALUNBRİG tedavisi sırasında CPK seviyeleri düzenli olarak takip edilmelidir. CPKartışının şiddetine bağlı olarak; ALUNBRİG tedavisi durdurulmalı ve bu doğrultuda doz ayarlamasıyapılmalıdır (bkz. bölüm 4.2.). Pankreatik enzimlerde artışALUNBRİG ile tedavi edilen hastalarda amilaz ve lipaz artışı meydana gelmiştir (bkz. bölüm 4.8.). ALUNBRİG tedavisi sırasında lipaz ve amilaz düzenli olarak takip edilmelidir. Laboratuvaranormalliklerinin şiddetine bağlı olarak; ALUNBRİG durdurulmalı ve bu doğrultuda dozayarlaması yapılmalıdır (bkz. bölüm 4.2.). Hepatotoksisite:ALUNBRİG tedavisi alan hastalarda hepatik enzimler (aspartat aminotransferaz, alanin aminotransferaz) ve bilirubin seviyelerinde artış meydana gelmiştir (bkz. bölüm 4.8.). ALUNBRİGtedavisine başlanmadan önce ve tedavinin ilk 3 ayı boyunca her 2 haftada bir karaciğer fonksiyonu(AST, ALT ve total bilirubin dahil) değerlendirilmelidir. Daha sonra periyodik izlemeye devamedilmelidir. Laboratuvar anormalliklerinin ciddiyet durumuna göre tedaviye ara verilmeli ve dozmodifikasyonu yapılmalıdır (bkz. bölüm 4.2.) HiperglisemiALUNBRİG ile tedavi edilen hastalarda serum glukoz seviyelerinde artış meydana gelmiştir. ALUNBRİG tedavisine başlamadan önce açlık serum glukoz seviyesi değerlendirilmeli ve akabindeperiyodik olarak takip edilmelidir. Gerekirse, antihiperglisemik tedaviler başlatılmalı veya optimizeedilmelidir. Optimum tıbbi yönetim ile yeterli hiperglisemik kontrol sağlanamaması durumunda;ALUNBRİG yeterli hiperglisemik kontrol sağlanana kadar durdurulmalıdır. İyileşme olduğunda,Tablo 1'de açıklandığı şekilde dozun azaltılması değerlendirilebilir veya ALUNBRİG kalıcı olarakkesilebilir. İlaç-ilaç etkileşimleriALUNBRİG'in güçlü CYP3A inhibitörleri ile eş zamanlı kullanımından kaçınılmalıdır. Güçlü CYP3A inhibitörlerinin birlikte kullanımmın kaçınılmaz olduğu durumunda; ALUNBRİG dozu 180mg'dan 90 mg'a veya 90 mg'dan 60 mg'a düşürülmelidir. Güçlü CYP3A inhibitörü kesildiktensonra; ALUNBRİG tedavisine, güçlü CYP3A inhibitörü başlatılmadan önce tolere edilen dozdadevam edilmelidir. ALUNBRİG'in güçlü ve orta dereceli CYP3A indükleyicileri ile eşzamanlı kullanımından kaçınılmalıdır (bkz. bölüm 4.5.). Orta dereceli CYP3A indükleyicilerinin eşzamanlı kullanımındankaçınılamazsa, ALUNBRİG dozu, tolere edilen mevcut ALUNBRİG dozu ile 7 günlük tedavidensonra 30 mg'lık artışlarla, orta dereceli CYP3A indükleyicisinin başlatılmasından önce tolere edilenALUNBRİG dozunun maksimum iki katına kadar artırılabilir. Orta derecede bir CYP3Aindükleyicisinin kesilmesinden sonra, ALUNBRİG'e orta dereceli CYP3A indükleyicisininbaşlatılmasından önce tolere edilen dozda devam edilmelidir. Fotosensitivite ve fotodermatozALUNBRİG ile tedavi edilen hastalarda güneş ışığına karşı ışığa duyarlılık oluşmuştur (bkz. bölüm 4.8). Hastalara ALUNBRİG alırken ve tedavinin kesilmesinden sonra en az 5 gün boyunca güneşeuzun süre maruz kalmaktan kaçınmaları tavsiye edilmelidir. Dışarıdayken, hastalara şapka vekoruyucu giysi giymeleri ve olası güneş yanığına karşı korunmaya yardımcı olmak için genişspektrumlu Ultraviyole A (UVA)/ Ultraviyole B (UVB) güneş kremi ve dudak kremi (SPF >30)kullanmaları tavsiye edilmelidir. Şiddetli fotosensitivite reaksiyonları (> Derece 3) içinALUNBRİG, başlangıç düzeyine geri dönene kadar durdurulmalıdır. Doz buna göredeğiştirilmelidir (bkz. bölüm 4.2). FertiliteÇocuk doğurma potansiyeli olan kadınlara ALUNBRİG ile tedavi sırasında ve son dozu takiben en az 4 ay boyunca etkili, hormonal olmayan kontrasepsiyon kullanımı tavsiye edilmelidir. Çocukdoğurma potansiyeline sahip kadın partneri olan erkeklere, tedavi sırasında ve son ALUNBRİGdozundan sonra en az 3 ay süreyle etkili kontrasepsiyon kullanımı tavsiye edilmelidir (bkz. bölüm4.6.). LaktozALUNBRİG laktoz monohidrat içerir. Nadir kalıtımsal galaktoz intoleransı, total laktaz yetmezliği ya da glukoz-galaktoz malabsorpsiyon problemi olan hastaların bu ilacı kullanmamaları gerekir. SodyumBu tıbbi ürün her sodyum içermez. dozunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriBrigatinib plazma konsantrasyonlarını arttırabilen ajanlarCYP3A inhibitörleriİn vitroçalışmalar brigatinibin CYP3A4/5'in bir substratı olduğunu göstermiştir. Sağlıklı gönüllülerde; güçlü bir CYP3A inhibitörü olan itrakonazolün günde iki defa 200 mg'lık çoklu dozuile 90 mg'lık tekli brigatinib dozunun eş zamanlı uygulanması, tek başına uygulanan 90 mg'lıkbrigatinib dozuna göre, brigatinib Cmaks'ını % 21, EAAo -sonsuz u % 101 (2 kat) ve EAAo-i2o'yi % 82(<2 kat) oranında artırmıştır. Bunlarla sınırlı olmamak üzere belirli antiviral ajanlar (örn. indinavir,nelfinavir, ritonavir, sakinavir), makrolid antibiyotikler (örn. klaritromisin, telitromisin,troleandomisin), antifungal ajanlar (örn. ketokonazol, vorikonazol) ve nefazodon dahil olmak üzeregüçlü CYP3A inhibitörlerinin ALUNBRİG ile eş zamanlı kullanımından kaçınılmalıdır. GüçlüCYP3A inhibitörlerinin birlikte kullanımının kaçınılmaz olduğu durumunda; ALUNBRİG dozuyaklaşık % 50 oranında (yani 180 mg'dan 90 mg'a veya 90 mg'dan 60 mg'a) düşürülmelidir. Güçlübir CYP3A inhibitörü