Nibtu 100 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA URUN BILGISI¡ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERI TIBBİ ÜRÜNÜN ADINİBTU 100 mg film kaplı tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Bir film kaplı tablet, 100 mg imatinib (119,5 mg imatinib mesilat olarak) içerir. Yardımcı maddeler:Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tablet. NİBTU tabletler; açık kahve renkli, bir tarafı çentikli, yuvarlak film kaplı tabletlerdir. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarNİBTU'nun endikasyonları: Yeni tanı konmuş Philadelphia kromozomu pozitif kronik faz kronik miyeloid lösemi(KML) hastalarında, Akselere faz Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML)hastalarında, Blastik faz Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML)hastalarında, Diğer tedavilere dirençli Philadelphia kromozomu pozitif kronik miyeloid lösemi(KML) hastalarında, İlk tanısı Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML) olan ancaktedavi ile Philadelphia kromozomu negatif hale gelen kronik/akselere/blastik faz kronikmiyeloid lösemi hastalarında, Kronik miyeloid lösemili (KML) olan 3 yaş ve üzerindeki çocuklarda birinci basamaktedavide, Erişkin hastalarda rezeke edilemeyen ve/veya metastatik malign C-KIT reseptörütaşıyan gastrointestinal stromal tümör (GIST) hastalarında, Opere edilmiş, C-KIT reseptörü pozitif bulunan erişkin GIST (gastrointestinal stromaltümör) hastalarında AFIP* kriterlerine göre yüksek risk** taşıyanlarda adjuvantedavide üç yıl süre ile, Belge DoBelge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys Yeni tanı konulmuş Philadelphia kromozomu pozitif yetişkin ve pediyatrik akutlenfoblastik lösemi (Ph+ ALL) hastalarında klinik yararı gösterilmiş çoklu ajanlıkemoterapi şemaları ile kombine olarak remisyon indüksiyonu amacıyla, Relaps-refrakter Philadelphia kromozomu pozitif akut lenfoblastik lösemi (Ph+ALL)hastalarında klinik yararı gösterilmiş çoklu ajanlı kemoterapi şemaları ile kombineolarak remisyon indüksiyonu amacıyla, FIP1L1-PDGFRA füzyon geni laboratuvar incelemeleriyle gösterilen hipereozinofiliksendrom ve sistemik mastositoz hastalarında kullanılabilir. (* Armed Forces Institute of Pathology (AFIP) kriterleri bölüm 5.1 Farmakodinamik özellikler'de verilmiştir. **AFIP kriterlerine göre yüksek riskli grupların tanımı; 1- Mide yerleşimli alanlarda tümör büyüklüğü 6 cm'nin üzerinde olan ve mitotik indeksi 5'in üzerinde bulunanlar 2- Mide dışındaki yerleşimlerde 10 cm ve üzerinde tümör büyüklüğü olan ya da mitotik indeksi 5'in üzerindebulunanlar) 4.2. Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresiTedavi, hematolojik malign hastalıklar ve malign sarkomlar bulunan hastaların tedavisinde deneyimi olan bir doktor tarafından başlatılmalıdır. Tedavi, hasta yarar sağladığı sürece devam ettirilmelidir. Kronik Miyeloid Lösemi de (KML) DozajNİBTU'nun önerilen dozu, kronik fazdaki erişkin KML hastaları için 400 mg/gündür. Kronik faz KML, aşağıdaki tüm kriterlerin karşılandığı durum olarak tanımlanır: kanda ve kemikiliğinde blast < %15, periferik kan bazofilleri < %20, trombosit > 100 x 109/L. NİBTU'nunönerilen dozu, akselere fazdaki erişkin hastalar için 600 mg/gündür. Akselere faz,aşağıdakilerden herhangi birinin varlığı olarak tanımlanır: kanda ve kemik iliğinde blast > %15fakat < %30, kanda ve kemik iliğinde blast artı promiyelosit > %30 (< %30 blasta neden olur), periferik kan bazofilleri > %20, trombosit <100 x 109/L (tedaviden ilişkisiz olarak).NİBTU'nun önerilen dozu, blast krizindeki erişkin hastalar için 600 mg/gündür. Blast krizi,kanda veya kemik iliğinde ~ %30 blast ya da hepatosplenomegali harici bir ekstramedüllerhastalık olarak tanımlanır. İlaca bağlı oluşan ciddi advers etki ve ağır lösemiyle ilişkili nötropeni veya trombositopeni gelişmemiş olması koşuluyla, hastalığın ilerlemesi (herhangi bir zamanda), en az 3 aylıktedaviden sonra tatmin edici bir hematolojik yanıt alınamaması, 12 aylık tedaviye rağmensitogenetik cevap elde edilmemesi veya daha önce elde edilmiş olan hematolojik ve/veyasitogenetik yanıtın kaybolması gibi durumlarda; kronik fazda hastalık bulunanlarda dozun 400mg'dan 600 mg'a yükseltilmesi, ya da hızlanmış faz veya blast krizi bulunan hastalarda dadozun 600 mg'dan maksimum 800 mg günlük doza yükseltilmesi düşünülebilir. Philedelphia kromozomu pozitif, akut lenfoblastik lösemide (Ph+ ALL) dozajPh+ ALL hastalarında önerilen NİBTU dozajı, remisyon indüksiyon kemoterapi şemaları çerçevesinde belirlenir. Belge Do Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebysHipereozinofilik sendrom ve sistemik mastositozda dozajYetişkin hipereozinofilik sendrom ve sistemik mastositoz hastalarında önerilen NİBTU dozajı, günde 100 mg'dır. Yanıtsız hallerde 400 mg'a dek çıkılabilir. Bu doz aşılamaz. Gastrointestinal Stromal Tümörlerde (GIST) dozajYetişkin, rezeke edilemeyen ve/veya metastatik malign GIST hastalarında önerilen NİBTU dozajı, günde 400 mg'dır. Değerlendirmelerin tedaviye yetersiz yanıtı ortaya koymaları durumunda, advers ilaç reaksiyonları göstermeyen hastalarda dozun 400 mg'dan 600 mg veya 800 mg'a yükseltilmesidüşünülebilir. GIST rezeksiyonunu takiben yetişkin hastaların adjuvan tedavisinde önerilen NİBTU dozu 400mg/gün'dür. Önerilen tedavi süresi 36 aydır. Adjuvan tedavi ortamında, NİBTU ileoptimum tedavi süresi bilinmemektedir. Advers reaksiyonlar için doz ayarlamalarıHematolojik olmayan advers reaksiyonlarNİBTU kullanıldığında eğer ciddi hematolojik olmayan advers reaksiyon gelişirse, tedavi bu olay ortadan kalkıncaya kadar durdurulmalıdır. Daha sonra, olayın ilk ciddiyetine göredeğişecek şekilde tedavi devam ettirilir. Eğer bilirubin, normal sınırın üst limitini (NSÜL) 3 kattan fazla aşacak şekilde yükselirse ya da karaciğer transaminazlarında NSÜL değerinin 5 katından fazla artış olursa, NİBTU, bilirubindüzeyleri < 1,5 x NSÜ^ ve transaminaz düzeyleri < 2,5 x NSÜL seviyesine ininceye kadardurdurulmalı ve daha sonra da azaltılmış günlük dozlarla devam ettirilmelidir. Yetişkinlerdedoz 400 mg'dan 300 mg'a veya 600 mg'dan 400 mg'a veya 800 mg'dan 600 mg'a, çocuklardaise 260 mg/m2/gün'den 200 mg/m2/gün'e veya 340 mg/m2/gün'den 260 mg/m2/gün'edüşürülmelidir. Hematolojik advers reaksiyonlarAğır nötropeni ve trombositopeni geliştiği takdirde dozun azaltılması ya da tedavinin kesilmesi aşağıdaki tabloda belirtildiği şekilde düzenlenmelidir.
Uygulama şekli:Reçetedeki doz, gastrointestinal etkileri en aza indirmek için yemek sırasında ve büyük bir bardak suyla yutulmalıdır. Günde 400 veya 600 miligramlık dozlar bir defada. 800 miligramlıkise her birinde 400'er miligram olmak üzere sabah ve akşam iki bölümde alınmalıdır. Film kaplı tabletleri yutamayan hastalarda tablet, bir bardak suda veya elma suyunda dağıtılabilir. İhtiyaç duyulan sayıda tablet, uygun hacimde içeceğin (100 miligramlık tablet içinyaklaşık 50, 400 miligramlık tablet için yaklaşık 200 mL) içerisine konarak bir kaşıklakarıştırılır. Meydana gelen süspansiyon, tablet(ler)in tam olarak dağılmasından sonra derhaliçilmelidir. Özel popülasyonlara ilişkin ek bilgiler:Karaciğer yetmezliği:İmatinib, temel olarak karaciğer yoluyla metabolize olur. Hafif, orta şiddette veya şiddetli karaciğer fonksiyon bozukluğu olan hastalara, önerilen minimal doz olan günde 400 mgverilmelidir. Bu doz, tolere edilemediği takdirde azaltılabilir (bkz. bölüm 4.4 Özel kullanımuyarıları ve önlemleri, 4.8 İstenmeyen etkiler, 5.1 Farmakodinamik özellikler ve 5.2Farmakokinetik özellikler). Böbrek yetmezliği:İmatinib ve metabolitleri böbrek yoluyla önemli miktarda atılmazlar. Böbrek yetmezliği olan veya diyaliz uygulanan hastalara böbrek bozukluğu başlangıç dozu olarak, önerilen minimumdoz, günlük 400 mg verilebilir. Bununla birlikte, bu hastalarda dikkatli olunması önerilir. Tolereedilememesi halinde doz azaltılabilir ya da etki görülmemesi halinde doz artırabilir (bkz 4.4Özel kullanım uyarıları ve önlemleri). Belge Do Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebysPediyatrik popülasyon:NİBTU'nun KML endikasyonunda 2 yaşın altındaki çocuklarda ve Ph+ ALL endikasyonunda 1 yaşın altındaki çocuklarda kullanımıyla ilgili herhangi bir deneyim bulunmamaktadır.NİBTU'nun diğer endikasyonlarda çocuklarda kullanılması konusundaki deneyim çoksınırlıdır. İmatinibin 18 yaşın altındaki MDS/MPD, DFSP, GIST ve HES/CEL hastası çocuklardaki güvenliliği ve etkililiği klinik çalışmalarda belirlenmemiştir. Halihazırda mevcut yayımlanmışveriler bölüm 5.1'de özetlenmektedir fakat pozoloji ile ilişkili bir öneride bulunulamamaktadır(bkz. bölüm 5.1. Farmakodinamik özellikler). Çocuklarda doz uygulaması vücut yüzey alanını (mg/m2) temel almalıdır. Kronik faz ve ilerlemiş faz KML'si ve Ph+ A^L'si olan çocuklar için 340 mg/m2 günlük doz önerilmektedir(toplam doz günde 600 mg'ı geçmemelidir). Tedavi KML ve Ph+ ALL'de günde bir kere dozuygulaması yoluyla verilebilir. KML'de alternatif olarak günlük doz iki uygulamayabölünebilir - sabah bir ve akşam bir (bkz. bölüm 5.1. Farmakodinamik özellikler). Geriyatrik popülasyon:Yaşlılarda spesifik olarak imatinib farmakokinetiği araştırılmamıştır. Katılan hastaların % 20'sinden fazlasının 65 ve daha yukarı yaşlarda olduğu klinik çalışmalarda, yetişkin hastalarda yaşla ilişkili anlamlı farmakokinetik farklılıklargözlenmemiştir. Yaşlılarda, özel bir doz önerisi gerekli değildir. 4.3. KontrendikasyonlarAktif maddeye veya eksipiyanlardan herhangi birine karşı aşırı duyarlılık. 4.4. Özel kullanım uyarıları ve önlemleriNİBTU, başka ilaçlarla eşzamanlı olarak kullanıldığında önemli ilaç etkileşimleri görülme potansiyeli bulunmaktadır. NİBTU, proteaz inhibitörleri, azol antifungaller, belirli makrolitler(bkz. bölüm 4.5), dar terapötik pencereye sahip CYP3A4 substratları (örn. siklosporin, pimozid,takrolimus, sirolimus, ergotamin, diergotamin, fentanil, alfentanil, terfenadin, bortezomib,dosetaksel, kinidin) veya varfarin ve diğer kumarin türevleri ile birlikte verildiğinde dikkatliolunmalıdır (bkz. bölüm 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri). İmatinib ve CYP3A4 enzimini indükleyen tıbbi ürünlerin (örn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital veya Hypericum perforatum [Sarı kantaron]) eşzamanlıkullanımı, NİBTU maruziyetini önemli ölçüde azaltarak terapötik başarısızlık riskini artırabilir.Bu nedenle kuvvetli CYP3A4 indükleyicilerinin ve imatinibin eşzamanlı uygulamasındankaçınılmalıdır (bkz. bölüm 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri). Hipotiroidizm: NİBTU tedavisi sırasında levotiroksin replasmanı yapılan tiroidektomi hastalarında klinik hipotiroidizm olguları bildirilmiştir. Bu , tür , hastalarda tiroid stimule edici hormon (TSH) ^ Bu belge^^Belge düzeyleri yakından izlenmelidir. Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebysHepatotoksisite: NİBTU temel olarak karaciğerde metabolize olur ve atılımın yalnızca %13'ü böbrekler aracılığıyladır. Karaciğer disfonksiyonu (hafif, orta şiddette ve şiddetli) olan hastalarda,periferik kan sayımları ve karaciğer enzimleri dikkatli bir şekilde izlenmelidir (bkz 4.2 Pozolojive uygulama şekli, 4.8 İstenmeyen etkiler, 5.1 Farmakodinamik özellikler, 5.2 Farmakokinetiközellikler). GIST hastalarında karaciğer yetmezliğine sebebiyet verebilecek karaciğermetaztazları görülmesi olasıdır. İmatinib ile karaciğer yetmezliği ve hepatik nekroz dahil karaciğer hasarı vakaları gözlenmiştir. İmatinib, yüksek doz kemoterapi rejimleri ile kombine edildiğinde ciddi hepatik reaksiyonlardabir artış bildirilmiştir. İmatinib, yüksek doz kemoterapi kürleriyle birlikte kullanıldığındatransaminaz ve bilirubin düzeylerinin yükselmesi şeklinde, geçici karaciğer toksisitesigörülmüştür. Buna ilave olarak yaygın olmayan akut karaciğer yetmezliği rapor edilmiştir.İmatinib, karaciğer fonksiyon bozukluğu potansiyeli olan kemoterapi kürleriyle birliktekullanılacaksa, karaciğer fonksiyonlarının izlenmesi tavsiye edilir (bkz 4.8 İstenmeyen etkiler). Sıvı retansiyonu: İmatinib alan yeni tanı konulmuş KML hastalarının yaklaşık % 2,5'inde ciddi sıvı retansiyonu (plevral efüzyon, ödem, pulmoner ödem, asit, yüzeysel ödem) ortaya çıktığı bildirilmiştir. Bunedenle, hastalarda düzenli aralıklarla kilo kontrolü önerilir. Beklenmedik, ani bir kilo artışıdikkatli araştırılmalı ve gerektiğinde uygun destek tedavisi uygulanmalı ve terapötik önlemleralınmalıdır. Klinik çalışmalarda, yaşlı hastalarda ve daha önceden kardiyak hastalık hikayesibulunanlarda bu olayların insidanslarının arttığı saptanmıştır. Kardiyak disfonksiyonu olanhastalarda dikkatli olunmalıdır. Kalp hastalığı ya da böbrek yetmezliği olan hastalar: Kalp hastalığı, kalp yetmezliği açısından risk faktörleri bulunan veya böbrek yetmezliği hikayesi olan hastalar dikkatlice takip edilmeli, kalp veya böbrek yetmezliğini düşündürenbelirti ve semptomları olan her hasta değerlendirilmeli ve tedavi edilmelidir. Miyokardiyum içinde hipereozinofili sendromu (HES) hücrelerinin gizli sızdırmasının görüldüğü hastalarda izole kardiyojenik şok/sol ventrikül disfonksiyonu olguları, imatinibtedavisine başlanmasıyla beraber oluşan HES hücre degranülasyonu ile ilişkilendirilmiştir. Budurumun sistemik steroidler kullanılarak, dolaşımı destekleyen önlemler alarak ve imatinibtedavisini geçici olarak durdurarak düzeltilebileceği bildirilmiştir. Yaygın olmayan kardiyakyan etkiler bildirildiği için, HES/CEL (kronik eozinofilik lösemi) popülasyonunda imatinibtedavisine başlamadan önce dikkatli bir yarar/zarar (risk) değerlendirmesi yapılmalıdır. Miyelodisplastik/miyeloproliferatif hastalıklar (MDS/MPD) ve sistemik mastositoz yüksek eozinofil düzeyleri ile ilişkili olabilir. Bu nedenle, eozinofil düzeylerinin yüksek olduğuMDS/MPD vakalarında, sistemik mastositoz (SM) vakalarında ve HES vakalarında imatinibuygulanmadan önce kardiyoloji uzmanı tarafından değerlendirme yapılmalı, ekokardiyografikinceleme yapılmalı ve serum troponin düzeyleri ölçülmelidir. Bunlardan birinde anormalliktespit edilirse kardiyoloji uzmanı ile beraber takip edilmeli ve tedavi başlangıcında imatinible Gastrointestinal kanama: Rezeke edilemeyen ve/veya metastatik GIST'li hastalarda yürütülen bir çalışmada gerek gastrointestinal gerekse tümör içi hemorajiler bildirilmiştir (bkz. bölüm 4.8 İstenmeyen etkiler).Eldeki verilere dayanılarak, GIST'li hastaları her iki hemoraji tipi açısından daha yüksek riskaltına sokan herhangi bir predispozan faktör tanımlanmamıştır (örn. tümör büyüklüğü, tümöryeri, pıhtılaşma bozuklukları). Vaskülarite artışı ve kanamaya yatkınlıkta artış, GIST'indoğasında yer aldığından ve hastalığın klinik seyrinin parçası olduğundan, tüm hastalardahemoraji izlemi ve kontrolüne yönelik standart uygulamalar ve prosedürler uygulanmalıdır. Ayrıca, KML, ALL ve diğer hastalıkları olan hastalarda pazarlama sonrası deneyimde nadir bir gastrointestinal hemoraji nedeni olarak gastrik antral vasküler ektazi (GAVE) bildirilmiştir(bkz. Bölüm 4.8 İstenmeyen etkiler). Gerektiğinde, NİBTU tedavisinin bırakılmasıdüşünülmelidir. Tümör lizis sendromu: TLS meydana gelme olasılığı nedeniyle, NİBTU başlatılmadan önce klinik açıdan anlamlı dehidrasyonun düzeltilmesi ve yüksek ürik asit düzeylerinin tedavisi önerilmektedir (bkz.Bölüm 4.8 İstenmeyen etkiler). Hepatit B reaktivasyonu: Hepatit B virüsü (HBV) kronik taşıyıcısı olan hastalarda, BCR-ABL tirozin kinaz inhibitörleri ile tedavi sonrası, Hepatit B reaktivasyonu ortaya çıkmıştır. Bazı vakalar, karaciğer nakli veyaölüme sebep olan akut karaciğer yetmezliği veya fulminan hepatit ile sonuçlanır. NİBTU tedavisine başlanmadan önce, hastalar HBV enfeksiyonu açısından test edilmelidir. Pozitif HBV serolojisine sahip (aktif hastalığı olanlar dahil) ve tedavi sırasında HBVenfeksiyonu için pozitif test sonucu veren hastalarda, tedavi başlatılmadan önce karaciğerhastalığı ve HBV tedavisi konusunda uzman hekimlere danışılmalıdır. NİBTU ile tedaviyeihtiyaç duyan HBV taşıyıcıları, tedavi boyunca ve tedavi sonlandırıldıktan sonra birkaç ayboyunca aktif HBV enfeksiyonu bulgu ve belirtileri için yakından izlenmelidir (bkz. Bölüm4.8). Fototoksisite: NİBTU tedavisi ile ilişkili fototoksisite riski nedeniyle doğrudan güneş ışığına maruziyetten kaçınılmalı ya da maruziyet en aza indirilmelidir. Hastalara, koruyucu kıyafetler ya da yüksekgüneş koruma faktörüne (SPF) sahip güneş kremlerinin kullanımı gibi önlemler almalarısöylenmelidir. Trombotik mikroanjiyopati: BCR-ABL tirozin kinaz inhibitörleri (TKI'lar), NİBTU için bireysel vaka raporları dahil olmak üzere trombotik mikroanjiyopati (TMA) ile ilişkilendirilmiştir (bkz. Bölüm 4.8). Eğer NİBTUalan bir hastada TMA ile ilişkili laboratuar ya da klinik bulgular meydana gelirse, tedavibırakılmalı ve ADAMTS13 .aktivitesi ve anti-ADAMTS13-antikorunun belirlenmesi dahil Belge DowTMAYi^nnkapsHmlı3biıtJ5değerlendinMe yaprlmalı^^.//EğeruridüşüktrADAMTS^13aktivitesi ile birlikte anti-ADAMTS13-antikoru yükselmişse, NİBTU tedavisi yeniden başlatılmamalıdır. Laboratuvar testleri: NİBTU ile tedavi sırasında düzenli olarak tam kan sayımları yapılmalıdır. KML hastalarında NİBTU tedavisine, nötropeni ya da trombositopeni eşlik etmiştir. Bununla birlikte, busitopenilerin ortaya çıkışı, hastalığın tedavi edildiği evreye bağlıdır ve kronik fazda KMLbulunan hastalarla karşılaştırıldığında, hızlanmış fazda KML ya da blast krizinde bulunanhastalarda daha sık olmaktadır. Bu durumda, 4.2 Pozoloji ve uygulama şekli bölümündeönerildiği gibi imatinib tedavisi kesilebilir ya da dozu azaltılabilir. NİBTU alan hastalarda karaciğer fonksiyonu (transaminazlar, bilirubin, alkalen fosfataz) düzenli olarak takip edilmelidir. Böbrek fonksiyonu bozuk olan hastalarda, imatinib plazma maruziyetinin, böbrek fonksiyonu normal olan hastalara kıyasla daha yüksek olduğu görülmektedir; bunun olası nedeni imatinibibağlanan bir protein olan alfa-asit glikoproteinin (AGP) plazma düzeylerinin bu hastalarda dahayüksek olmasıdır. Böbrek bozukluğu olan hastalarda en düşük başlangıç dozu verilmelidir.Şiddetli böbrek bozukluğu olan hastalar dikkatle tedavi edilmelidir. Doz, tolere edilmiyorsaazaltılabilir (bkz. bölüm 4.2 ve 5.2). Uzun süreli imatinib tedavi, böbrek fonksiyonunda klinik olarak anlamlı azalma ile ilişkili olabilir. Bu nedenle imatinib tedavisine başlanmadan önce böbrek fonksiyonu değerlendirilmelive tedavi sırasında yakından izlenmeli, böbrek fonksiyon bozukluğu açısından risk faktörlerigösteren hastalara özellikle dikkat edilmelidir. Böbrek fonksiyon bozukluğu gözlenirse,standart tedavi kılavuzları uyarınca uygun kontrol ve tedavi reçete edilmelidir. Pediyatrik popülasyonİmatinib alan çocuklarda ve ergenlik öncesi çocuklarda görülen büyüme geriliğine ilişkin vaka raporları alınmıştır. KML pediatrik popülasyonundaki gözlemsel bir çalışmada iki küçük altkümede pubertal durum veya cinsiyet fark etmeksizin medyan boy standart sapma skorlarında12 ve 24 ay sonra istatistiksel olarak anlamlı (fakat klinik anlamlılığı belirsiz) bir azalmabildirilmiştir. İmatinib tedavisi görmekte olan çocuklarda büyümenin yakından izlenmesiönerilir (bkz. Bölüm 4.8). 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriİmatinibin plazma konsantrasyonlarını değiştiren ilaçlar İmatinibin plazma konsantrasyonlarını arttırabilen ilaçlar: Sitokrom P450 izoenzimlerinden CYP3A4 aktivitesini inhibe eden maddeler (örn. indinavir, lopinavir/ritonavir, ritonavir, sakinavir, telaprevir, nelfinavir ve boseprevir gibi proteazinhibitörleri; ketokonazol, itrakonazol, posakonazol ve varikonazol gibi azol antifungal ajanlar;eritromisin, klaritromisin ve telitromisin gibi belirli makrolidler) metabolizmayı azaltabilir veimatinib konsantrasyonlarını arttırabilirler. Sağlıklı deneklere tek doz ketokonazol (birCY^3A4 inhibitörü) ile birlikte uygulandığında, imatinibe maruz kalma durumunda anlamlı birartış ortaya çıkmıştır (imatinibin ortalama Cmaks,ve EAA değerleri sırasıyla %26 ve %40 İmatinibin plazma konsantrasyonlarını azaltabilen ilaçlar: CYP3A4 aktivitesini indükleyen maddeler (örn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital, fosfenitoin, pirimidon ya da St. John's Wort olarak da bilinen Hypericum perforatumile eşzamanlı uygulama NİBTU'ya maruz kalmayı belirgin şekildeazaltabilir ve bu durum tedavinin başarısız olma riskini arttırabilir. Tedavi öncesi verilen birdenfazla 600 mg rifampisin dozunun ardından tek bir 400 mg NİBTU dozunun uygulanması,Cmaks ve EAA(0-ro) değerlerinde, rifampisin tedavisinin olmadığı durumdaki ilgili değerlerinen az %54 ve %74'ü oranında düşüşe neden olmuştur. İmatinib ile tedavi edilen malign gliomalıhastalarda karbamazepin, okskarbazepin ve fenitoin gibi enzim indükleyici antiepileptik ilaçlar(EIAED'ler) alırken benzer sonuçlar gözlenmiştir. İmatinib için plazma EAA, EIAEDkullanmayan hastalara kıyasla %73 oranında azalmıştır. Rifampisin veya diğer güçlü CYP3A4indükleyicileri ile imatinibin birlikte kullanımından kaçınılmalıdır.İmatinib ile plazma konsantrasyonu değişebilen ilaçlar: İmatinib, simvastatinin (CYP3A4 substratı) ortalama Cmaks ve EAA değerlerini sırasıyla 2 ve 3,5 kat arttırmaktadır ve bu durum CYP3A4'ün imatinib tarafından inhibe edildiğinigöstermektedir. Bu nedenle imatinib, dar bir terapötik pencereye sahip CYP3A4 substratlarıyla(örn. siklosporin, pimozid, takrolimus, sirolimus, ergotamin, diergotamin, fentanil, alfentanil,terfenadin, bortezomib, dosetaksel, kinidin) birlikte uygulandığında dikkatli olunmalıdır.İmatinib, CYP3A4 tarafından metabolize edilen diğer ilaçların da plazma konsantrasyonunuartırabilir (örn. triazolo-benzodiazepinler, dihidropiridin kalsiyum kanal blokörleri, bazı HMG-KoA redüktaz inhibitörleri, örn. statinler, vs.). İmatinib kullanımı ile birlikte bilinen artmış kanama riski nedeniyle (örn. hemoraji), anti-koagülasyon gerektiren hastalar varfarin gibi kumarin türevleri yerine düşük molekül ağırlıklı ya da standart heparin ile tedavi edilmelidir. In vitroortamda imatinib, CYP3A4 aktivitesini etkileyen konsantrasyonların benzeri konsantrasyonlarda sitokrom P450 izoenzimlerinden CYP2D6 aktivitesini de inhibeetmektedir. Günde iki kez 400 mg dozda uygulanan imatinibin CYP2D6-aracılı metoprololmetabolizması üzerinde zayıf bir inhibitör etkisi vardır; metoprolol Cmaks ve EAA değerleriyaklaşık %23 kadar artar (%90 GA [1,16-1,30]). İmatinib, CYP2D6 substratları ile bir aradauygulandığında doz ayarlamalarının gerekli olmadığı görülmektedir ancak metoprolol gibi darterapötik pencereye sahip CYP2D6 substratları ile dikkatli olunması tavsiye edilir. Metoprololile tedavi edilen hastalarda klinik izlem göz önünde bulundurulmalıdır.İmatinib in vitroin vivokoşullarda, 400 mg imatinib ve 1000 mg parasetamoluygulamasının ardından görülmemiştir. Daha yüksek imatinib ve parasetamol dozlarıçalışılmamıştır. Bu nedenle yüksek dozda NİBTU ve parasetamol eşzamanlı uygulanırkendikkatli olunmalıdır.Levotiroksin kullanan tiroidektomi , hastalarında NİBTU eşzamanlı kullanıldığında önlemleri). Bu nedenle dikkat önerilir. Bununla birlikte gözlenen etkileşimin mekanizması halen bilinmemektedir. Tüm Ph+ ALL hastalarında kemoterapiyle eşzamanlı olarak imatinib uygulanmasıyla ilgili klinik deneyim vardır (bkz Bölüm 5.1 Farmakodinamik özellikler), ancak imatinib vekemoterapi rejimleri arasındaki ilaç-ilaç etkileşimleri iyi tanımlanmamıştır. İmatinibin adversetkileri, örn. hepatotoksisite, miyelosupresyon ya da diğerleri artış gösterebilir ve L-asparaginazile eşzamanlı kullanımın hepatatoksite artışıyla ilişkili olabileceği bildirilmiştir (bkz. Bölüm 4.8İstenmeyen etkiler). Bu nedenle, NİBTU'nun kombinasyonda kullanımı özel dikkatgerektirmektedir. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin klinik etkileşim çalışması yürütülmemiştir. Pediyatrik popülasyonPediyatrik popülasyona ilişkin klinik etkileşim çalışması yürütülmemiştir. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi D'dir. Çocuk doğurma potansiyeli bulunan kadınlar/ Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli bulunan kadınlara tedavi sırasında ve tedavi durdurulduktan sonra en az 15 gün boyunca etkili bir kontrasepsiyon uygulamaları önerilmelidir. Gebelik dönemiImatinibin gebelik ve/veya fetus/yeni doğan üzerinde zararlı farmakolojik etkileri bulunmaktadır. NİBTU, gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Hayvanlarüzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. 5.3 Kliniköncesi güvenlilik verileri). İmatinibin gebe kadınlarda kullanımına ilişkin klinik çalışmalarmevcut değildir. İmatinib alan kadınlarda spontan düşükler ve bebekte konjenital anomalilerleilgili pazarlama sonrası raporlar mevcuttur. NİBTU, beklenen fayda, potansiyel riske ağırbasmadığı sürece gebelik sırasında kullanılmamalıdır. Gebelik sırasında kullanılmasıdurumunda, hastaya fötüs üzerindeki potansiyel riskleri hakkında bilgi verilmelidir. Laktasyon dönemiİmatinibin insan sütüne geçişi hakkında sınırlı bilgi vardır. Emziren iki kadında yapılan çalışmalar hem imatinibin hem de aktif metabolitinin anne sütüne geçebileceğini ortayakoymuştur. Tek bir hastada incelenen süt/plazma oranı, imatinib için 0,5, metabolit için ise 0,9olarak belirlenerek metabolitin süte daha fazla geçtiğini düşündürmüştür. İmatinib vemetabolitinin toplam konsantrasyonu ve bebeklerin maksimum günlük süt alımıdüşünüldüğünde, toplam maruziyetin düşük olması beklenir (bir terapötik dozun ~%10'u).Bununla birlikte, bebeğin imatinibe düşük dozlarda maruz kalmasının etkileri bilinmediğinden, Üreme yeteneği / FertiliteYapılan klinik dışı çalışmalarda, üreme parametreleri üzerinde etkiler gözlenmiş olsa da dişi ve erkek farelerin fertiliteleri etkilenmemiştir (bkz. Bölüm 5.3). İmatinib alan hastalarda ilacınfertilite ve gametogenez üzerindeki etkileri ile ilgili klinik çalışmalar yapılmamıştır. NİBTUtedavisi gören ve fertilite konusunda endişe duyan hastalar hekimlerine danışmalıdır. 4.7. Araç ve makina kullanmaya etkisiHastalara imatinib ile tedavi sırasında baş dönmesi, somnolans ya da bulanık görme gibi istenmeyen etkiler yaşayabilecekleri bildirilmelidir. Bu nedenle, araba ya da araç kullanırkendikkatli olunması önerilmelidir. 4.8. İstenmeyen etkilerİleri aşamalarda maligniteleri olan hastalarda, altta yatan hastalık, progresyon ve sayısız tıbbi ürünün eşzamanlı uygulanması ile bağlantılı çeşitli semptomlar nedeniyle advers reaksiyonlarınnedensellik ilişkisinin değerlendirilmesini zorlaştıran sayısız karmaşıklaştırıcı tıbbi durummevcut olabilir. KML klinik çalışmalarında ilaçla ilişkili advers reaksiyonlar nedeniyle ilacın kesilmesi durumu, yeni tanı konan hastaların %2,4'ünde, interferon tedavisinin başarısız olmasından sonra geçkronik fazdaki hastaların %4'ünde, interferon tedavisinin başarısız olmasından sonra hızlanmışfazdaki hastaların %4'ünde ve interferon tedavisinin başarısız olmasından sonra blastkrizindeki hastaların %5'inde gözlenmiştir. GIST çalışmasında ilaç, hastaların %4'üne ilaçlailişkili advers reaksiyonlar nedeniyle kesilmiştir. İki istisna haricinde advers reaksiyonlar tüm endikasyonlarda benzer olmuştur. GIST ile karşılaştırıldığında KML hastalarında daha fazla miyelosüpresyon görülmüştür, bu durumolasılıkla altta yatan hastalık ile ilişkilidir. Rezekte edilemeyen ve/veya metastatik GIST'lihastalarda yürütülen bir çalışmada 7 (%5) hasta CTC derece 3/4 GI kanamalar (3 hasta), tümöriçi kanamalar (3 hasta) ya da ikisini birden (1 hasta) yaşamıştır. GI tümör bölgeleri GIkanamaların kaynağı olmuş olabilir (bkz. bölüm 4.4). GI ve tümör kanamaları ciddi ve bazenölümcül olabilmektedir. Her iki endikasyonda en sık bildirilen (>%10) ilaçla ilişkili adversreaksiyonlar hafif bulantı, kusma, ishal, abdominal ağrı, yorgunluk, kas ağrısı, kas krampları vedöküntü olmuştur. Yüzeysel ödemler tüm çalışmalarda yaygın bir bulgu olmuş ve temeldeperiorbital ya da alt uzuv ödemleri şeklinde tarif edilmiştir. Bununla birlikte, bu ödemlernadiren şiddetli olmuş ve diüretiklerle, diğer destekleyici önlemlerle veya imatinib dozuazaltılarak kontrol edilebilmiştir. İmatinib Ph+ ALL hastalarında yüksek doz kemoterapi ile kombine edildiğine transaminaz yükselmesi ve hiperbilirubinemi formunda geçici karaciğer toksisitesi gözlenmiştir. Sınırlıgüvenlilik veritabanı göz önünde bulundurulduğunda, çocuklarda şu ana kadar bildirilen adversolaylar, erişkin Ph+ ALL hastalarında bilinen güvenlilik profili ile uyumludur. Ph+ ALL hastasıçocuklardaki güvenlilik veritabanı çok .sınırlı . olmakla birlikte herhangi bir yeni güvenlilik ^endişesi tanımlanmamıştır. Plevral efüzyon, assit, pulmoner ödem ve yüzeysel ödemin eşlik ettiği ya da etmediği hızlı kilo artışı gibi çeşitli advers reaksiyonlar kolektif olarak sıvı tutulumu şeklinde tarif edilebilir. Bureaksiyonlar genellikle imatinib tedavi geçici olarak durdurularak ve diüretiklerle ya da diğeruygun destekleyici bakım önlemleriyle kontrol edilebilmektedir. Diğer yandan, bu reaksiyonların bazıları şiddetli ya da yaşamı tehdit edici olabilmektedir ve blast krizi olan çeşitli hastalar plevral efüzyon, konjestif kalp yetmezliği ve böbrekyetmezliğinden oluşan kompleks bir klinik öykü ile yaşamlarını kaybetmiştir. Pediatrik klinikçalışmalarda özel bir güvenlilik bulgusu söz konusu olmamıştır. İstenmeyen etkiler MedDRA sistem organ sınıfına göre listelidir. Sıklıklar şu şekilde tanımlanmıştır: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan(>1/1000 ila<1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerdenhareketle tahmin edilemiyor). Aşağıda bildirilen advers reaksiyonlar ve sıklıkları, KML veGIST için yürütülen çalışmalara dayanmaktadır. Gözlenen advers reaksiyonlar Enfeksiyonlar ve enfestasyonlarYaygın olmayan: Herpes zoster, herpes simplex, nazofarenjit, pnömoni1, sinüzit, selülit, üst solunum yolu enfeksiyonu, influenza, idrar yolu enfeksiyonu, gastroenteritis, sepsisSeyrek: Fungal enfeksiyonBilinmiyor: Hepatit B reaktivasyonu* (Kist ve polipler de dahil olmak üzere) iyi huylu ve kötü huylu neoplazmalarSeyrek: Tümör lizis sendromu Bilinmiyor: Tümör kanaması/tümör nekrozu* Kan ve lenf sistemi hastalıklarıÇok yaygın: Nötropeni, trombositopeni, anemi Yaygın: Pansitopeni, febril nötropeni Yaygın olmayan: Trombositemi, lenfopeni, kemik iliği depresyonu, eozinofili, lenfadenopati Seyrek: Hemolitik anemi, trombotik mikroanjiyopati Bağışıklık sistemi hastalıkları:Bilinmiyor: Anafilaktik şok* Metabolizma ve beslenme hastalıklarıYaygın: Anoreksi Yaygın olmayan: Hipokalemi, iştah artışı, hipofosfatemi, iştah azalması, dehidrasyon, gut, hiperürisemi, hiperkalsemi, hiperglisemi, hiponatremiSeyrek: Hiperkalemi, hipomagnezemi Psikiyatrik hastalıklarYaygın olmayan: Depresyon, libido azalması, anksiyete Seyrek: Konfüzyon Sinir sistemi hastalıklarıÇok yaygın: Baş ağrısı2 Yaygın: Baş dönmesi-sersemlik, parestezi, tat duyusu bozuklukları, hipoestezi Yaygın olmayan: Migren, somnolans, senkop, periferik nöropati, bellek bozukluğu, siyatik, huzursuz ayak sendromu, tremor, beyin kanaması Seyrek: Kafa-içi basıncının artması, konvülziyon, optik nörit Bilinmiyor: Serebral ödem* Göz hastalıklarıYaygın: Göz kapağı ödemi, lakrimasyon artışı, konjunktiva kanaması, konjunktivit, göz kuruması, bulanık görme Yaygın olmayan: Göz tahrişi, göz ağrısı, orbita ödemi, sklera kanaması, retina kanaması, blefarit, maküla ödemi Seyrek: Katarakt, glokom, papilödem Bilinmiyor: Vitröz kanama* Kulak ve iç kulak hastalıklarıYaygın olmayan: Vertigo, kulak çınlaması, işitme kaybı Kardiyak hastalıkları3Yaygın olmayan: Palpitasyonlar, taşikardi, konjestif kalp yetmezliği , pulmoner ödem Seyrek: Aritmi, atriyal fibrilasyon, kardiyak arest, miyokart enfarktüsü, angina pektoris,perikardiyal efüzyonBilinmiyor: Perikardit*, kalp tamponadı* Vasküler hastalıkları4Yaygın: Al basması, kanama Yaygın olmayan: Hipertansiyon, hematom, subdural hematom, periferik soğukluk, hipotansiyon, Raynaud fenomeni Bilinmiyor: Tromboz/emboli* Solunum, göğüs bozuklukları ve mediastinal hastalıklarıYaygın: Dispne, burun kanaması, öksürük Yaygın olmayan: Plevral efüzyon5, faringolaringeal ağrı, farenjit Seyrek: Plevra ağrısı, pulmoner fibroz, pulmoner hipertansiyon, pulmoner kanama * Bilinmiyor: Akut respiratuvar yetmezlik11 , interstisyal akciğer hastalığı* Gastrointestinal hastalıklarıÇok yaygın: Bulantı, ishal, kusma, dispepsi, karın ağrısı6 Belge DoBelge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebysYaygın: Aşırı miktarda barğırsak gazları, karında gerilme, gastro-özofageal reflü, kabızlık, ağız kuruması, gastrit Yaygın olmayan: Stomatit, ağız ülserasyonu, gastrointestinal kanama7, geğirme, melena, özofajit, asit, gastrik ülser, kan kusma, dudak iltihabı, disfaji, pankreatitSeyrek: Kolit, ileus, enflamatuar barsak hastalığı Bilinmiyor: İleus/intestinal obstrüksiyon*, gastrointestinal perforasyon*, divertikülit*, gastrik antral vasküler ektazi (GAVE)* Hepato-bilier hastalıklarıYaygın: Karaciğer enzimlerinde artış Yaygın olmayan: Hiperbilirübinemi, hepatit, sarılık Seyrek: Karaciğer yetmezliği8, hepatik nekroz Deri ve deri altı doku hastalıklarıÇok yaygın: Periorbital ödem, dermatit/egzama/deri döküntüsü Yaygın: Kaşıntı, yüz ödemi, deride kuruma, eritem, alopesi, gece terlemeleri, ışığa duyarlılık reaksiyonu Yaygın olmayan: Püstüler döküntü, kontüzyon, terlemede artış, ürtiker, ekimoz, çürük eğiliminde artış, hipotrikoz, deride hipopigmentasyon, eksfoliyatif dermatit, tırnak kırılması,folikülit, peteşiler, psoriazis, purpura, deride hiperpigmentasyon, büllöz erupsiyonlarSeyrek: Akut febril nötrofilik dermatoz (Sweet's hastalığı), tırnakta renk kaybı, anjiyonörotiködem, veziküler döküntü, eritem multiform, lökositoklastik vaskülit, Stevens-Johnsonsendromu, akut jeneralize ekzantematöz püstülozis (AGEP) Bilinmiyor: Palmar-plantar eritrodisestezi sendromu (el-ayak sendromu)*, likenoid keratoz*, liken planuz*, toksik epidermal nekroliz*, eozinofili ve sistemik semptomlarla ilaç döküntüsü(DRESS sendromu)*, psödoporfiri* Kas-iskelet bozukluklar, bağ doku ve kemik hastalıklarıÇok yaygın: Kas spazmları ve krampları, miyalji9, artralji, kemik ağrısı10 da dahil olmak üzere kas-iskelet ağrıları Yaygın: Eklemlerde şişme Yaygın olmayan: Kaslarda ve eklemlerde sertlik Seyrek: Kas zayıflığı, artrit, rabdomiyoliz/miyopati Bilinmiyor: Avasküler nekroz/kalça osteonekrozu*, çocuklarda büyüme geriliği* Böbrek ve idrar yolu hastalıklarıYaygın olmayan: Böbrek ağrısı, hematüri, akut böbrek yetmezliği, idrar sıklığında artış Bilinmiyor: Kronik böbrek yetmezliği Üreme sistemi ve meme hastalıklarıYaygın olmayan: Jinekomasti, erektil disfonksiyon, menoraji, düzensiz menstrüasyon, cinsel disfonksiyon, meme başında ağrı, memelerde büyüme, skrotum ödemiÇok seyrek: Hemorajik korpus luteum, hemorajik over kisti Belge DoBelge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebysGenel bozukluklar ve uygulama bölgesine ilişkin hastalıklarıÇok yaygın: Sıvı retansiyonu ve ödem, yorgunluk Yaygın: Güçsüzlük, pireksi, anazarka, titreme nöbetleri, kaslarda sertlikler Yaygın olmayan: Göğüs ağrısı, keyifsizlik Laboratuvar bulgularıÇok yaygın: Vücut ağırlığı artışı Yaygın: Vücut ağırlığı azalması Yaygın olmayan: Kanda kreatinin düzeyinin yükselmesi, kandaki kreatin fosfokinaz düzeyinin yükselmesi, kandaki laktat dehidrojenaz düzeyinin yükselmesi, kanda alkalin fosfatazdüzeyinin yükselmesi Seyrek: Kanda amilaz düzeyinin yükselmesi *Bu tür reaksiyonlar, esas olarak imatinib ile edinilen pazarlama sonrası deneyimlerden rapor edilmiştir. Bu veriler, spontan vaka raporlarının yanı sıra devam eden çalışmalardan alınan ciddi advers olayları,genişletilmiş erişim programlarını, klinik farmakoloji çalışmalarını ve onaylanmamış endikasy onlardakeşif çalışmalarını içermektedir. Bu reaksiyonlar belirsiz büyüklükteki bir popülasyondanbildirildiğinden, sıklıklarını güvenilir bir şekilde tahmin etmek veya imatinib maruziyetiyle nedensel bir ilişki kurmak her zaman mümkün değildir. 1Transforme KML hastalarında ve GIST hastalarında en sık pnömoni bildirilmiştir. 2GIST hastalarında en sık baş ağrısı görülmüştür. 3Bir hasta yılı esasında, konjestif kalp yetmezliği de dahil olmak üzere kardiyak olaylar transforme KML hastalarında kronik KML hastalarından daha sık gözlemlenmiştir. 4GIST hastalarında en sık kızarma görülmüştür; GIST ve transforme KML (KML-AF ve KML-BK) hastalarında en sık görülen ise kanamadır (hematom, hemoraji). 5Plevral efüzyon GIST hastalarında ve transforme KML (KML- AF ve KML- BK) hastalarında kronik KML hastalarından daha yaygın olarak bildirilmiştir. 6/7Abdominal ağrı ve gastrointestinal kanama en sık GIST hastalarında görülmüştür. 8Bazı ölümcül hepatik yetmezlik ve hepatik nekroz vakaları bildirilmiştir. 9Pazarlama sonrasında, imatinib ile tedavi sırasında veya bırakılmasından sonra müsküloskeletal ağrı gözlenmiştir. 10KML hastalarında kas iskelet ağrısı ve ilişkili olaylar GIST hastalarından çok daha sık gözlemlenmiştir. 11ileri evrede hastalığı, ağır enfeksiyonları, şiddetli nötropenisi ve diğer ciddi eşlik eden rahatsızlıkları olan hastalarda fatal vakalar bildirilmiştir. Laboratuvar testi anormallikleri HematolojiKML'de başta nötropeni ve trombositopeni olmak üzere sitopeniler tüm çalışmaların devamlı bir bulgusu olmuş, > 750 mg gibi daha yüksek dozlarda daha sık oldukları düşünülmüştür (fazI çalışma). Bununla birlikte, sitopenilerin ortaya çıkışı, aynı zamanda açıkça hastalığın evresinede bağlı olmuştur. Sitopeniler, yeni tanı konulan KML vakalarında, diğer vakalara kıyasla dahaseyrektir. Evre 3 veya 4 nötropenilerin (ANC<1,0x109/L) ve trombositopenilerin (trombosit