kesildikten sonra; ALUNBRİG tedavisine, güçlü CYP3A inhibitörübaşlatılmadan önce tolere edilen dozda devam edilmelidir.Orta dereceli CYP3A inhibitörleri (örn. diltiazem ve verapamil), fizyolojik-bazlı bir farmakokinetik modelden elde edilen simülasyonlara dayanarak, brigatinib EAA'sini yaklaşık % 40 oranındaartırabilir. Orta dereceli CYP3A inhibitörleri ile kombinasyonda, ALUNBRİG için doz ayarlamasıgerekli değildir. ALUNBRİG'in orta dereceli CYP3A inhibitörleri ile eş zamanlı uygulanmasıdurumunda, hastalar yakından takip edilmelidir. Greyfurt veya greyfurt suyu da brigatinibin plazma konsantrasyonlarını arttırabilir ve bu nedenle kullanımından kaçınılmalıdır (bkz. bölüm 4.2.). CYP2C8 İnhibitörleriİn vitroçalışmalar brigatinibin CYP2C8'in bir substratı olduğunu göstermiştir. Sağlıklı gönüllülerde; güçlü bir CYP2C8 inhibitörü olan gemfibrozilin günde iki defa 600 mg'lık çokludozu ile 90 mg'lık tekli brigatinib dozunun eş zamanlı uygulanması, tek başına uygulanan 90mg'lık brigatinib dozuna göre, brigatinib Cmaks'ını % 41, EAAo -sonsuz u % 12 ve EAAo-i2o'yi % 15oranında azaltmıştır. Gemfibrozilin brigatinibin farmakokinetiği üzerindeki etkisi klinik olarakanlamlı değildir ve azalmış brigatinib maruziyetinin altta yatan mekanizması bilinmemektedir.Güçlü CYP2C8 inhibitörleri ile eş zamanlı kullanım sırasında doz ayarlaması gerekli değildir.P-gp ve BCRP inhibitörleriBrigatinib, in vitroP-glikoprotein (P-gp) ve meme kanseri direnç proteininin (BCRP) bir substratıdır. Brigatinibin yüksek çözünürlük ve yüksek geçirgenlik gösterdiği göz önündebulundurulduğunda; P-gp ve BCRP inhibisyonunun, brigatinibin sistemik maruziyetinde klinikolarak anlamlı bir değişikliğe yol açması beklenmemektedir. P-gp ve BCRP inhibitörleri ile eşzamanlı kullanım sırasında ALUNBRİG dozunun ayarlanması gerekli değildir.Brigatinib plazma konsantrasyonlarını azaltabilen ajanlarCYP3A indükleyicileriSağlıklı gönüllülerde; güçlü bir CYP3A indükleyicisi olan rifampisinin günlük 600 mg'lık çoklu dozu ile 180 mg'lık tekli brigatinib dozunun eş zamanlı uygulanması, tek başına uygulanan 180mg'lık brigatinib dozuna göre, brigatinib Cmaks'ını % 60, EAAo -sonsuz u % 80 (5 kat) ve EAAo-i2o'yi% 80 (5 kat) oranında azaltmıştır. Bunlarla sınırlı olmamak üzere rifampisin, karbamazepin,fenitoin, rifabutin, fenobarbital ve St. John's Wort dahil güçlü CYP3A indükleyicileri ileALUNBRİG'in eş zamanlı kullanımından kaçınılmalıdır. Orta dereceli CYP3A indükleyicileri, fizyolojik-bazlı bir farmakokinetik modelden elde edilen simülasyonlara dayanarak, brigatinib EAA'sini yaklaşık % 50 oranında azaltabilir. Bunlarla sınırlıolmamak üzere efavirenz, modafinil, bosentan, etravirin ve nafsilin dahil olmak üzere orta dereceliCYP3A indükleyicileri ile ALUNBRİG'in eş zamanlı kullanımından kaçınılmalıdır. Orta dereceliCYP3A indükleyicilerinin eşzamanlı kullanımından kaçınılamazsa, ALUNBRİG dozu, tolere edilenmevcut ALUNBRİG dozu ile 7 günlük tedaviden sonra 30 mg'lık artışlarla, orta dereceli CYP3Aindükleyicisinin başlatılması önce tolere edilen ALUNBRİG dozunun maksimum iki katına kadarartırılabilir. Orta derecede bir CYP3A indükleyicisinin kesilmesinden sonra, ALUNBRİG''e ortadereceli CYP3A indükleyicisinin başlatılmasından önce tolere edilen dozda devam edilmelidir. Plazma konsantrasyonları brigatinib ile değişebilen ajanlarCYP3A substratlarıHepatositlerle gerçekleştirilen in vitroO-INF'yi %26 ve AUCo-last'ı %30 oranında azaltmıştır. Brigatinib; büyük ölçüde CYP3A ile metabolize edilen, eş zamanlıuygulanan tıbbi ürünlerin plazma konsantrasyonlarını azaltabilir. Bu nedenle, ALUNBRİG'in darterapötik indekse sahip CYP3A substratları (örn: alfentanil, fentanil, kinidin, siklosporin, sirolimus,takrolimus) ile birlikte kullanımından kaçınılmalıdır zira etkinliklerinde azalma olabilir.ALUNBRİG ayrıca CY^3A indüksiyonundan sorumlu aynı mekanizmalar (örn. pregnane X reseptör aktivasyonu) ile diğer enzimleri ve taşıyıcıları (örn. CYP2C, P-gp) da indükleyebilir. Taşıyıcı substratlarBrigatinibin P-gp substratları (örn. digoksin, dabigatran, kolşisin, pravastatin), BCRP (örn. metotreksat, rosuvastatin, sülfasalazin), organik katyon taşıyıcısı 1 (OCT1), çoklu ilaç ve toksinekstrüzyon proteini 1 (MATE1) ve 2K (MATE2K) substratları ile eş zamanlı uygulanması, busubstratların plazma konsantrasyonlarını arttırabilir. ALUNBRİG dar terapötik indekse sahiptaşıyıcı substratlarla (örn: digoksin, dabigatran, metotreksat) birlikte kullanılırken hastalar yakındantakip edilmelidir. 4.6. Gebelik ve LaktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Kadın ve Erkeklerde Doğum kontrolü (kontrasepsiyon)ALUNBRİG ile tedavi edilen, çocuk doğurma potansiyeli bulunan kadınların hamile kalmamaları ve ALUNBRİG ile tedavi edilen erkeklerin ise tedavi sırasında çocuk sahibi olmamaları tavsiyeedilmelidir. Üreme potansiyeli olan kadınlara ALUNBRİG ile tedavi sırasında ve son dozu takibenen az 4 ay boyunca etkili, hormonal olmayan kontrasepsiyon kullanmaları tavsiye edilmelidir.