ki laRıp
sıklığı, yeni tanı konulan kronik faz p Aare«^mtps://www.fumye.gov.ınsagTık-fıfcK-ebys
eki v <
st BelgeelgeKML vakalarındakinin 4-6 katıdır. Yeni tanı kronik faz KML vakalarında % 16,7 nötropeni ve % 8,9 trombositopeni görülürken, bu oranlar hızlanmış ve blastik fazda sırasıyla, % 59-64 ve% 44-63 olarak bildirilmiştir. Yeni tanı konulmuş olan kronik faz KML vakalarında evre 4nötropeni (ANC < 0,5x109/L) ve trombositopeni (trombosit sayısı<10x109/L), sırasıylayalnızca % 3,6 ve < %1 oranında görülmüştür. Nötropenik ve trombositopenik periyotlarınortalama süresi genellikle sırasıyla 2 ve 3. haftalar arasında ve 3 ve 4. haftalar arasında yeralmıştır. Bu olaylar, genellikle imatinib ile tedavinin dozu azaltılarak ya da tedavi kesilerekkontrol edilebilir, ancak bazı nadir vakalarda kalıcı olarak tedavinin bırakılmasına nedenolabilir. Pediyatrik KML hastalarında en sık gözlenen toksisiteler; nötropeni, trombositopenive anemi dahil olmak üzere 3 ya da 4. derece sitopeniler olmuştur. Bunlar genellikle ilk birkaçay içerisinde gerçekleşmektedir. Rezeke edilemeyen ya da metastatik malign GIST (çalışma B2222) bulunan hastalarda, sırasıyla hastaların % 5,4 ve % 0,7'sinde evre 3 ve 4 anemi bildirilmiştir ve bu durum en azındanbazı hastalarda gastrointestinal ya da intra-tümöral kanamayla ilişkili olabilir. Sırasıylahastaların % 7,5 ve %2,7'sinde evre 3 ve 4 nötropeni ve hastaların % 0,7'sinde evre 3trombositopeni görülmüştür. Hiçbir hastada evre 4 trombositopeni gelişmemiştir. Özellikletedavinin ilk 6 haftasında beyaz kan hücresi ve nötrofil sayılarında azalmalar ortaya çıkmış, budeğerler daha sonra nispeten sabit kalmıştır. BiyokimyaKML hastalarında transaminazlarda (<%5) ya da bilirubinde (< %1) ciddi artışlar olmuştur ve genellikle doz azaltılarak ya da kesilerek (bu epizodların ortalama süresi yaklaşık 1 haftaolmuştur) kontrol altına alınmıştır. KML hastaların % 1'inden azında karaciğer laboratuaranormallikleri nedeniyle tedavi sürekli olarak kesilmiştir. GIST hastalarının (çalışma B2222)%6,8'inde 3. veya 4. evre SGPT (serum glutamik piruvik transferaz); %4,8'inde 3. veya 4. evreSGOT (serum glutamik oksaloasetik transferaz) yükselmeleri kaydedilmiş; bilirübin düzeyiyükselen hastaların oranı %3'ün altında kalmıştır. Nadir sitolitik ve kolestatik hepatit ve karaciğer yetmezliği olguları söz konusu olmuştur; yüksek doz parasetamol kullanan bir hasta dahil olmak üzere bunların bazıları ölümlesonuçlanmıştır. Seçili advers reaksiyonların tanımlanmasıHepatit B reaktivasyonuBCR-ABL TKI'lerle ilişkili olarak hepatit B reaktivasyonu bildirilmiştir. Bazı vakalarda, karaciğer nakliyle veya ölümle sonuçlanan akut karaciğer yetmezliği veya fulminan hepatitortaya çıkmıştır (bkz. Bölüm 4.4). Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck. gov.tr;e-posta: [email protected],; tel: 0 800. .31.4 00 08; faks:0 312 218 35 99)^ '--Bu belge ^Belge Do Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys4.9. Doz aşımı ve tedavisiTerapötik dozlardan daha yüksek dozlarla deneyim sınırlıdır. İmatinib doz aşımı ile ilgili bireysel vakalar spontan olarak ve literatürde bildirilmiştir. Genellikle, bu vakalarda bildirilensonuçlar düzelme ya da iyileşme şeklinde olmuştur. Doz aşımı halinde, hasta gözlem altındatutulmalı ve uygun semptomatik tedavi uygulanmalıdır. Farklı doz aralıklarında bildirilen olaylar aşağıda verilmiştir: Erişkinlerde doz aşımı: 1200 ila 1600 mg (1 ila 10 gün arasında değişen sürelerle): Bulantı, kusma, diyare, döküntü, eritem, ödem, şişme, yorgunluk, kas spazmları, trombositopeni, pansitopeni, karın ağrısı, başağrısı, iştahta azalma. 1800 ila 3200 mg (6 gün boyunca günde 3200 mg'a kadar dozlar): Güçsüzlük, miyalji, CPK düzeyinde yükselme, bilirubin düzeyinde yükselme, gastrointestinal ağrı. 6400 mg (tek doz): Literatürde yer alan bir vakada, bulantı, kusma, karın ağrısı, pireksi, yüzde şişme, nötrofil sayısında azalma, transaminaz düzeylerinde yükselme görülen bir hastabildirilmiştir. 8 ila 10 g (tek doz): Kusma ve gastrointestinal ağrı bildirilmiştir. Pediyatrik doz aşımı: 400 mg'lık tek doza maruz kalan 3 yaşındaki bir erkek çocukta kusma, diyare ve anoreksi; 980 mg'lık tek doza maruz kalan 3 yaşındaki diğer bir erkek çocukta ise lökosit sayısında azalmave diyare görülmüştür. Doz aşımı durumunda hasta gözlemlenmeli ve uygun destek tedavisi verilmelidir. 5. Farmakolojik özellikler5.1. Farmakodinamik özellikler:Farmakoterapötik grup: Antineoplastik ajan, protein-tirozin kinaz inhibitörü, BCR-ABL tirozin kinaz inhibitörleriATC kodu: L01EA01 Etki mekanizması: İmatinib küçük bir molekül yapısına sahip bir protein-tirozin kinaz inhibitörüdür; Bcr-Abl tirozin kinaz (TK) aktivitesini ve birçok reseptör TK'yı kuvvetli bir şekilde inhibe etmektedir:KIT, c-KIT proto-onkogen tarafından kodlanan kök hücre faktörü (Stem cell factor - SCF)reseptörü, diskoidin etki bölgesine ait reseptörler (DDR1 ve DDR2), koloni uyarıcı faktörreseptörü (CSF-1R), trombosit kökenli büyüme faktörü (Platelet derived growth factor -PDGF) reseptörleri alfa ve beta (PDGFR-alfa ve PDGFR-beta). İmatinib aynı zamanda bureseptör kinazların aktivasyonunun aracılık ettiği hücresel olayları da inhibe edebilmektedir. Farmakodinamik etkiler: İmatinib, in vitro,in vi-yodüzeylerde kırılma noktalarının yoğunlaştığı bölge-Abelson (Bcr-Abl) tirozin^kinazı güçlü, bir şekilde inhibe eden bir protein-tirozin kinaz^ ^Bu belge, gü^nlr elektronik imza ile imzalanmıştır.^Belge odi^iıbitösüdüry BiieştkyB'öröAbb ıpoz^i^hüc^e dizilerinde,,phüadelphia\kramozomıpozitifcKMLve ALL hastalarının yeni lösemi hücrelerinde selektif olarak proliferasyonu inhibe etmekte ve apopitozisi uyarmaktadır. Bileşik, in vivoolarak, Bcr-Abl pozitif tümör hücreleri kullanılan hayvan modellerinde tek ajan olarak anti-tümör aktivite gösterir.İmatinib, aynı zamanda trombosit türevi büyüme faktörü (Platelet derived growth factor -PDGF) ve kök hücre faktörü (Stem cell factor - SCF), c-KIT için reseptör tirozin kinazların bir inhibitörüdür ve PDGF- ve SCF- tarafından yönlendirilen hücresel olayları inhibe eder. In vitroKronik Miyeloid Lösemide Klinik Çalışmalarİmatinibin etkinliği, bir bütün olarak elde edilen hematolojik ve sitogenetik yanıt oranlarını ve hastalıksız sağkalım süresini temel alır. Yeni tanı almış KML harici, hastalık ilişkilisemptomların iyileşmesi veya sağkalım süresinin artması gibi klinik faydaların olduğunugösteren kontrollü çalışma yoktur. İleri evre, blast veya hızlandırılmış faz hastalıkta Philadelphia kromozomu pozitif (Ph +) KML, diğer Ph + lösemiler veya kronik fazda KML'si olan fakat daha önce interferon-alfa (IFN)tedavide başarısız olunan hastalarda üç büyük, uluslararası, açık etiketli, kontrollü olmayan FazII çalışma yapılmıştır. Yeni tanı almış Ph + KML hastalarında büyük, açık etiketli, çokmerkezli, uluslararası, randomize bir Faz III çalışma yürütülmüştür. Ek olarak, iki Faz Içalışmada ve bir Faz II çalışmada çocuklar tedavi edilmiştir. Bütün klinik çalışmalarda hastaların %38-40'ının en az 60, %10-12'sinin en az 70 yaşında olduğu bildirilmiştir. Kronik faz, yeni tanı konulmuş:Bu faz III çalışmasında, imatinib monoterapisi, interferon-alfa (IFN) + sitarabin (ARA-C) kombinasyonuyla karşılaştırılmıştır. Yanıtsızlık (6 ayda tamhematolojik yanıt (CHR) olmaması, artan WBC, 24 ayda majör sitogenetik yanıt (MCyR)olmaması), yanıt kaybı (CHR veya MCyR kaybı) veya tedaviye şiddetli intolerans gösterenhastaların alternatif tedavi koluna geçmelerine izin verilmiştir. İmatinib grubundaki hastalardagünde 400 miligramlık doz kullanılmıştır. IFN grubundaki hastalar, 10 gün / ay boyuncasubkutan Ara-C 20 mg / m2 / gün ile kombinasyon halinde subkutan olarak 5 MIU / m2 / günhedef IFN dozu ile tedavi edilmiştir.Toplam 1106 (her grupta 553) hasta, randomize edilmiştir. İki kol arasında çalışma başlangıcı ^ ^^ Bu belge»Belge Do | ||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||
|
* pM<0,001, Fischer's exact test **Moleküler yanıt oranları erişilebilir verilere bağlıdır. Hematolojik yanıt kriterleri (bütün yanıtlar >4 hafta sonra doğrulanmalıdır):Kandaki lökosit sayısı < 10 x109/L, trombosit sayısı < 450 x109/L, miyelosit+metamiyelosit < %5; kanda blast hücresi veya promiyelosit yok, bazofiller <%20, kemik iliği dışında hastalık yok Sitogenetik yanıt kriterleri:tam (%0 Ph+ metafazlar), kısmi (%1-35), minör (%36-65) veya minimal (%66-95). Majör yanıt (%0-35), hem kısmi hem tam yanıtları içerir [1].Majör moleküler yanıt kriterleri:Gerçek-zaman kantitatif revers kriptaz polimeraz zincir reaksiyonuyla ölçülen Bcr-Abl transkriptlerinin periferik kanda, başlangıç düzeyine göre en az 3 log azalması. | ||||||||||||||||||||||||||||||
Birinci basamak tedavide tam hematolojik yanıt, majör sitogenetik yanıt ve tam sitogenetik yanıt oranları, son muayene tarihinde yanıtsızlıkların sansürlendiği Kaplan-Meier yaklaşımıkullanılarak hesaplanmıştır. Bu yaklaşım kullanıldığında, imatinib ile birinci basamak tedaviiçin hesaplanan kümülatif yanıt oranları 12 aylık tedaviden 84 aylık tedaviye şu şekilde düzelmegöstermiştir: THY %96,4'ten %98,4'e ve TSY %69,5'ten %87,2'ye.
7 yıllık takipte, imatinib grubunda 93 (%16,8) olay olmuştur: 37 (%6,7) hızlanmış faz/blastik kriz (AF/BK) ilerleme, 31 (%5,6) major sitogenetik yanıt (MSY) kaybı, 15 (%2,7) tamhematolojik yanıt (THY) kaybı ya da white blood cell (beyaz kan hücresi) (WBC) artışı ve 10(%1,8) KML ile ilişkisiz ölüm. Buna karşılık IFN+Ara-C grubunda 165 (%29,8) olay olmuş vebunların 130'u birinci seçenek IFN+Ara-C tedavisi sırasında meydana gelmiştir.
Tedavide geçen süre ile birlikte hızlandırılmış faza veya blast krizine yıllık progresyon oranı azalmış ve dördüncü ve beşinci yıllarda yıllık % 1'den az olmuştur. 84 ayda tahmin edilenprogresyonsuz sağkalım imatinib grubunda %81,2 ve kontrol grubunda %60,6 olmuştur(p < 0,001). İmatinib için herhangi bir türdeki yıllık progresyon oranları da zamanla azalmıştır.
İmatinib ve IFN+Ara-C gruplarında, sırasıyla, toplam 71 (%12,8) ve 85 (%15,4) hasta ölmüştür. 84 ayda randomize imatinib ve IFN+Ara-C gruplarında tahmin edilen genel sağkalım, sırasıyla%86,4 (83, 90) ve %83,3 (80, 87) düzeyindedir (p=0,073, log-rank testi). Bu olaya kadar geçenzaman sonlanım noktası, IFN + Ara-C'den İmatinibe yüksek geçiş oranından büyük ölçüdeetkilenir.