Üreme potansiyeline sahip kadın partneri olan erkeklere, tedavi sırasında ve son ALUNBRİGdozundan sonra en az 3 ay süreyle etkili kontrasepsiyon kullanımı önerilmelidir. Gebelik dönemiALUNBRİG gebe bir kadına uygulandığında ölümcül sonuçlar doğurabilir. Hayvanlar üzerinde yapılan çalışmalar, üreme toksisitesini göstermektedir (bkz. bölüm 5.3.). ALUNBRİG'in gebekadınlardaki kullanımına ilişkin klinik veri bulunmamaktadır. Annenin klinik durumu tedavigerektirmiyorsa; ALUNBRİG gebelik dönemi sırasında kullanılmamalıdır. ALUNBRİG gebeliksırasında kullanılırsa veya hasta bu tıbbi ürünü alırken hamile kalırsa; hastaya potansiyel olarakfetüse zarar verebileceği bilgisi verilmelidir. Laktasyon dönemiALUNBRİG'in anne sütüne geçip geçmediği bilinmemektedir. Mevcut veriler ilacın anne sütüne geçme potansiyelini hariç tutamaz. ALU^^RİG tedavisi sırasında emzirme durdurulmalıdır. Üreme yeteneği/FertiliteALUNBRİG'in insan fertilitesi üzerindeki etkilerine ilişkin veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar esas alındığında; ALUNBRİG erkeklerde fertilitede azalmaya nedenolabilir (bkz. bölüm 5.3.). Bu bulguların insan fertilitesine yönelik klinik anlamı bilinmemektedir. 4.7. Araç ve makine kullanımı üzerindeki etkilerALUNBRİG araç ve makine kullanma yeteneği üzerinde minör bir etkiye sahiptir. Ancak; hastalar ALUNBRİG'i kullanırken görme bozukluğu, baş dönmesi veya yorgunluk yaşayabileceği için, araçveya makine kullanırken dikkatli olmalıdırlar. 4.8. İstenmeyen etkilerGüvenlilik profilinin özetiTavsiye edilen doz rejiminde ALUNBRİG ile tedavi edilen hastalarda raporlanan en yaygın advers reaksiyonlar (> %25); AST artışı, CPK artışı, hiperglisemi, lipaz artışı, hiperinsülinemi, diyare,ALT artışı, amilaz artışı, anemi, mide bulantısı, bitkinlik, hipofosfatemi, lenfosit sayısında azalma,öksürük, alkalin fosfataz artışı, döküntü, APTT artışı, miyalji, baş ağrısı, hipertansiyon, akyuvarsayısında azalma, dispne ve kusmadır. Neoplazma progresyonu ile ilişkili olaylar dışında; tavsiye edilen doz rejiminde ALUNBRİG ile tedavi edilen hastalarda rapor edilen en yaygın ciddi advers reaksiyonlar (> %2) pnömoni,pnömonit, dispne ve pireksi olmuştur. Advers reaksiyonların tablo halinde listesi:Aşağıda tanımlanan veriler üç klinik çalışmada tavsiye edilen doz rejiminde ALUNBRİG'e maruziyeti yansıtmaktadır: Daha önce bir ALK-inhibitörü ile tedavi görmemiş ileri evre ALK-pozitif KHDAK hastalarında bir Faz 3 çalışma (ALTA 1L) (N=136), daha önce krizotinibtedavisine progresyon göstermiş olup ALUNBRİG'le tedavi edilen ALK-pozitif KHDAKhastalarında bir faz 2 çalışma (N=110) ve ilerlemiş malignansili hastalarda bir faz 1/2 dozeskalasyon/ekspansiyon çalışması (N=28). Bu çalışmalarda, tavsiye edilen doz rejimindeALUNBRİG kullanan hastalarda medyan maruziyet süresi 21,8 ay olmuştur. Rapor edilen advers reaksiyonlar Tablo 3'te sunulmaktadır ve Sistem Organ Sınıfına, tavsiye edilen terime ve görülme sıklığına göre listelenmiştir. Sıklık kategorileri: (çok yaygın >1/10, yaygın(>1/100 ila <1/10), yaygın olmayan >1/1.000 ila <1/100, seyrek >1/10.000 ila <1/1.000; bilinmiyor(eldeki verilerden hareketle tahmin edilemiyor). Her bir sıklık grubu içinde istenmeyen etkilerazalan şiddete göre verilmiştir. Tablo 3: ALUNBRIG ile 180 mg rejiminde tedavi edilen hastalarda (N= 274) raporedilen advers reaksiyonlar (Advers Olaylar için Ortak Terminoloji Kriterleri (CTCAE) versiyon 4.03'e göre)
aAtipik pnömoni, pnömoni, pnömoni, pnömoni aspirasyonu, kriptokok pnömonisi, alt solunum yolu enfeksiyonu, alt solunum yolu viral enfeksiyonu, akciğer enfeksiyonunu içerir.b5. derece olayları içerir.cDerece geçerli değildir. dBaş ağrısını, sinüs kaynaklı baş ağrısını, baş rahatsızlığını, migren ve tansiyon kaynaklı baş ağrısını içerir. ePareztezi, periferik duyusal nöropati, disestezi, hiperestezi, hipoestezi, nevralji, periferal nöropati, nörotoksisite,periferal motor nöropatisi, polinöropati, yanma hissi ve herpetik nevraljiyi içerir. fGörsel derinlik algısının değişmesi, katarakt, kazanılmış renk körlüğü, diplopi, glokom, göz içi basıncında artış, maküler ödem, fotofobi, fotopsi, retinal ödem, bulanık görme, görme keskinliğinin azalması, görme alanı defekti, görmebozukluğu, vitröz yırtılma, vitroz flatör, geçici amorozu içerir. gBradikardi, sinüs bradikardisini içerir. hSinüs taşikardisi, taşikardi, atriyal taşikardi, kalp atım hızımda artışı içerir. 'Kan basıncında artış, diyastolik hipertansiyon, hipertansiyon, sistolik hipertansiyonu içerir. jDispne, efor dispnesini içerir. kİnterstisyel akciğer hastalığını, pnömoniti içerir. 'Abdominal rahatsızlık hissi, abdominal distansiyon, karın ağrısı, karnın alt kısmında ağrı, karnın üst kısmında ağrı, epigastrik rahatsızlığı içerir. mAftöz stomatit, stomatit, aftöz ülser, ağız ülserleri, ağız mukozası kabarmasını içerir. nDermatit akneiform, eritem, eksfolyatif döküntü, döküntü, eritematöz döküntü, maküler döküntü, makülopapüler döküntü, papüler döküntü, kaşıntılı döküntü, püstüler döküntü, dermatit, alerjik dermatit, kontakt dermatit, jeneralize eritem, foliküler döküntü, ürtiker, ilaç döküntüsü ve toksik deri döküntüsünü içerir. oKaşıntı, alerjik kaşıntı, jeneralize kaşıntı, genital bölgede kaşıntı, vulvovajinal bölgede kaşıntıyı içerir. p Işığa duyarlılık reaksiyonu, polimorf ışık erüpsiyonu, solar dermatiti içerir. sGöz kapağı ödemi, yüz ödemi, periferik ödem, periorbital ödem, yüz şişmesi, jeneralize ödem, periferik şişme, anjiyoödem, dudaklarda şişme, periorbital şişlik, ciltte şişlik, göz kapağında şişmeyi içerir. tKan kolesterol seviyesinde artış, hiperkolesterolemiyi içerir._Seçili advers reaksiyonların tanımıPulmoner advers reaksiyonlarALTA IL'de; hastaların % 2,9'unda tedavinin başlangıcında (8 gün içinde) herhangi bir Derece ILD/pnömonit, hastaların % 2,2'sinde Derece 3-4 ILD/pnömonit görülmüştür. ÖlümcülILD/pnömonit mevcut değildir. İlave olarak, hastaların % 3,7'sinde pnömonit tedavinin ilerleyenzamanlarında görülmüştür. ALTA'da, hastaların % 6,4'ünde tedavinin başlarında (9 gün içinde, medyan başlangıç: 2 gün) ILD/pnömonit, pnömoni ve dispne dahil olmak üzere, herhangi bir derecede pulmoner adversreaksiyonlar görülmüştür; hastaların % 2,7'sinde Derece 3-4 pulmoner advers reaksiyon ve 1hastada (% 0,5) ölümcül pnömoni görülmüştür. Derece 1-2 pulmoner advers reaksiyonları takiben;ALUNBRİG ile tedavi kesilmiştir ve daha sonra yeniden başlatılmış veya doz azaltılmıştır. Üçölümcül vaka (hipoksi, akut solunum güçlüğü sendromu ve pnömoni) dahil olmak üzere, erkenpulmoner advers reaksiyonlar hastalarda (N= 137) (Çalışma 101)bir doz yükseltme çalışmasında dameydana gelmiştir. İlave olarak, ALTA'da hastaların % 2,3'ünde tedavinin ilerleyen zamanlarındapnömonit görülmüştür, 2 hastada Derece 3 pnömonit görülmüştür (bkz. bölüm 4.2 ve 4.4.). Yaşlı hastalarErken pulmoner advers reaksiyon, 65 yaş altı hastaların %3,1'inde, 65 yaş ve üzeri hastaların ise %10,1'inde rapor edilmiştir. HipertansiyonALUNBRİG ile 180 mg rejiminde tedavi edilen hastaların % 30'unda hipertansiyon bildirilmiştir olup; bu hastaların % 11'i Derece 3 hipertansiyona sahiptir. 180 mg rejiminde % 1,5 oranındahipertansiyon için doz azaltılması gerçekleştirilmiştir. Tüm hastalarda ortalama sistolik vediyastolik kan basıncı zamanla artmıştır (bkz. bölüm 4.2 ve 4.4.). BradikardiALUNBRİG ile 180 mg rejiminde tedavi edilen hastaların % 8,4'ünde bradikardi rapor edilmiştir. 180 mg tedavi rejiminde hastaların % 8,4'ünde dakikada 50 kalp atım hızından az kalp atım hızı rapor edilmiştir (bkz. bölüm 4.2. ve 4.4.). Görme bozukluğuALUNBRİG ile 180 mg rejiminde tedavi edilen hastaların % 14'ünde görme bozukluğu advers reaksiyonları rapor edilmiş olup; bunlardan 3 tanesi, maküler ödem ve katarakt dahil olmak üzereDerece 3 advers reaksiyon (% 1,1) olarak rapor edilmiştir. 180 mg rejiminde iki hastada (% 0,7) görme bozukluğu için doz azaltılması gerçekleştirilmiştir (bkz. bölüm 4.2 ve 4.4.). Periferal nöropati180 mg rejiminde tedavi edilen hastaların % 20'sinde periferal nöropati advers reaksiyonları rapor edilmiştir. Hastaların % 33'ünde tüm periferal nöropati advers reaksiyonları ortadan kalkmıştır.Periferal nöropati advers reaksiyonlarının medyan süresi 6.6 ay ve maksimum süre 28,9 aydır. Kreatin _ fosfokinaz (CPK) artışıALTA 1L ve ALTA'da, ALUNBRİG ile 180 mg rejiminde tedavi edilen hastaların % 64'ünde CPK artışı rapor edilmiştir. Derece 3-4 CPK artışı insidansı % 18 olmuştur. CPK artışının başlangıcınakadar geçen medyan süre 28 gün olmuştur. 180 mg rejiminde hastaların % 10'unda CPK artışı için doz azaltılması gerçekleştirilmiştir (bkz. bölüm 4.2. ve 4.4.). Pankreatik enzimlerin artışıALUNBRİG ile 180 mg rejiminde tedavi edilen hastaların sırasıyla % 47 ve % 54'ünde amilaz ve lipaz artışları rapor edilmiştir. Derece 3 ve 4 artışları için amilaz ve lipaz insidansı sırasıyla % 7,7ve % 15 olmuştur. Amilaz ve lipaz artışının başlangıcına kadar geçen medyan süre sırasıyla 16 günve 29 gün olmuştur. 180 mg rejiminde hastaların sırasıyla % 4,7 ve % 2,9'unda lipaz ve amilaz artışı için doz azaltılması gerçekleştirilmiştir (bkz. bölüm 4.2. ve 4.4.). Hepatik enzimlerin artışıALUNBRİG ile 180 mg rejiminde tedavi edilen hastaların sırasıyla % 49 ve % 68'inde ALT ve AST artışları rapor edilmiştir. Derece 3 ve 4 artışları için, insidanslar ALT ve AST için sırasıyla %4,7 ve % 3,6 olmuştur. 180 mg rejiminde hastaların sırasıyla % 0,7 ve % 1,1'inde ALT ve AST artışı için doz azaltılması gerçekleştirilmiştir (bkz. bölüm 4.2. ve 4.4.). HiperglisemiHastaların % 61'inde hiperglisemi görülmüştür. Hastaların % 6,6'sında Derece 3 hiperglisemi meydana gelmiştir. Hiçbir hastada hiperglisemi nedeniyle doz azaltılması gerçekleştirilmemiştir. Fotosensitivite ve fotodermatozALUNBRİG ile farklı doz rejimlerinde tedavi edilen 804 hastadan alınan verilerle yedi klinik çalışmanın havuzlanmış analizi, hastaların %5,8'inde fotosensitivite ve fotodermatozun raporedildiğini ve hastaların %0,7'sinde Derece 3-4 meydana geldiğini göstermiştir. Hastaların %0,4'ündedoz azaltılmıştır (bkz. bölüm 4.2 ve 4.4). Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlıkmesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi(TÜFAM)'ne bildirmeleri gerekmektedir ([email protected]:tel: 0 800 31400 08: faks: 0 312 218 35 99).4.9. Doz aşımı ve tedavisiALUNBRİG'in doz aşımına ilişkin spesifik herhangi bir antidot mevcut değildir. Doz aşımı meydana gelmesi durumunda; hastalar advers reaksiyonlar (bkz. bölüm 4.8) bakımından takip edilirve uygun bir destek tedavisi sağlanır. 