İmatinib tedavisinin kronik fazdaki, yeni tanı konulmuş KML'deki sağkalım etkisi, aynı rejimde IFN+Ara-C (n=325) kullanılan başka bir Faz III çalışmadan elde edilen birincilverilerle birlikte yukarıda belirtilen imatinib verilerinin retrospektif analizinde ayrıntılı olarakincelenmiştir. Bu yayında, genel sağkalım bakımından imatinib'in IFN+Ara-C karşısındakiüstünlüğü kanıtlanmıştır (p<0,001); 42 ay içinde 47 (%8,5) imatinib hastası ve 63 (%19,4)IFN+Ara-C hastası ölmüştür.
İmatinib tedavisindeki hastalarda sitogenetik yanıt ve moleküler yanıt derecesi, uzun dönem sonuçlar üzerinde açık bir etkiye sahip olmuştur. 12 ayda TSY'si (KSY) olan hastaların tahmini%96'sında (%93) akselere faza/blast krizine progresyon olmazken 12 ayda MSY'si olmayanhastaların sadece %81'inde 84 ayda ilerlemiş KML'ye progresyon olmadığı görülmüştür (genelp<0,001, TSY ile KSY arasında p=0,25). 12 ayda Bcr-Abl transkriptlerinde en az 3 logaritmalıkazalması olan hastalarda akselere faza/blast krizine progresyonsuz kalma olasılığı 84 ayda %99bulunmuştur. 18 aylık dönüm noktası analizine dayanılarak benzer bulgular tespit edilmiştir.
Bu çalışmada günde 400 mg'dan 600 mg'a, ardından günde 600 mg'dan 800 mg'a doz artırımlarına izin verilmiştir. 42 aylık izlem sonrasında 11 hasta sitogenetik yanıtlarındadoğrulanmış bir kayıp (4 hafta içinde) deneyimlemiştir. Bu 11 hastanın 4'ünde doz günde 800mg'a artırılmış olup hastaların 2'si sitogenetik yanıtı tekrar elde etmiş (1'inde kısmi, 1'indetam; tam yanıt elde eden ayrıca moleküler yanıta da ulaşmıştır), diğer yandan dozlarıartırılmayan 7 hastanın sadece biri tam sitogenetik yanıtı tekrar elde etmiştir. Doz artırımıöncesindeki hasta popülasyonu (n=551) ile karşılaştırıldığında, dozun günde 800 mg'ayükseltildiği 40 hastada bazı advers reaksiyonların yüzdesi daha yüksek olmuştur. Daha sıkgörülen advers reaksiyonlar gastrointestinal hemorajileri, konjonktivit ve transaminazlar veyabilirubinde yükselmeyi içermiştir. Diğer advers olaylar daha düşük ya da eşit sıklıklabildirilmiştir.
Kronik faz, interferon tedavisinin başarısız kaldığı hastalar:
532 hasta, 400 miligramlık başlangıç dozuyla tedavi edilmiştir. Bu hastalar; hematolojik başarısızlık (%29), sitogenetikbaşarısızlık (%35) veya interferon intoleransı (%36) olmak üzere başlıca 3 gruba ayrılmıştır.Hastalar daha önce medyan 14 ay boyunca >25x106 lU/hafta dozlarda IFN tedavisi görmüştürve hepsi de geç kronik fazdadır; tanıdan itibaren geçen medyan süre 32 aydır. Çalışmanın^ *a>>
Belge Do
Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys
birincil etkililik değişkeni majör sitogenetik yanıt oranıdır (tam + kısmı yanıt, kemik iliğinde %0 ila %35 Ph+ metafaz).
Bu çalışmada hastaların %65'i bir majör sitogenetik yanıta ulaşmıştır; hastaların %53'ünde (doğrulanmış %43) yanıt tamdır (Tablo 3). Hastaların %95'inde tam hematolojik yanıtaulaşılmıştır.
Hızlanmış faz:
Bu fazdaki 235 KML vakasının ilk 77'sinde tedaviye günde 400 mg ile başlanmıştır; daha sonra çalışma protokolü, daha yüksek imatinib dozlarının kullanılmasınaolanak tanıyacak şekilde tadil edilmiştir ve geriye kalan 158 hasta, başlangıçta 600 mg imatinibkullanmıştır.Tam hematolojik yanıt, hiçbir lösemi kanıtının mevcut olmaması (kemik iliğindeki ve kandaki blast hücrelerinin kaybolması, ancak periferik kan tablosunda, tam yanıt için gerekendüzelmenin gerçekleşmemesi) veya kronik faz kronik miyeloid lösemiye dönüş olaraktanımlanan tam hematolojik yanıt ede edilme oranı, bu çalışmanın etkililik konusundakideğerlendirilen primer parametresi olmuştur. Doğrulanmış hematolojik yanıt, hastaların %71,5'inde elde edilmiştir. Bu hastalardan % 27,7'sinde ayrıca majör sitogenetik yanıt ( %20,4'ünde tam sitogenetik yanıt) alınmış olması önemlidir. 600 mg imatinib kullanan hastalardabugünkü saptamalara göre tahmini ortanca medyan hastalıksız sağkalım ve genel sağkalımoranları, sırasıyla 22,9 ay ve 42,5 ay olarak hesaplanmıştır.
Mi^yeloid blast krizi:
Bu çalışma, blast krizi gelişmiş olan 260 hasta üzerinde yapılmıştır. Bu hastaların 95'i (%37'si), hızlanmış faz veya yine blast krizi nedeniyle daha önce de kemoterapigörmüştür (önceden tedavi edilmiş olan hastalar), 165 (%63) hastada ise daha öncekemoterapi uygulanmamıştır (önceden tedavi edilmemiş olan hastalar). Başlangıç dozu, ilk37 hastada 400 miligramdı; daha sonra yapılan protokol tadilatı, daha yüksek dozlarınkullanılmasına olanak verdiğinden, diğer 223 hasta, başlangıçta 600 mg imatinib kullanmıştır.Primer etkililik parametresi, hızlanmış faz çalışmasında olduğu gibi yine tam hematolojik yanıt, lösemi kanıtının mevcut olmaması veya kronik faza dönüş olarak tanımlanan, hematolojik yanıtoranı olmuştur. Hastaların %31'inde hematolojik yanıt elde edilmiştir (daha önce tedavigörmemiş hastalarda %36, daha önce tedavi görmüş hastalarda %22). 600 mg imatinib kullananhastalardaki hematolojik yanıt oranı, 400 mg imatinib kullanmış olanlara kıyasla daha yüksektir(%16'ya karşılık %33, p=0,0220). Daha önceden tedavi edilmemiş ve tedavi edilmiş hastalarınmevcut tahmini ortalama sağkalımı sırasıyla 7,7 ve 4,7 aydır.
Lenfoid blast krizi:Tablo 3 KML vakalarında elde edilen yanıtlar
|
|
Çalışma 0110 37 aylık veriKronik faz,IFN başarısızlığı(n=532) |
Çalışma 0109 40.5 aylık veriHızlanmış faz(n=235) |
Çalışma 0102 38 aylık veriMiyeloid blast krizi(n=260) |
|
Hastaların yüzdesi (%95 güven aralığı) | |||
|
Hematolojik yanıt1 Tam hematolojik yanıt (THY)Lösemi kanıtı yok (NEL)Kronik faza dönüş (RTC) |
% 95 (92,3-96,3) % 95 |
%71 (65,3-77,2) % 42% 12% 17 |
% 31 (25,2-36,8) % 8% 5% 18 |
|
Majör sitogenetik yanıt2 Tam (Onaylanmış3) [%95 CI] Kısmi |
% 65 (61,2-69,5) % 53 % 43 (38,6-47,2) %12 |
% 28 (22,0-33,9) % 20 % 16 (11,3-21,0) % 7 |
% 15 (11,2-20,4) % 7 % 2 (0,6-4,4) % 8 |
^Hematolojik yanıt kriterleri (bütün yanıtlar >4 hafta sonra doğrulanmış olmalıdır):
THY: çalışma 0110 [kandaki WBC <10 x109/L, trombosit sayısı <450 x109/L, miyelosit + metamiyelosit <%5 ; kanda blast veya promiyelosit yok; bazofiller < %20, kemik iliği dışında hastalık yok] ve çalışma 0102 ve 0109[ANC>1.5 x109/L, trombosit sayısı >100 x109/L, kanda blast hücresi yok, BM blast hücresi oranı <%5 ve BMdışında hastalık yok]
NEL: THY ile aynı kriterler; yalnızca ANC >1 x109/L ve trombosit sayısı >20 x 109/L (çalışma 0102 ve 0109'da) RTC: BM ve PB blast hücresi oranı <%15; PM ve PB blast hücresi + promiyelosit oranı <%30, PB bazofil oranı<%20, dalak ve karaciğer hariç BM dışında hatalık yok (çalışma 0102 ve 0109'da).
ANC = mutlak nötrofil sayısı, BM = kemik iliği, PB = periferik kan, WBC = lökosit sayısı
2Sitogenetik yanıt kriterleri:
Majör yanıt = tam (%0 Ph+ metafaz) + kısmi (%1-35) yanıt
3İlk kemik iliği çalışmasından en az bir ay sonra gerçekleştirilen ikinci kemik iliği sitogenetik değerlendirmesiyle doğrulanan tam sitogenetik yanıt.
Pediyatrik hastalar:
Kronik faz KML'si (n=11) veya blast krizi aşamasında KML'si ya da Ph+ akut lösemileri (n=15) olan, 18 yaş altı toplam 26 pediatrik hasta bir faz I doz yükseltmeçalışmasına kaydedilmiştir. Bu, yoğun ön tedavi görmüş hastalardan oluşan bir popülasyondur:hastaların %46'sı önceden BMT ve %73'ü önceden çoklu ajanlı kemoterapi görmüştür.Hastalar 260 mg/m2/gün (n=5), 340 mg/m2/gün (n=9), 440 mg/m2/gün (n=7) ve 570 mg/m2/gün(n=5) imatinib dozları ile tedavi edilmiştir. Sitogenetik verileri mevcut olan 9 kronik faz KMLhastasının 4'ünde (%44) ve 3'ünde (%33) sırasıyla tam ve kısmi sitojenik yanıt elde edilmişolup bu oranlar %77 MCyR değeri ile sonuçlanmıştır.Yeni tanı almış ve tedavi edilmemiş, kronik fazda KML'si olan toplam 51 pediatrik hasta açık-etiketli, çok merkezli, tek kollu bir faz II çalışmaya kaydedilmiştir. Hastalar 340 mg/m2/gün imatinib ile tedavi edilmiş, doz sınırlayıcı toksisitesi hariç ara verilmemiştir. İmatinib tedavisiyeni tanı konmuş pediatrik KML hastalarında, 8 haftalık tedavi sonrasında %78 CHR oranı ilehızlı yanıt sağlamaktadır. Yüksek CHR oranına, hastaların %65'inde tam sitojenik yanıt(CCyR) gelişimi eşlik etmiş olup bu oran, erişkinlerde gözlenen sonuç ile karşılaştırılabilirniteliktedir. Ek olarak, hastaların %16'sında kısmı sitoj enik yanıt (PCyR) gözlenmiş, bu da %81MCyR değerini vermiştir. CCyR'ye ulaşan hastaların büyük çoğunluğu, Kaplan-Meier tahminedayalı 5,6 aylık yanıta kadar geçen medyan süre ile CCyR'ye 3 ila 10'uncu aylar arasındaulaşmıştır.
Avrupa İlaç Ajansı, Philadelphia kromozomu (bcr-abl translokasyon) pozitif kronik faz kronik miyeloid lösemide pediatrik popülasyonun tüm alt kümelerinde imatinib ile çalışmalarınsonuçları sunma zorunluluğunu iptal etmiştir. (pediatrik kullanım ile ilgili bilgi için bkz. bölüm4.2).
Ph+ ALL için klinik çalışmalar
Yeni teşhis edilen Ph+ ALL:
Kontrol grubuna yer vererek yapılan ve imatinibin, 55 yaş ve üzeri yeni tanı almış 55 hastada kemoterapi indüksiyonuyla karşılaştırıldığı bir çalışmada (ADE10), tek ajan olarak kullanılanimatinib, kemoterapiye kıyasla anlamlı derecede daha yüksek tam hematolojik yanıt oranı ilesonuçlanmıştır (%50'ye karşılık %96,3, p=0,0001). Kemoterapiye yanıt vermeyen veya zayıfyanıt veren hastalarda imatinib kurtarma tedavisi olarak kullanıldığında, 11 hastanın 9'unda(%81,8) tam hematolojik yanıt elde edilmiştir. Bu klinik etki, 2 haftalık tedaviden sonra,kemoterapi kolu ile karşılaştırıldığında imatinib ile tedavi edilen hastalarda, bcr-abltranskriptlerinde daha büyük bir azalmayla ilişkilendirilmiştir (p=0,02). Tüm hastalarindüksiyon sonrasında imatinib ve konsolidasyon kemoterapisi almış (bkz. Tablo 4) ve bcr-abltranskriptlerinin düzeyleri sekizinci haftada iki kolda aynı olmuştur. Çalışma tasarımıdoğrultusunda beklendiği üzere, iki grup arasında remisyon süresi, hastalıksız sağkalım veyagenel sağkalım açısından herhangi bir fark gözlenmemiş, ancak tam moleküler yanıt elde edilenve minimal rezidüel hastalık düzeyinde kalan hastalarda gerek remisyon süresi (p=0,01)gerekse hastalıksız sağkalım (p=0,02) bakımından sonuçlar daha iyi olmuştur.
Kontrol gruplarına yer verilmeyen dört klinik çalışmada (AAU02, ADE04, AJP01 ve AUS01) yeni tanı almış 211 Ph+ ALL hastasından oluşan bir popülasyonda gözlenen sonuçlar, yukarıdatarif edilen sonuçlar ile uyumludur. Kemoterapi indüksiyonu ile kombinasyon halindekiimatinib (bkz. Tablo 4) %93'lük bir tam hematolojik yanıt oranı (değerlendirilebilir 158hastanın 147'si) ve %90'lık bir majör sitogenetik yanıt oranı (değerlendirilebilir 21 hastanın19'u) sonuçlarını vermiştir. Tam moleküler yanıt oranı %48 bulunmuştur (değerlendirilebilir102 hastanın 49'u). Hastalıksız sağkalım (DFS) ve genel sağkalım (OS) her durumda 1 yılıgeçmiştir ve iki çalışmadaki (AJP01 ve AUS01) geçmiş kontrolden üstün olmuştur (DFSp<0,001; OS p<0,0001).