5.FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: ATC kodu: Antineoplastik ilaç, protein kinaz inhibitörleri L01ED04 Etki mekanizmasıBrigatinib, insülin-benzeri büyüme faktörü 1 reseptörü (IGF-1R) ve ALK'yı, c-ros onkogen 1(ROS1), hedefleyen bir tirozin kinaz inhibitörüdür. Brigatinib, in vitroin vivotayinlerdeALK'nin otofosforilasyonunu ve aşağı yönlü sinyal proteini STAT3'ün ALK aracılıfosforilasyonunu inhibe etmiştir.Brigatinib, farelerde EML4-ALK ve NPM-ALK füzyon proteinlerini eksprese eden hücre hatlarının in vitroproliferasyonunu inhibe etmiştir ve EML4-ALK-pozitif KHDAK ksenograft büyümesinindoza bağlı inhibisyonunu göstermiştir.Brigatinib; G1202R ve L1196M dahil, ALK inhibitörlerine direnç ile ilişkilendirilen EML4-ALK'nın mutant formlarını eksprese eden hücrelerin canlılığını in vitroinvivoda inhibe etmiştir.Kardiyak elektrofizyolojiÇalışma 101'de, ilerlemiş malignansili 123 hastada 30 mg ila 240 mg'lık günlük brigatinib dozu ile ALUNBRİG'in QT aralığını uzatma potansiyeli değerlendirilmiştir. Başlangıç noktasındanmaksimum ortalama QTcF (Friedericia yöntemi ile düzeltilmiş QT) değişimi 10 milisaniyeden kısaolmuştur. Bir maruziyet-QT analizi, konsantrasyona bağlı herhangi bir QTc aralığı uzamasıolmadığını öne sürmüştür. Klinik etkililik ve güvenlilikALTA 1LALUNBRİG'in güvenliliği ve etkililiği; daha önceden ALK-hedefli bir tedavi almamış ve ilerlemiş ALK-pozitif KHDAK'lı 275 yetişkin hastada gerçekleştirilen randomize (1:1), açık etiketli, çokmerkezli bir çalışmada (ALTA 1L) değerlendirilmiştir. Uygunluk kriterleri; lokal bir bakım standardı testine dayalı olarak belgelenen bir ALK düzenlemesine sahip ve ECOG performans skoru 0-2 olan hastaların dahil edilmesine izin vermiştir.Hastaların lokal olarak ilerlemiş veya metastatik ortamda önceden 1 taneye kadar kemoterapi rejimialmasına izin vermiştir. Leptomeningeal metastazlar da dahil olmak üzere tedavi edilmiş veyatedavi edilmemiş merkezi sinir sistemi (CNS) metastazları olan nörolojik olarak stabil hastalaruygundur. Pulmoner interstisyel hastalığı, ilaca bağlı pnömonit veya radyasyon pnömoniti öyküsüolan hastalar çalışma dışı bırakılmıştır. Hastalar; günde bir kez 90 mg'lık dozda 7 günlük bir tedavi sonrası günde bir kez 180 mg (N=137) ALUNBRİG veya oral olarak günde iki defa krizotinib 250 mg (N= 138) almak üzere 1:1 oranındarandomize edilmiştir. Randomizasyon beyin metastazı (var, yok) ve lokal olarak ilerlemiş veyametastatik hastalık için önceden kemoterapi kullanımı (evet, hayır) ile katmanlara ayrılmıştır. Hastalık progresyonu yaşayan krizotinib kolundaki hastalara ALUNBRİG ile tedavi görmeleri için çapraz geçiş önerildi. Krizotinib koluna randomize edilen ve son analiz zamanına kadar çalışmatedavisini bırakan 121 hastanın tamamı arasında, 99 (%82) hastaya müteakip ALK tirozin kinazinhibitörleri (TKI'ler) verildi. Krizotinib koluna randomize edilen seksen (%66) hasta, daha sonraALUNBRİG tedavisi aldı; buna, çalışmada çapraz geçiş yapan 65 (%54) hasta da dahildir. Majör sonuç ölçümü, Çift Kör Hakemli Bağımsız İnceleme Komitesi (BIRC) tarafından değerlendirilen Solid Tümörlerdeki Yanıt Değerlendirme Kriteri'ne (RECIST v1.1) göreprogresyonsuz sağkalım (PFS) olmuştur. BIRC tarafından değerlendirilen ilave sonuç ölçümleri,doğrulanmış objektif yanıt oranını (ORR), yanıt süresini (DOR), yanıt verilene kadar geçen süreyi,hastalık kontrol oranını (DCR), intrakraniyal ORR'yi, intrakraniyal PFS'yi ve intrakraniyal DOR'uiçermiştir. Araştırmacı tarafından değerlendirilen sonuçlar arasında PFS ve genel sağkalım yeralmıştır. ALTA 1L'deki temel demografik özellikler ve hastalık özellikleri şu şekildedir: medyan yaş 59 (% 32'si 65 yaş ve üzeri olmak üzere 27 -89 arası), % 59 beyaz ırk ve % 39 Asyalı, % 55 kadın, % 39ECOG PS 0 ve % 56 ECOG PS 1, % 58 hiç sigara kullanmamış, % 93 hastalık evresi IV, % 96 adenokarsinoma histolojisine sahip, beyin için radyoterapi almış ve % o 30 başlangıçta CNS metastazına sahip, % 14 daha önce 27 daha önce kemoterapi almış. Ekstratorasik metastazbölgelerine beyin (hastaların % 30'u), kemik (hastaların % 31'i) ve karaciğer (hastaların % 20'si)dahildir. Medyan bağıl doz yoğunluğu ALUNBRİG için %97, krizotinib içinse %99 idi. ALUNBRİG kolunda 11 aylık medyan takip süresinde gerçekleştirilen primer analizde; ALTA 1L çalışması, BIRC tarafından PFS'de istatistiksel olarak anlamlı bir iyileşme gösteren primersonlanma noktasını karşılamıştır. ALUNBRİG kolunda 24,9 aylık medyan takip süresinde 28 Haziran 2019'da protokole göre belirlenmiş bir ara analiz gerçekleştirilmiştir. ITT popülasyonunda BIRC'ye göre medyan PFS,ALUNBRİG kolunda 24 ay ve krizotinib kolunda 11 aydı (HR =0,49 [%95 GA (0,35, 0,68)], p<0,0001). ALUNBRİG kolunda medyan 40.4 aylık takip süresinde gerçekleştirilen son hasta son temas tarihi olan 29 Ocak 2021 ile protokolde belirtilen nihai analizin sonuçları aşağıda sunulmuştur.
Başlangıçta herhangi bir beyin metastazı olan hastalarda ve ölçülebilir beyin metastazları (en uzun çap > 10 mm) olan hastalarda RECIST vl.l'e göre intrakraniyal etkililiğim BIRC değerlendirmesiTablo 5'te özetlenmiştir. Tablo 5: ALTA IL'deki Hastalarda BIRC-Değerlendirmeli İntrakraniyal Etkililik
CI= Güven aralığı; NE= Tahmin edilebilir değildir. cİlk doğrulanmış intrakraniyal yanıt tarihinden intrakraniyal hastalık progresyonuna (yeni intrakraniyal lezyonlar, en alt noktadan itibaren > % 20 intrakraniyal hedef lezyon çapı büyümesi veya hedef olmayan intrakraniyal lezyonların kesinprogresyonu) veya ölüm veya sansürleme tarihine kadar ölçülmüştür. dRandomizasyon tarihinden intrakraniyal hastalık progresyonuna (yeni intrakraniyal lezyonlar, en alt noktadan itibaren > % 20 intrakraniyal hedef lezyon çapı büyümesi veya hedef olmayan intrakraniyal lezyonların kesin progresyonu) veyaölüm veya sansürleme tarihine kadar ölçülmüştür. ^Beynine palyatif radyoterapi alan 1 hasta dahildir fBeynine palyatif radyoterapi alan 3 hasta dahildir ALTAALUNBRİG'in güvenliliği ve etkililiği; krizotinib tedavisine progresyon gösteren, lokal olarak ilerlemiş veya metastatik ALK-pozitif KHDAK'lı 222 yetişkin hastada gerçekleştirilen randomize(1:1), açık etiketli, çok merkezli bir çalışmada (ALTA) değerlendirilmiştir. Uygunluk kriterleri; valide edilmiş bir teste dayalı olarak belgelenen bir ALK düzenlemesine sahip öncesinde kemoterapi almış, ECOG Performans Durumu 0-2 olan ve hastaların dahil edilmesineizin vermiştir. İlave olarak, merkezi sinir sistemi (CNS) metastazı olan hastalar, nörolojik olarakstabil olmaları ve kortikosteroid dozunda artış gerektirmedikleri sürece, dahil edilmiştir. Pulmonerinterstiyal hastalık veya ilaç ile ilişkili pnömoni geçmişi olan hastalar hariç tutulmuştur. Pulmonerinterstisyel hastalığı veya ilaca bağlı pnömonit öyküsü olan hastalar çalışma dışı bırakılmıştır. Hastalar; günde bir kez 90 mg (90 mg'lık rejim, N=112) veya günde bir kez 90 mg'lık dozda 7 günlük bir tedavi sonrası günde bir kez 180 mg (180 mg'lık rejim, N=110) ALUNBRİG almaküzere 1:1 oranında randomize edilmiştir. Medyan takip süresi 22,9 ay olmuştur. Randomizasyonbeyin metastazı (var, yok) ve krizotinib tedavisine en iyi cevap (tam veya kısmi yanıt, diğerherhangi bir yanıt/bilinmeyen) ile katmanlara ayrılmıştır. Majör sonuç ölçümü, araştırmacı tarafından değerlendirilen Solid Tümörlerdeki Yanıt Değerlendirme Kriteri'ne (RECIST v1.1) göre objektif yanıt oranını (ORR) doğrulamıştır. İlavesonuç ölçümleri; Bağımsız İnceleme Komitesi (IRC) tarafından değerlendirilen doğrulanmışORR'yi, yanıt verilene kadar geçen süreyi; progresyonsuz sağkalımı (PFS); yanıt süresini (DOR);genel sağkalımı ve IRC tarafından değerlendirilen intrakraniyal ORR, intrakraniyal DOR'yiiçermiştir. ALTA'daki temel demografik özellikler ve hastalık özellikleri şu şekildedir: medyan yaş 54 (% 23'ü 65 yaş ve üzeri olmak üzere 18 -82 arası), % 67 beyaz ırk ve % 31 Asyalı, % 57 kadın, % 36ECOG PS 0 ve % 57 ECOG PS 1, % 7 ECOG PS2, % 60 hiç sigara kullanmamış, % 35 daha evvelsigara kullanmış, %5 mevcut durumda sigara içen, % 98 Evre IV, % 97 adenokarsinom ve % 74daha önce kemoterapi almış. Ekstratorasik metastazların en yaygın olduğu bölgelere % 69 beyin (%62'si daha önce beynine radyasyon almış), % 39 kemik ve % 26 karaciğer dahildir. ALTA analizinden elde edilen etkililik sonuçları Tablo 6'da özetlenmektedir ve araştırmacı tarafından değerlendirilen PFS için Kaplan-Meier (KM) eğrisi Şekil 2'de gösterilmektedir.
Araştırmacının Değerlendirdiği Sistemik Progresyonsuz Sağkalım: Tedavi Kolu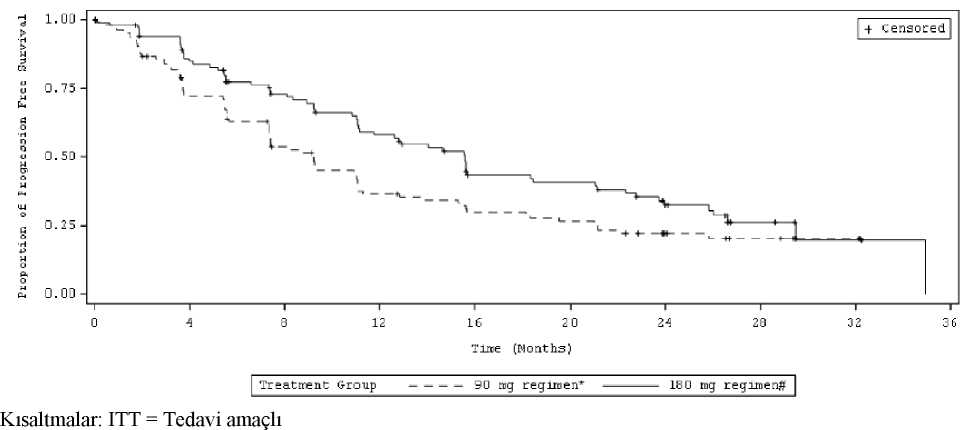 Şekil 2:Not: Progresyonsuz sağkalım tedavinin başlangıcından, ilk olarak hangisinin meydana geldiğine bakılmaksızın hastalık progresyonunun ilk olarak kanıtlandığı veya ölüme kadar olan tarihe kadar geçen süre olarak belirlenmiştir. *90 mg günlük tedavi rejimi i7 gün boyunca 90 mg/günlük tedaviyi takiben günde bir kez 180 mg ALTA'da başlangıçtaki ölçülebilir beyin metastazları (en uzun çap >10 mm) olan hastalardaki intrakraniyal ORR ve intrakraniyal yanıt süresinin IRC değerlendirmeleri Tablo 7'deözetlenmektedir. TabloBaşlangıçta Ölçülebilir Beyin Metastazı olan Hastalardaki İntrakraniyalEtkililik