Tablo 4 Imatinible kombinasyon halinde kullanılan kemoterapi rejimi
Çalışma ADE10
DEX 10 mg/m2 oral, gün 1-5; CP 200 mg/m2 i.v., gün 3, 4, 5
MTX 12 mg intratekal, gün 1
DEX 10 mg/m2 oral, gün 6-7, 13-16;
VCR 1 mg/m2 i.v., gün 7, 14;
IDA 8 mg/m2 i.v. (0.5 h), gün 7, 8, 14, 15; CP 500 mg/m2 i.v. (1 h), gün 1;
Ara-C 60 mg/m2 i.v., gün 22-25, 29-32
MTX 500 mg/m2 i.v. (24 h), gün 1, 15;
6-MP 25 mg/m2 oral, gün 1-20
_Konsolidasyon tedavisi I, III, V
Ara-C 75 mg/m2 i.v. (1 h), gün 1-5;
VM26 60 mg/m2 i.v. (1 h), gün 1-5
|
İndüksiyon tedavisi (de novoPh+ ALL) |
Daunorubisin 30 mg/m2 i.v., gün 1-3, 15-16; VCR 2 mg toplam doz i.v., gün 1, 8, 15, 22; CP 750 mg/m2 i.v., gün 1, 8; Prednizon 60 mg/m2 oral, gün 1-7, 15-21; IDA 9 mg/m2 oral, gün 1-28; MTX 15 mg intratekal, gün 1, 8, 15, 22; Ara-C 40 mg intratekal, gün 1, 8, 15, 22; Metilprednizolon 40 mg intratekal, gün 1, 8, 15, 22 |
|
Konsolidasyon (de novoPh+ ALL) |
Ara-C 1000 mg/m2/12 h i.v. (3 h), gün 1-4; Mitoksantron 10 mg/m2 i.v., gün 3-5; MTX 15 mg intratekal, gün 1; Metilprednizolon 40 mg intratekal, gün 1 |
Çalışma ADE04 | |
|
Faz öncesi |
DEX 10 mg/m2 oral, gün 1-5; CP 200 mg/m2 i.v., gün 3-5;MTX 15 mg intratekal, gün 1 |
|
İndüksiyon tedavisi I |
DEX 10 mg/m2 oral, gün 1-5; VCR 2 mg ,.v., gün 6, 13, 20; Daunorubisin 45 mg/m2 i.v., gün 6-7, 13-14 |
|
İndüksiyon tedavisi II |
CP 1 g/m2 i.v. (1 h), gün 26, 46; Ara-C 75 mg/m2 i.v. (1 h), gün 28-31, 35-38, 42-45; 6-MP 60 mg/m2 oral, gün 26-46 |
|
Konsolidasyon tedavisi |
DEX 10 mg/m2 oral, gün 1-5; Vindesine 3 mg/m2 i.v., gün 1; MTX 1.5 g/m2 i.v. (24 h), gün 1; Etoposide 250 mg/m2 i.v. (1 h) gün 4-5;Ara-C 2 x 2 g/m2 i.v. (3 h, q 12 h), gün 5 |
Çalışma AJP01 | |
|
İndüksiyon tedavisi |
CP 1.2 g/m2 i.v. (3 h), gün 1; Daunorubisin 60 mg/m2 i.v. (1 h), gün 1-3; Vinkristin 1.3 mg/m2 i.v., gün 1, 8, 15, 21;Prednizolon 60 mg/m2/gün oral |
|
Konsolidasyon tedavisi |
Değişimli kemoterapi kürü: MTX 1 g/m2 i.v. (24 h) gün 1 ile yüksek kemoterapi ve 4 siklus boyunca Ara-C 2 g/m2 i.v. (q 12 h), gün 2-3 |
|
İdame |
VCR 1.3 g/m2 i.v., gün 1; Prednizolon 60 mg/m2 oral, gün 1-5 |
Çalışma AUS01 | |
|
İndüksiyon-konsolidasyon tedavisi |
Hyper-CVAD rejimi: CP 300 mg/m2 i.v. (3 h, q 12 h), gün 1-3; Vinkristin 2 mg i.v., gün 4, 11; Doksorubisin 50 mg/m2 i.v. (24 h), gün 4; Değişimli olarak DEX 40 mg/gün gün 1-4 ve 11 -14 ya da MTX 1 g/m2 i.v. (24 h) gün 1 ve Ara-C 1 g/m2 i.v. (2 h, q 12 h), gün 2-3 (toplam 8 kür). |
|
İdame |
13 ay boyunca aylık olarak VCR 2 mg i.v.; Prednizolon 200 mg oral, 13 ay boyunca ayda 5 gün |
|
Tüm tedavi rejimleri CNS profilaksisi için steroid uygulaması içermelidir. | |
|
Ara-C: sitozin arabinozid; CP: siklofosfamid; DEX: deksametazon; MTX: metotreksat; 6-MP: 6-merkaptopürin; VM26: Teniposid; VCR: vinkristin; IDA: idarubisin; i.v.:intravenöz. | |
Pediyatrik hastalar:
I2301 çalışmasında, Ph+ ALL'si olan toplam 93 pediyatrik, ergen ve genç yetişkin hasta (1 ila 22 yaşları arasında) açık etiketli, çok merkezli, sıralı gruplu, randomizeolmayan bir faz III çalışmaya kaydedilmiş ve indüksiyon tedavisinden sonra yoğun kemoterapiile kombinasyon halinde imatinib (340 mg/m2/gün) ile tedavi edilmiştir. İmatinib 1. gruptan 5.gruba doğru artan süre ve daha erken imatinib tedavisi olacak şeklilde aralıklı olarakuygulanmıştır; en düşük imatinib yoğunluğu grup 1'de ve en yüksek imatinib yoğunluğu grup5'tedir (ilk kemoterapi tedavi ,kürlerjesıreaifflda ı§örekliâüfliük. imatinib doz uygulaması ile gün
Belge DcoiarakeenüZunesörg)peGrupe5ihasrafa#B«a](n=50) ekemoterapieileekowb'i&dsyonvhalğnde<edavi
kürünün erken dönemlerinde imatinibe sürekli günlük maruziyet, imatinibsiz standart kemoterapinin uygulandığı tarihsel kontrollerle (n=120) karşılaştırıldığında 4 yıllık olaysızsağkalımı (EFS) artırmıştır (sırasıyla %69,6'ya karşılık %31,6). Grup 5 hastalarında tahmini 4yıllık GS, tarihsel kontrollerdeki %44,8 değeri ile karşılaştırıldığında %83,6 olmuştur.
Kohort 5'teki 50 hastadan 20'si (% 40) hematopoietik kök hücre nakli almıştır.
Tablo 5 Çalışma I2301'de imatinib ile kombinasyon halinde kullanılan kemoterapi rejimi
|
Konsolidasyon bloğu 1 (3 hafta) |
VP-16 (100 mg/m2/gün, IV): 1-5. günler İfosfamid (1,8 g/m2/gün, IV): 1-5. günler MESNA (3 saatte bir 360 mg/m2/doz, x 8 doz/gün, IV): 1-5. günler G-CSF (5 gg/kg, SC): 6-1. günler veya en düşük değer sonrası ANC > 1500olana kadar IT Metotreksat (yaşa düzeltilmiş): SADECE 1. gün Üçlü IT tedavisi (yaşa düzeltilmiş): 8., 15. gün |
|
Konsolidasyon bloğu 2 (3 hafta) |
Metotreksat (24 saatte 5 g/m2, IV): gün 1 Leucovorin (36 saatte 75 mg/m2, IV; 15 mg/m2 IV veya PO 6 saatte bir x 6 doz)iii: 2. ve 3. günler Üçlü IT tedavisi (yaşa düzeltilmiş): gün 1 ARA-C (3 g/m2/doz q 12 h x 4, IV): 2. ve 3. günler G-CSF (5 gg/kg, SC): 4-13 günler en düşük değer sonrası ANC > 1500 olana kadar |
|
Yeniden indüksiyon bloğu 1 (3 hafta) |
VCR (1,5 mg/m2/gün, IV): 1, 8 ve 15. Günler DAUN (45 mg/m 2/gün bolus, IV): 1 ve 2. GünlerCPM (250 mg/m2/doz 12 saatte bir x 4 doz, IV): 3 ve 4. günler PEG-ASP (2500 IU/m2, IM): gün 4 G-CSF (5 gg/kg, SC): 5-14. günler veya en düşük değer sonrası ANC > 1500 olana kadar Üçlü IT tedavisi (yaşa düzeltilmiş): 1 ve 15. Günler DEX (6 mg/m2/gün, PO): 1-7 ve 15-21. günler |
|
Yoğunlaştırma bloğu 1 (9 hafta) |
Metotreksat (24 saatte 5 g/m2, IV): 1 ve 15. günler Leucovorin (36. saatte 75 mg/m2, IV; 15 mg/m2 IV veya PO 6 saatte bir x 6 doz)iii: 2, 3, 16 ve 17. günler Üçlü IT tedavisi (yaşa düzeltilmiş): 1 ve 22. günler VP-16 (100 mg/m2/gün, IV): 22-26. günler CPM (300 mg/m2/gün, IV): 22-26. günler MESNA (150 mg/m2/gün, IV): 22-26. günler G-CSF (5 gg/kg, SC): 27-36. günler veya en düşük değer sonrası ANC > 1500 olana kadar ARA-C (3 g/m2, 12 saatte bir, IV): 43, 44. günler L-ASP (6000 IUnit/m2, IM): gün 44 |
| ||||||||||
|
G-CSF = granülosit koloni uyarıcı faktör, VP-16 = etoposid, MTX = metotreksat, IV = intravenöz, SC = subkutan, IT = in 6akr |
firaJ«IM=i^^?.!i'IİA-C=^i'#,«n:£PS=i!Hofosfaffld,VC6»Vi^nkfS
= deksametazon, DAUN = daunorubisin, 6-MP = 6-merkaptopurin, E.Coli L-ASP = Lasparaginaz, PEG-ASP = PEG asparaginaz, MESNA = 2-merkaptoetan sülfonat sodyum, iii = veya MTX düzeyi <0,1 pM olana kadar, 6saatte bir = her 6 saatte bir, Gy = Gray
Çalışma AIT07, kemoterapi ile kombinasyon halinde imatinib ile tedavi edilen 128 hastayı (1 ila <18 yaş) içeren çok merkezli, açık etiketli, randomize, Faz II / III bir çalışmadır. Buçalışmadan elde edilen güvenlilik verilerinin, imatinibin Ph + ALL hastalarında güvenlilikprofili ile uyumlu olduğu görülmektedir.
Nüksetmiş/tedaviye dirençli Ph+ ALL
İmatinib, yineleyen/refraktör Ph+ ALL hastalarında tek ajan olarak kullanıldığında, 411 hastanın 53'ünde yanıt değerlendirilebilmiş, hematolojik yanıt oranı %30 (%9'u tam) ve majörsitogenetik yanıt oranı ise %23 olarak bulunmuştur (Not: 411 hastanın 353'ü, primer yanıtverileri toplanmaksızın genişletilmiş erişim çalışmasında tedavi edilmiştir). 411yineleyen/refraktör Ph+ ALL hastasından oluşan toplam popülasyonda progresyona kadargeçen medyan süre 2,6 ile 3,1 ay aralığında olurken, değerlendirilebilir 401 hastada medyangenel sağkalım 4,9 ile 9 ay aralığında bulunmuştur. Bu veriler, sadece 55 yaş ve üzeri hastalardahil edilecek şekilde yeniden analiz yapıldığında da benzer olmuştur.
SM ile İlgili Klinik Çalışmalar
ABL, KIT ya da PDGFR protein tirozin kinazlarla ilişkili yaşamı tehdit edici hastalıkları olan farklı hasta popülasyonlarında imatinib'in test edildiği açık-etiketli, çok merkezli bir faz IIklinik çalışma (çalışma B2225) yürütülmüştür. Bu çalışmada tedavi edilen ve 45'indehematolojik hastalıklar, 140'ında da çeşitli solid tümörler bulunan 185 hastadan 5'inde SMsaptanmıştır. SM hastaları günlük 100 mg ila 400 mg imatinib ile tedavi edilmiştir. Yayınlanmış10 vaka raporu ve vaka serisinde, yaşları 26 ila 85 arasında değişen 25 SM hastası dahabildirilmiştir. Bu hastalara da günlük 100 mg ila 400 mg dozda imatinib uygulanmıştır. SM içintedavi edilen toplam popülasyonun (30 hasta) 10'unda (%33) tam hematolojik yanıt, 9'unda(%30) kısmi hematolojik yanıt elde edilmiştir (toplam yanıt oranı %63). Sitogenetikanormallikler yayınlanmış raporlarda ve çalışma B2225'te tedavi edilen 30 hastanın 21'indedeğerlendirilmiştir. Bu 21 hastanın sekizinde FIP1L1-PDGFR-alfa füzyon kinaz saptanmıştır.Çalışma B2225'te tedavi edilen hastalarda ortanca medyan tedavi süresi 13 ay olmuş (aralık:1,4-22,3 ay), yayınlanmış literatürde yanıt veren hastalarda ise aralık 1 ay ila 30 ayın üzerindebir süre arasında değişmiştir.
HES/CEL ile İlgili Klinik Çalışmalar
ABL, KIT ya da PDGFR protein tirozin kinazlarla ilişkili yaşamı tehdit edici hastalıkları olan farklı hasta popülasyonlarında imatinibin test edildiği açık-etiketli, çok merkezli bir faz II klinikçalışma (çalışma B2225) yürütülmüştür. Bu çalışmada, HES'i olan 14 hasta günde 100 mg ila1000 mg dozda imatinib ile tedavi edilmiştir. Yayınlanmış 35 vaka raporu ve vaka serisinde,HES/CEL'li 162 hasta daha günde 75 mg ila 800 mg dozlarında imatinib almıştır. 176 hastadanoluşan toplam popülasyonun 117'sinde sitogenetik anormallikler değerlendirilmiştir. Bu 117hastanın 61'inde, FIP1L1-PDGFR-alfa füzyon kinaz tanımlanmıştır. Diğer 3 yayınlanmış
tıttus://www.turkiye.gov.tr/sagliK-titcK-ebys'.dört-HErt-HE
Belge Do
.W56aMU'(
tr/sagri
bys
PDGFRa füzyon kinaz pozitif hastanın tümü, aylarca sürdürülen bir CHR elde etmiştir (raporlama sırasında sansürlenen 1+ ila 44+ ay arasında). Yakın tarihli bir yayında bildirildiğigibi, bu 65 hastadan 21'i, 28 aylık (aralık 13-67 ay) ortalama bir takip süresiyle tam molekülerremisyona ulaşmıştır. Bu hastaların yaşları 25 ile 72 aralığında olmuştur. Ek olarak, olguraporlarında araştırmacılar tarafından semptomatolojide ve diğer organ disfonksiyonanormalliklerindeki gelişmeler bildirilmiştir. Kalp, sinir, deri/derialtı doku,solunum/göğüs/mediastinal, kas-iskelet/bağ dokusu/vasküler ve gastrointestinal organsistemlerinde gelişmeler bildirilmiştir.
HES/CEL'li pediyatrik hastalarda kontrollü çalışma yoktur. 3 yayında PDGFR gen yeniden düzenlemeleri ile ilişkili HES ve CEL'li üç (3) hasta bildirilmiştir. Bu hastaların yaşları 2 ila 16yıl arasında değişmiştir ve imatinib günde 300 mg / m2 veya günlük 200 ila 400 mg arasındadeğişen dozlarda verilmiştir. Tüm hastalar tam hematolojik yanıt, tam sitogenetik yanıt ve/veyatam moleküler yanıt elde etmiştir.