Başlangıçta beyin metastazı olan hastalarda, intrakraniyal hastalık kontrol oranı 90 mg kolunda (N= 81) % 77.8 (% 95 CI: 67,2 - 86,3) ve 180 mg kolunda (n= 74) % 85,1 (% 95 CI: 75-92,3) olmuştur. Çalışma 101Ayrı bir doz bulma çalışmasında, ALK-pozitif KHDAK'ı olan ve krizotinibde progresyon gösteren 25 hastaya günde bir defa 90 mg'lık dozda 7 günlük bir tedavi sonrası günde 1 defa 180 mgALUNBRİG uygulanmıştır. Bu hastaların 19'u araştırmacı değerlendirmesi ile doğrulanmış objektifbir yanıt göstermiştir (% 76; % 95 CI: 55, 91) ve yanıt veren 19 hasta arasında yanıtın KM tahminimedyan süresi 26,1 ay (% 91 CI: 7,9, 26,1) olmuştur. KM medyan PFS 16,3 aydır (% 95 CI: 9,2,NE) ve 12 aylık genel sağkalım olasılığı % 84.0 (% 95 CI: 62,8, 93,7) olmuştur. Pediyatrik popülasyonAvrupa İlaç Ajansı, akciğer karsinomasında (küçük hücreli ve küçük hücreli dışı karsinoma) pediyatrik popülasyonun tüm alt gruplarında ALUNBRİG ile gerçekleştirilen çalışmalarınsonuçlarını sunma yükümlülüğünden muaf tutmuştur (pediyatrik kullanıma ilişkin bilgi için bölüm4.2'ye bakınız). 5.2. Farmakokinetik özelliklerGenel ÖzelliklerEmilim:Çalışma 101'de, hastalara brigatinibin tekli oral doz (30 - 240 mg) uygulamasını takiben pik konsantrasyonu için medyan süre (Tmaks), dozlama sonrası 1-4 saat aralığında değişmiştir. Tekli birdozdan sonra ve kararlı durumda, sistemik maruziyet 60 mg - 240 mg/gün doz aralığı boyunca dozorantısaldır. Tekrarlı dozlama sonrasında makul bir akümülasyon gözlenmiştir (geometrik ortalamaakümülasyon oranı: 1,9 - 2,4). 90 mg ve 180 mg'lık günlük dozlarda brigatinibin geometrikortalama kararlı durum Cmaks'ı sırasıyla 552 ve 1452 ng/mL'dir ve ilgili EAA O-Tsırasıyla 8,165 ve20,276 h.ng/mL'dir. Brigatinib P-gp ve BCRP taşıyıcı proteinlerinin bir substratıdır.Yüksek-yağlı öğün sonrası brigatinib uygulanan sağlıklı gönüllülerde; bir gece boyunca açlık sonrası Cmaks ve EAA ile karşılaştırıldığında; EAA üzerinde bir etkisi olmaksızın brigatinib Cmaks'ı% 13'e indirgenmiştir. Brigatinib yemeklerle birlikte veya yemekten bağımsız olarak uygulanabilir. Dağılım:Brigatinib insan plazma proteinlerine orta derecede (% 91) bağlanmıştır ve bağlanma konsantrasyona bağlı değildir. Kan-plazma konsantrasyonu oranı 0,69'dur. Hastalara günde bir defa180 mg brigatinib uygulanmasını takiben; kararlı durumda brigatinib dağılımının görünür geometrikhacim ortalaması (Vz/F) 307 L olmuştur ve bu durum dokulara orta derecede bir dağılım olduğunugöstermektedir. Biyotransformasyon:İn vitroçalışmalar; brigatinibin primer olarak CYP2C8 ve CYP3A4 ile, ve çok daha düşük bir düzeyde CYP3A5 ile metabolize olduğunu göstermiştir.Oral olarak 180 mg'lık tekli [14C]-brigatinib dozunun sağlıklı gönüllülere uygulanmasını takiben; N-demetilasyon ve sistein konjugasyonu iki majör metabolik klerens yolu olmuştur. Kombineedilmiş idrar ve dışkıda, radyoaktif dozun % 48, % 27 ve % 9,1'i sırasıyla değişmemiş brigatinib,N-desmetik brigatinib (AP26123) ve brigatinib sistein konjugatı olarak atılmıştır. Değişmemişbrigatinib, AP26123 (% 3,5) ile birlikte dolaşımdaki majör radyoaktif bileşenlerdir (% 92), ayrıcaprimer metabolit in vitrodaIn vitrokinaz ve hücresel tayinlerde,AP26123 metaboliti brigatinibden yaklaşık 3-kat daha düşük bir potens ile ALK'yı inhibe etmiştir.Eliminasyon:Günde bir defa 180 mg brigatinib verilen hastalarda; kararlı durumda brigatinibin görünür geometrik ortalama oral klerensi (CL/F), 8,9 L/saat ve medyan plazma eliminasyon yarılanmasüresi 24 saattir. Brigatinibin primer atılım yolu dışkı yoluyladır. 6 sağlıklı erkek gönüllüye [14C] brigatinibin tekli 180 mg oral dozunun uygulanmasını takiben; uygulanan dozun % 65'i dışkıda ve % 25'i idrardasaptanmıştır. Değişmemiş brigatinib dışkı ve idrarda toplam radyoaktivitenin sırasıyla % 41 ve %86'sını temsil etmiştir, geri kalan kısım metabolitler olmuştur. Hastalardaki karakteristik özelliklerKaraciğer yetmezliği:Brigatinibin farmakokinetiği; normal karaciğer fonksiyonuna sahip sağlıklı gönüllülerde (N=9), hafif karaciğer yetmezliği olan (Child-Pugh sınıf A, N=6), orta derecede karaciğer yetmezliği olan(Child-Pugh sınıf B, N=6) veya şiddetli karaciğer yetmezliği olan (Child-Pugh sınıf C, N=6)hastalarda karakterize edilmiştir. Brigatinib farmakokinetiği; normal karaciğer fonksiyonuna sahipsağlıklı gönüllüler ve hafif (Child-Pugh sınıf A) veya orta dereceli (Child-Pugh sınıf B) karaciğeryetmezliği olan hastalar arasında benzerdir. Normal karaciğer fonksiyonuna sahip sağlıklıgönüllüler ile karşılaştırıldığında, şiddetli karaciğer yetmezliği (Child-Pugh sınıf C) olan hastalardabağlanmamış EAAo-sonsuz % 37 daha yüksek olmuştur (bkz. bölüm 4.2.). Böbrek yetmezliği:Brigatinib farmakokinetiği; popülasyon farmakokinetik analizleri esas alındığında, normal böbrek fonksiyonuna sahip hastalar ve hafif - orta dereceli böbrek yetmezliği (eGFR > 30 mL/dk) olanhastalarda benzerdir. Bir farmakokinetik çalışmada; normal böbrek fonksiyonuna sahip hastalar(eGFR > 90 mL/dk, N=8) ile karşılaştırıldığında, şiddetli böbrek yetmezliği (eGFR <30 mL/dk,N=6) olan hastalarda bağlanmamış EAAo-sonsuz % 94 daha yüksek olmuştur (bkz. bölüm 4.2.). Irk ve cinsiyet:Popülasyon farmakokinetik analizleri; ırk veya cinsiyetin brigatinib farmakokinetikleri üzerinde herhangi bir etkisi olmadığını göstermiştir. Yaş, vücut ağırlığı ve albümin konsantrasyonları:Popülasyon farmakokinetik analizleri; vücut ağırlığı, yaş ve albümin konsantrasyonunun brigatinib farmakokinetikleri üzerinde klinik olarak anlamlı bir etkisi olmadığını göstermiştir. 