Rezeke edilemeyen ya da metastatik GIST'de yapılan klinik çalışmalar
Rezektabl olmayan veya metastatik malign gastrointestinal stromal tümörleri (GIST) olan hastalarda faz II, açık etiketli, randomize, kontrolsüz çok uluslu bir çalışma yürütülmüştür. Buçalışmaya 147 hasta kaydedilmiş ve 36 ay boyunca günde bir kez oral olarak 400 mg veya 600mg kullanımına randomize edilmiştir. Bu hastalar 18 ila 63 yaşında olup, rezektabl olmayanve/veya metastatik Kit-pozitif malign GIST patolojik tanısına sahiptir. İmmünohistokimya Kitantikoru ile (A-4502, tavşan poliklonal antiserumu, 1:100; DAKO Corporation, Carpinteria,CA) antijen geri kazanımı sonrası avidin-biotin-peroksidaz kompleksi yöntemi ile analize görerutin olarak yürütülmüştür.
Birincil etkililik kanıtı objektif yanıt oranlarını temel almıştır. Tümörlerin en az bir hastalık bölgesinde ölçülebilir olması gerekmiş olup, yanıt karakterizasyonu Güneybatı Onkoloji Grubu(SWOG) kriterlerini temel almıştır. Bulgular Tablo 6'da sunulmaktadır.
Tablo 6 STIB2222 kodlu GIST çalışmasında en iyi tümör yanıtı
Yanıtlar |
Tüm dozlar (n=147) 400 mg n= 73600 mg n=74n (%) |
|
Tam yanıt |
1 (0,7) |
|
Kısmi yanıt |
98 (66,7) |
|
Stabil hastalık |
23 (15,6) |
|
İlerleyici hastalık |
18 (12,2) |
|
Değerlendirilemeyen |
5 (3,4) |
|
Bilinmeyen |
2 (1,4) |
İki doz grubu arasında yanıt oranları bakımından farklılıklar söz konusu olmamıştır. Ara analiz tarihinde önemli sayıda stabil hastalığa sahip hasta, daha uzun süreli tedavi ile kısmi yanıtaulaşmıştır (medyan takip süresi 31 ay). Yanıta kadar geçen medyan süre 13 hafta olmuştur (%95GA 12-23). Yanıt veren olgu'laEdaütedaviitbaşar^^sız'liğrriaıkıadar geçen medyan süre 122 hafta
Belge Do Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys
(%95 GA 106-147), genel çalışma popülasyonunda ise 84 hafta (%95 GA 71-109) bulunmuştur. Medyan genel sağkalım noktasına ulaşılamamıştır. 36 aylık izlem sonrasında Kaplan-Meiersağkalım tahmini %68'dir.
İki klinik çalışmada (çalışma B2222 ve gruplar arası çalışma S0033), günlük imatinib dozu, 400 mg veya 600 mg daha düşük günlük dozlarında progrese olan hastalarda 800 mg'ayükseltilmiştir. Doz, toplam 103 hastada 800 mg'a çıkarılmıştır; doz yükseltildikten sonra 6hasta kısmi yanıta ve 21 hasta hastalık stabilizasyonuna ulaşarak %26'lık genel klinik yanıtsonucunu vermiştir. Eldeki güvenlilik verilerinden yola çıkılarak, 400 mg veya 600 mg dahadüşük günlük dozlarında progrese olan hastalarda dozun günde 800 mg'a çıkarılmasının,imatinibin güvenlilik profilini etkilemediği görülmektedir.
Adjuvan GIST için klinik çalışmalar
Adjuvan tedavi koşullarında imatinib, 773 hasta ile yürütülen çok merkezli, çift kör, uzun süreli, plasebo kontrollü bir faz III çalışmada (Z9001) araştırılmıştır. Bu hastaların yaşları 1891 aralığında olmuştur. İmmünhistokimya ile KIT proteini eksprese eden primer GISTyönünde histolojik tanısı bulunan ve en geniş yerinde >3 cm tümör büyüklüğüne sahip olan,çalışmaya kayıt öncesindeki 14-70 gün içersinde primer GIST'i tam gross rezeksiyon ile alınanhastalar dahil edilmiştir. Primer GIST rezeke edildikten sonra hastalar şu iki koldan birinerandomize edilmiştir: bir yıl süreyle imatinib 400 mg/gün veya plasebo.
Çalışmanın birincil sonlanma noktası, randomizasyon tarihinden rekürense ya da herhangi bir nedene bağlı ölüme kadar geçen süre şeklinde tanımlanan rekürenssiz sağkalım (RFS)olmuştur.
İmatinib RFS'de anlamlı uzama sağlamış, imatinib grubunda hastaların %75'i 38. ayda rekürenssiz iken plasebo grubundaki hastaların %75'i 20. ayda rekürenssiz kalmıştır (sırasıyla%95 GA [30-hesaplanamaz]; [14-hesaplanamaz]); (tehlike oranı = 0,398 [0,259-0,610],p<0,0001). Bir yıl sonunda genel RFS, plasebo (%82,3) karşısında imatinib için anlamlıdüzeyde daha iyi bulunmuştur (%97,7) (p<0,0001). Bu şekilde rekürens riski plaseboya oranla%89 azaltılmıştır (tehlike oranı = 0,113 [0,049-0,264]).
Primer GIST'lerine yönelik ameliyatları sonrasında hastalardaki rekürens riski, şu prognoz faktörleri esas alınarak retrospektif şekilde değerlendirilmiştir: tümör büyüklüğü, mitotikindeks, tümör yeri. Mitotik indeks verileri, tedavi amaçlı (ITT) popülasyonu oluşturan 713hastanın 556'sı için mevcut idi. Birleşik Devletler Ulusal Sağlık Enstitüleri (NIH) ve SilahlıKuvvetler Patoloji Enstitüsü (AFIP) risk sınıflandırmalarına göre yapılan alt grup analizlerininsonuçları Tablo 7'de gösterilmektedir. Düşük ve çok düşük risk gruplarında herhangi bir faydagözlenmemiştir. Genel bir sağkalım faydası gözlenmemiştir.
Tablo 7 NIH ve AFIP risk sınıflandırmasına göre Z9001 deneyi RFS analiz özeti | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Full takip periyodu- N E-Tahmin edilebilir değil |
İkinci bir çok merkezli, açık etiketli faz III çalışmada (SSG XV
n
i/AIO), cerrahi GIST rezeksiyonu sonrasında olan ve aşağıdaki durumlardan birinin bulunduğu hastalarda 400mg/gün imatinib ile 36 ay karşısında 12 aylık tedavi karşılaştırılmıştır: tümör çapı > 5 cm vemitotik sayım > 5/50 yüksek güç alanı (HPF); veya tümör çapı > 10 cm ve herhangi bir mitotiksayım veya mitotik sayımı > 10/50 HPF olan herhangi bir büyüklükteki tümör ya da peritonboşluğuna doğru rüptüre olan tümörler. Toplam 397 hastadan olur alınmış ve bu hastalarçalışmaya randomize edilmiştir (199 hasta 12 ay kolunda ve 198 hasta 36 ay kolunda) medyanyaş 61 idi [aralık 22 ila 84 yaş]). Medyan takip süresi 54 ay olup (randomizasyondan veri kesmetarihine kadar) ilk hastanın randomize edilişinden veri kesme tarihine kadar geçen medyan süre83 aydır.Çalışmanın birincil sonlanma noktası, randomizasyon tarihinden nükse ya da herhangi bir nedene bağlı ölüme kadar geçen süre şeklinde tanımlanan nükssüz sağkalım (RFS) olmuştur.
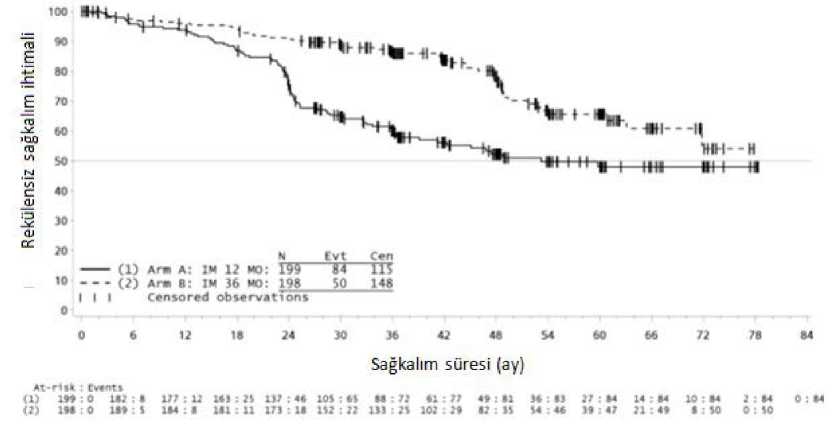
36 aylık imatinib tedavisi, 12 aylık imatinib tedavisi ile karşılaştırıldığında RFS'de anlamlı ölçüde uzama sağlamıştır (genel tehlike oranı (HR) = 0,46 [0,32, 0,65], p<0,0001) (Tablo 8,Şekil 1).
Buna ek olarak, 36 aylık imatinib tedavisi, 12 aylık imatinib tedavisi ile karşılaştırıldığında genel sağkalım (OS) süresini anlamlı ölçüde uzatmıştır (HR = 0,45 [0,22, 0,89], p=0,0187)(Tablo 8, şekil 2).
Daha uzun süreli tedavi (> 36 ay) yeni rekürenslerin oluşumunu geciktirebilmektedir; ancak, bu bulgunun genel sağkalım üzerindeki etkisi halen bilinmemektedir.
Belge Do Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys
Toplam ölüm sayısı 12 aylık tedavi kolu için 25 ve 36 aylık tedavi kolu için 12 şeklinde olmuştur.
İmatinib ile 36 ay süreli tedavi, ITT analizinde, yani tüm çalışma popülasyonun dahil edildiği analizde, 12 aylık tedaviden daha üstün bulunmuştur. Mutasyon tipine göre yapılan planlı biralt grup analizinde, ekson 11 mutasyonları olan hastalarda 36 aylık tedavide RFS için tehlikeoranı 0,35 olmuştur [%95 GA: 0,22, 0,56].
Gözlemlenen olay sayısının düşük olması sebebiyle, daha az yaygın olan mutasyon alt grupları için herhangi bir sonuç çıkartılamamaktadır.
Tablo 8. 12 aylık ve 36 aylık İmatinib Tedavisi (SSGXVni/AIO Çalışması) | |||||||||||||||
|
Şekil 1. Primer rekürenssiz sağkalım sonlanım noktası için Kaplan-Meier tahminleri (ITT popülasyonu)
Şekil 2. Genel sağkalım için Kaplan-Meier tahminleri (ITT popülasyonu) |
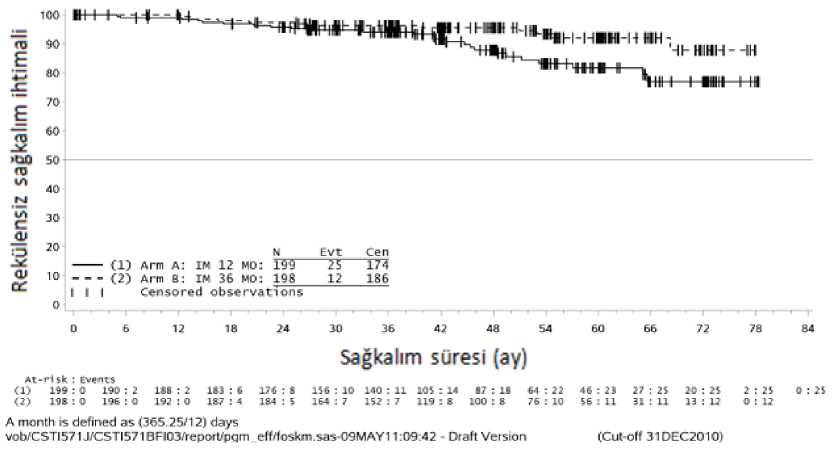 |
C-Kit pozitif GIST olan pediyatrik hastalarda kontrollü çalışma yoktur. 7 yayında GIST'li (Kit ve PDGFR mutasyonları olan veya olmayan) onyedi (17) hasta bildirilmiştir. Bu hastaların yaşı8 ila 18 aralığında olmuştur ve imatinib, hem adjuvan hem de metastatik koşullarda günde 300ila 800 mg arasında değişen dozlarda verilmiştir. GIST tedavisi gören pediyatrik hastalarınçoğunda c-kit veya PDGFR mutasyonlarını doğrulayan veriler bulunmamakta olup bu durumkarışık klinik sonuçlara yol açmış olabilir.
5.2. Farmakokinetik özelliklerGenel özellikler
NİBTU'nun farmakokinetiği 25 - 1000 mg'lık bir doz aralığında değerlendirilmiştir. Plazma farmakokinetik profilleri 1. günde ve plazmada kararlı düzeylerin elde edildiği 7. ya da 28.günde analiz edilmiştir.
Emilim:
Tablet formülünün ortalama mutlak biyoyararlanımı % 98'dir. Bir oral dozu takiben plazma imatinib eğri altında kalan alan (EAA) değerlerinde, yüksek oranda hastalar arası değişkenlikgörülmüştür. Yüksek yağ içeren bir gıda ile birlikte verildiğinde, imatinibin emilim oranıminimal düzeyde azalmış (Cmaks'da % 11 azalma ve tmaks'da 1,5 saatlik uzama), açlıkkoşullarına göre EAA değerinde küçük bir azalma (%7,4) olmuştur. Geçirilmiş gastrointestinalcerrahinin ilaç absorpsiyonu üzerindeki etkisi araştırılmamıştır.
Dağılım:
Klinik açıdan uygun konsantrasyonlarda kullanılan imatinibin plazma proteinlerine bağlanması yaklaşık % 95 olmuş,
in vitro
deneyler temelinde, daha çok albümin ve alfa-asit-glikoproteine,az miktarda da lipoproteine bağlanmıştır.Biyotransformasyon:
İnsanlarda, dolaşımdaki temel metaboliti ana ilaç ile
in vitro
benzer etki gücünde (potens) olduğu gösterilmiş N-demetillenmiş piperazin (CGP71588) türevidir. Bu metabolitin plazmaEAA değerinin imatinibin EAA değerinin sadece % 16'sı olduğu bulunmuştur. N-demetilemetabolitin plazma proteinlerine bağlanması, asıl bileşiğinkine benzerdir.İmatinib ve N-demetil metaboliti birlikte, dolaşımdaki radyoaktivitenin yaklaşık %65'ini oluşturmuştur (EAA (0-48saat)). Dolaşımdaki radyoaktivitenin kalan kısmı bir dizi minörmetabolitten oluşmuştur.
İn vitro sonuçlar CYP3A4'ün, imatinib biyotransformasyonunu katalize eden başlıca P450 enzimi olduğunu göstermiştir. Potansiyel eşzamanlı ilaçlardan (asetaminofen, asiklovir,allopurinol, amfoterisin, sitarabin, eritromisin, flulonazol, hidroksiüre, norfloksasin, penisilinV) oluşan bir panelde sadece eritromisin (IC50 50 ^M) ve flukonazol (IC50 118 ^M) imatinibmetabolizmasında klinik açıdan anlamlı olabilecek inhibisyon göstermiştir.