5.3. Klinik öncesi güvenlilik verileriBrigatinib ile gerçekleştirilen güvenlilik farmakoloji çalışmalarında pulmoner etkiler (solunum hızında değişim; insan Cmax'ının 1-2 katı), kardiyovasküler etkiler (kalp atım hızında ve kanbasıncında değişim; insan Cmax'ının 0,5 katı) ve renal etkiler (renal fonksiyonda azalma; insanCmax'ının 1-2,5 katı) için potansiyel tespit edilmiş; ancak QT uzaması veya nörofonksiyonal etkipotansiyeli görülmemiştir. Klinik kullanımla ilişkili olabilecek şekilde klinik maruziyet seviyelerine benzer maruziyet seviyelerinde hayvanlarda görülen advers reaksiyonlar gastrointestinal sistem, kemik iliği, gözler,testisler, karaciğer, böbrek, kemik ve kalple ilgilidir. Bu etkiler genellikle dozlamanın yapılmadığıiyileşme periyodu boyunca geri dönüşümlüdür; ancak, gözler ve testislerdeki etkiler iyileşmeninolmaması nedeniyle belirgin istisnalardır. Tekrarlı doz toksisite çalışmalarında, maymunlarda insanEAA'sının 0,2 katı ve üzerinde akciğer değişimleri (köpüksü alveolar makrofajlar) görülmüştür;ancak bunlar minimal düzeyde olup normal maymunlarda arka plan bulguları olarak raporedilenlere benzer özellik göstermiştir ve bu maymunlarda solunum rahatsızlığına dair herhangi birklinik kanıt bulunmamıştır. Brigatinib ile karsinojenisite çalışmaları gerçekleştirilmemiştir. Brigatinib, bakteriyel ters mutasyon (Ames) veya memeli hücre kromozomal aberasyon testlerinde in vitrodamutajenik değildir; ancak sıçan kemik iliği mikronukleus testinde mikronukleus sayısınıküçük miktarda arttırmıştır. Mikronukleus indüksiyonunun mekanizması anormal kromozomsegragasyonudur (aneugenisite) ve kromozomlar üzerinde klastojenik bir etkisi yoktur. Bu etkigünlük 180 mg dozda insan maruziyetinin yaklaşık 5 katında gözlemlenmiştir.Brigatinib erkek fertilitesini bozabilir. Tekrarlı-doz hayvan çalışmalarında testiküler toksisite gözlenmiştir. Sıçanlardaki bulgular; düşük ağırlıklı testisler, seminal veziküller ve prostat bezini vetestiküler tübüler dejenerasyonu içermektedir; bu etkiler, iyileşme dönemi boyunca geri dönüşümlüolmamıştır. Maymunlardaki bulgular; mikroskobik hipospermatogenez bulgusu ile birliktetestislerin boyutunda küçülmeyi içermektedir; bu etkiler, iyileşme dönemi boyunca geri dönüşümlüolmuştur. Genel olarak; sıçan ve maymunlarda erkek üreme organları üzerindeki bu etkiler, gündebir kez 180 mg'lık doz alan hastalarda gözlenen EAA değerinin 0,2 katı ve üzeri maruziyetlerdemeydana gelmiştir. Sıçan ve maymunlardaki genel toksikoloji çalışmalarında dişi üreme organlarıüzerinde belirgin bir advers etki gözlenmemiştir. Organogenez sırasında gebe sıçanlara günlük brigatinib dozlarının verildiği bir embryo-fötal gelişim çalışmasında; günde bir kez 180 mg dozda insan maruziyetindeki EEA değerinin yaklaşık0,7 katı gibi düşük dozlarda doza bağlı iskelet anormallikleri gözlenmiştir. Bulgular arasındaembriyo-letalite, fötal gelişimde yavaşlama ve iskelet değişimleri yer almaktadır. FARMASOTIK ÖZELLİKLER6.6.1. Yardımcı maddelerin listesiÇekirdek tabletLaktoz monohidrat (inek sütü) Mikrokristalin selüloz (PH-102) Sodyum nişasta glikolat (tip A) Hidrofobik kolloidal silika Magnezyum stearat Tablet kaplama(Opadry II Beyaz)Talk Polietilen glikol (Makrogol) Polivinil alkol Titanyum dioksit 6.2. GeçimsizliklerGeçerli değildir. 6.3. Raf ömrü36 ay 6.4. Saklamaya yönelik özel tedbirler30oC'nin altındaki oda sıcaklığında ve orijinal ambalajında ışıktan koruyarak saklayınız. Çocukların göremeyeceği ve erişemeyeceği yerlerde saklayınız. 6.5. Ambalajın niteliği ve içeriği28 film kaplı tablet, ısıl yapışmalı kağıt lamine folyo kaplamalı şeffaf termoform poli-kloro-tri-fluoro-etilen (PCTFE) blister ambalajda kullanma talimatı ile birlikte karton kutuda takdim edilir. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan tıbbi ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. Kullanılmamış olan ürünler ya da atık materyaller, yerel düzenlemelere uygun olarak atılmalıdır. Sitotoksik ve sitostatik beşeri tıbbi ürünlerin kullanımları sonucu boşalan iç ambalajlarının atıkları TEHLİKELİ ATIKTIR ve bu atıkların yönetimi 2/4/2015 tarihli ve 29314 sayılı Resmi Gazetedeyayımlanan Atık Yönetimi Yönetmeliği'ne göre yapılır. 7.RUHSAT SAHIBITakeda İlaç Sağlık Sanayi Ticaret Limited Şirketi Şişli/İstanbul RUHSAT NUMARASI
2019/345 9.ILK RUHSAT TARIHI/RUHSAT YENILEME TARIHIİlk ruhsat tarihi: 26.07.2019 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENILENME TARIHI1 Katkı sağlayan eş zamanlı kullanılan herhangi bir tıbbi ürününtanımlanmaması durumunda, ALUNBRİG tedavisi kalıcı olarakkesilmelidir. Nüksetme durumunda; ALUNBRİG tedavisi kalıcı olarakkesilmelidir. doz seviyesinde devam ettirilmelidir._ Katkı sağlayan eş zamanlı kullanılan bir tıbbi ürününtanımlanması ve kesilmesi veya dozunun ayarlanmasıdurumunda; ALUNBRİG klinik olarak gerekli sıklıkta takip ile,asemptomatik bradikardi veya dakikada 60 kalp atım hızı veyadaha yüksek dinlenme kalp hızı sağlandığında, Tablo 1'e göre birsonraki daha düşük doz seviyesinde devam ettirilmelidir. |
İlaç BilgileriAlunbrig 30 Mg Film Kaplı TabletEtken Maddesi: Brigatinib Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.