İn vitro koşullarda imatinibin CY^2C9, CYP2D6 ve CY^3A4/5'in markör substratlarının kompetitif bir inhibitörü olduğu gösterilmiştir. İnsan karaciğeri mikrozomlarında Ki değerlerisırasıyla 27, 7,5 ve 7,9 ^mol/l bulunmuştur. Hastalarda imatinibin maksimal plazmakonsantrasyonları 2-4 ^mol/l'dir, dolayısıyla bir arada uygulanan ilaçların CYP2D6 ve/veyaCY^3A4/5 aracılı metabolizmasında inhibisyon olasıdır. İmatinib,5-fluorourasil
biyotransformasyonuna müdahale etmemiştir fakat kompetitif CYP2C8 inhibisyonu (Ki = 34,7 ^M) sonucu paklitaksel metabolizmasını inhibe etmiştir. Bu Ki değeri, hastalarda beklenenimatinib plazma düzeylerinin çok üzerindedir, dolayısıyla 5-fluorourasil ya da paklitakselinimatinib ile bir arada uygulanması sonucu herhangi bir etkileşim beklenmemektedir.
Eliminasyon:
İmatinibin 14C-işaretli tek oral dozundan sonra, dozun yaklaşık % 81'i 7 gün içinde feçesle (dozun % 68'i) ve idrarla (dozun % 13'ü) itrah edilmiştir. Değişmemiş durumdaki imatinib,dozun % 25'ini (% 5 idrar, % 20 feçes) oluşturmuştur, geriye kalan kısım metabolitlerdir.
Doğrusallık / doğrusal olmayan durum:
Sağlıklı gönüllülerde oral uygulamanın ardından, imatinibin tı/2 değeri yaklaşık 18 saat olması günde tek doz şeklindeki pozolojinin uygun olduğu izlenimini vermektedir. Oral olarak 251000 mg imatinib uygulandıktan sonra artan dozla birlikte ortalama EAA artışı doğrusal birseyir izlemiştir. Tekrarlanan dozlarda imatinib kinetiğinde değişiklik olmamış ve günde bir kezuygulandığında birikim, kararlı ilaç konsantrasyonunun 1,5-2,5 katı olmuştur.
Farmakokinetik / farmakodinamik ilişkiler:
Popülasyon farmakokinetiği
KML hastalarındaki popülasyon farmakokinetiği analizlerine göre yaşın dağılım hacmi üzerinde küçük bir etkisi olmuştur (> 65 yaşındaki hastalarda % 12 artış). Bu değişimin klinikaçıdan anlamlı olmadığı düşünülmüştür. Vücut ağırlığının imatinib klerensi üzerindeki etkisine
Belge Dcbaıkııldı^ıin:da,w^ıfcgnHğı«lcğ^ndaki3b^üyki^e klerensinikŞ,idrl/8,tt@lması.tıbeklenirfcen),-tiW0b5kg
ağırlığındaki bir kişideki klerens 11,8 l/s'e yükselmektedir. Bu değişiklikler vücut ağırlığına göre bir doz ayarlaması yapılması için yeterli olarak kabul edilmemiştir. Cinsiyetin imatinibkinetiği üzerinde etkisi olmamıştır.
GIST hastalarında farmakokinetik
GIST hastalarında kararlı durum maruziyeti, aynı dozajda (400 mg/gün) KML hastaları için gözlenenden 1,5 kat daha yüksek olmuştur. GIST hastalarındaki ön popülasyon farmakokinetiğianalizine dayalı olarak, üç değişkenin (albümin, WBC ve bilirubin) imatinib farmakokinetiğiile anlamlı ilişkiye sahip olduğu bulunmuştur. Daha düşük albümin değerleri daha düşükklirense (CL/f) sebep olmuş ve daha yüksek WBC düzeyleri CL/f azalmasına neden olmuştur.Ancak bu ilişkiler, doz ayarlamasını gerektirecek ölçüde anlamlı şekilde ön plana çıkmamıştır.Bu hasta popülasyonunda hepatik metastazların varlığı potansiyel olarak karaciğeryetmezliğine ve azalmış metabolizmaya yol açabilir.
Çocuklarda farmakokinetik
Bir Faz I ve Faz II çalışmasında oral imatinib, pediyatrik hastalarda da, erişkin hastalardaki gibi hızla emilmiştir. Çocuklarda 260 ve 340 mg/m2 imatinible elde edilen EAA değerleri,erişkinlerde sırasıyla 400 ve 600 mg imatinible elde edilenler gibidir. 340 mg/m2 imatinibinbirinci ve sekizinci günlerdeki EAA(0-24 saat) değerleri bu ilacın, tekrarlanan dozlardan sonra 1,7kat biriktiğini göstermiştir.
Hematolojik bozuklukları (KML, Ph+ALL ya da imatinib ile tedavi edilen diğer hematolojik bozukluklar) olan pediyatrik hastalarda birleştirilmiş popülasyon farmakokinetiği analizinedayalı olarak imatinib klerensi vücut yüzey alanının (VYA) artmasına paralel olarakyükselmektedir. VYA etkisi için düzeltme yapıldıktan sonra, yaş, vücut ağırlığı ve vücut kitleindeksi gibi diğer demografik faktörler imatinib maruziyeti üzerinde klinik açıdan anlamlıetkiler yapmamıştır. Yapılan analiz, günde bir kere 260 mg/m2 (günde 400 mg'ı geçmemeküzere) ya da günde bir kere 340 mg/m2 alan (günde 600 mg'ı geçmemek üzere) pediyatrikhastalarda imatinib maruziyetinin, günde bir kere 400 mg ya da 600 mg imatinib alan yetişkinhastalardakine benzer olduğunu doğrulamıştır.
Organ fonksiyonu bozukluğu
İmatinib ve metabolitleri böbrek yoluyla anlamlı miktarda atılmaz. Böbrek fonksiyonlarında hafif ve orta şiddette bozukluk olan hastalar, böbrek fonksiyonları normal hastalardan dahayüksek plazma değerlerine sahip görünmektedir. Artış yaklaşık olarak 1,5-2 kattır ve imatinibingüçlü bir biçimde bağlandığı plazma alfa asit glikoprotein (AGP) değerinde 1,5 katlık bir artışakarşılık gelir. Böbrek bozukluğu olan hastalarda imatinibin serbest ilaç klerensi muhtemelenböbrek fonksiyonları normal hastalardakinin bir benzeridir çünkü böbrekler yoluyla atılımimatinib için minör bir eliminasyon yolunu oluşturmaktadır (bkz. 4.2 Pozoloji ve uygulamaşekli, 4.4 Özel kullanım uyarıları ve önlemleri).
Farmakokinetik analiz sonuçlarının kişiden kişiye değişikliklerin söz konusu olduğunu göstermesine rağmen, değişik derecelerde karaciğer yetersizliği olan hastalardaki imatinibeortalama maruz kalım, karaciğer fonksiyonları normal olan hastalara kıyasla yükselmemiştir(bkz 4.2 Pozoloji ve uygulama şekli, 4.4 Özel kullanım uyarıları ve önlemleri, 4.8 İstenmeyenEtkiler)
5.3. Klinik öncesi güvenlilik verileri
İmatinibin klinik öncesi güvenliliği sıçanlarda, değerlendirilmiştir.
Çoklu doz toksisite çalışmaları sıçanlarda, köpeklerde ve maymunlarda hafif ile orta dereceli hematolojik değişiklikler ortaya koymuş, sıçanlarda ve köpeklerde bu değişikliklere kemik iliğideğişiklikleri eşlik etmiştir.
Karaciğer, sıçanlarda ve köpeklerde hedef organ olmuştur. İki türde de transaminazlarda hafif ila orta dereceli artışlar ve kolesterol, trigliseritler, total protein ve albümin düzeylerinde hafifdüşüşler gözlenmiştir. Sıçan karaciğerinde herhangi bir değişiklik görülmemiştir. İki haftasüreyle tedavi edilen köpeklerde yükselmiş karaciğer enzimleri, hepatoselüler nekroz, safrakanalı nekrozu ve safra kanalı hiperplazisi ile şiddetli karaciğer toksisitesi gözlenmiştir.
İki hafta süreyle tedavi edilen maymunlarda fokal mineralizasyon ve renal tübüllerin dilatasyonu ve tübüler nekroz ile renal toksisite gözlenmiştir. Bu hayvanların birkaçında kanüre azotunda (BUN) ve kreatininde artış gözlenmiştir. 13 haftalık çalışmada sıçanlarda > 6mg/kg dozlarda serum veya idrar parametrelerinde değişiklikler olmaksızın mesane ve renalpapilla transisyonel epitelyum hiperplazisi gözlenmiştir. Kronik imatinib tedavisi ile fırsatçıenfeksiyonların oranında artış gözlenmiştir.
39 haftalık maymun çalışmasında NOAEL (advers etkinin görülmediği düzey), 15 mg/kg olan en düşük dozda saptanmış olup bu doz, beden yüzeyi bazında 800 mg'lık maksimum insandozunun yaklaşık üçte biridir. Tedavi, bu hayvanlarda normalde baskılanmış olan malaryalenfeksiyonlarda kötüleşme ile sonuçlanmıştır.
İmatinib, bir in vitro bakteriyel hücre testinde (Ames test), bir in vitro memeli hücre testinde (fare lenfoması) ve bir in vivo sıçan mikronükleus testinde test edildiğinde genotoksik etkigöstermemiştir. Metabolik aktivasyon varlığında klastojenisiteye (kromozom aberasyonu)yönelik bir in vitro memeli hücre testinde (Çin hamsterı overi) imatinib için pozitif genotoksiketkiler elde edilmiştir. Üretim prosesinin, son üründe de bulunan iki ara ürünü Ames testindemutajenisite açısından pozitiftir. Bu ara ürünlerden biri ayrıca fare lenfoma testinde de pozitifsonuç vermiştir.
Bir fertilite çalışmasında çiftleşmeden önce 70 gün süreyle dozlar uygulanan erkek sıçanlarda testiküler ve epididimal ağırlıklar ve hareketli sperm yüzdesi, beden yüzey alanı bazında 800mg/gün maksimum klinik dozu ile yaklaşık olarak eşit olan 60 mg/kg dozunda azalmıştır. Buetki < 20 mg/kg dozlarda görülmemiştir. Köpekte > 30 mg/kg oral dozlarda spermatogenezdede hafif ila orta dereceli bir azalma gözlenmiştir. Dişi sıçanlar çiftleşmeden önce 14 gün süreyleve 6. gestasyon gününe kadar dozlar uygulandığında çiftleşme ya da gebe hayvan sayısında
Belge D(heffhıangiiubiz\\etkk m®ajkanösuiOQlm:affli|tirFy60 mg/kge dozuadai :idişi//\sıçamJ)afda'önemliitölçüde
implantasyon sonrası fetal kayıp ve canlı fetüs sayısında azalma olmuştur. Bu etki < 20 mg/kg dozlarda görülmemiştir.
Sıçanlarla yürütülen bir oral prenatal ve postnatal gelişim çalışmasında 45 mg/kg/gün grubunda gestasyonun 14 ya da 15. gününde kırmızı vajinal akıntı kaydedilmiştir. Aynı dozda ölü doğanyavru sayısı ve ayrıca doğum sonrası 0-4 günler arasında ölen yavru sayısı da artmıştır. F1yavrularda aynı doz düzeyinde ortalama vücut ağırlıkları doğumdan öldürülene kadar geçensürede azalmıştır ve prepusyal ayrılma kriterine ulaşan yavru sayısı da hafif azalmıştır. 45mg/kg/gün dozunda F1 fertilitesi etkilenmemiş, diğer yandan rezorpsiyon sayısında artış vecanlı fetüs sayısında azalma tespit edilmiştir. Gerek anne hayvanlar gerekse F1 nesil için etkiningözlenmediği düzey (NOEL) 15 mg/kg/gün (800 mg'lık maksimum insan dozunun dörtte biri)olmuştur.
İmatinib organogenez sırasında, vücut yüzey alanı bazında 800 mg/gün maksimum klinik doz ile yaklaşık olarak eşit olan > 100 mg/kg dozlarda sıçanlara uygulandığında teratojen etkigöstermiştir. Teratojenik etkiler eksensefali veya ensefalosel, frontal kemiklerinolmaması/eksik olması ve parietal kemiklerin olmamasını içermiştir. Bu etkiler < 30 mg/kgdozlarda görülmemiştir.
Sıçanlarda juvenil gelişim toksikolojisi çalışmasında yeni hedef organ belirlenmemiştir (doğum sonrası 10 ila 70. gün). Juvenil toksikoloji çalışmasında, ortalama pediyatrik maruziyet olarakönerilen en yüksek doz olan 340 mg/m2 düzeyinin yaklaşık 0,3 ila 2 katı düzeylerde, büyümeüzerinde geçici etkiler ve vaginal açılma ve prepusyal ayrılmada gecikme gözlenmiştir. Ayrıca,ortalama pediyatrik maruziyet olarak önerilen en yüksek doz olan 340 mg/m2 düzeyininyaklaşık 2 katı düzeylerde, juvenil hayvanlarda (yaklaşık olarak sütten kesilme döneminde)mortalite gözlemlenmiştir.
2 yıllık sıçan karsinojenisite çalışmasında 15, 30 ve 60 mg/kg/gün olarak imatinib uygulanması, erkeklerde 60 mg/kg/gün dozunda ve dişilerde > 30 mg/kg/gün dozunda yaşam süresi üzerindeistatistiksel açıdan anlamlı azalmaya neden olmuştur. Ölenlerde yapılan histopatolojikinceleme, ölümün temel nedeni ya da öldürülme nedeni olarak kardiyomiyopati (her ikicinsiyet), kronik ilerleyici nefropati (dişiler) ve prepusyal bez papillomunu ortaya koymuştur.Neoplastik değişiklikler açısından hedef organlar böbrekler, mesane, üretra, prepusyal veklitoral bez, ince bağırsak, paratiroid bezleri, adrenal bezler ve glandüler-olmayan mideolmuştur.
Prepusyal/klitoral bezde papilloma/karsinoma sırasıyla 400 mg/gün veya 800 mg/gün dozlarında insan günlük maruziyetinin 0,5 veya 0,3 katına (EAA bazında) ya da çocuklarda 340mg/m2/gün dozunda günlük maruziyetin 0,4 katına karşılık gelen (EAA bazında) 30 mg/kg/gündozundan itibaren gözlenmiştir. Etki gözlenmeyen düzey (NOEL) 15 mg/kg/gün olmuştur. 400mg/gün veya 800 mg/gün dozlarında insan günlük maruziyetinin sırasıyla 1,7 veya 1 katına(EAA bazında) ya da çocuklarda 340 mg/m2/gün dozunda günlük maruziyetin 1,2 katınakarşılık gelen (EAA bazında) 60 mg/kg/gün dozunda renal adenoma/karsinoma, mesane ve
^ Bu'belge, güvfenlı ^KtroniK imza ile imzalanmıştır.
bezlerde benign ve malign medüller tümörler ve glandüler olmayan mide papillomaları/karsinomalan görülmüştür. Etki gözlenmeyen düzey (NOEL) 30 mg/kg/günolmuştur.
Sıçan karsinojenisite çalışmalarından elde edilen bu bulguların insanlar için anlamı bilinmemektedir.
Erken dönem klinik çalışmalarda tanımlanmayan non-neoplastik lezyonlar kardiyovasküler sistem, pankreas, endokrin organlar ve dişlerle ilgili olmuştur. En önemli değişiklikler bazıhayvanlarda kalp yetmezliği belirtilerine yol açan kardiyak hipertrofi ve dilatasyonu içermiştir.Etkin madde imatinib, tortul tabaka organizmaları için çevresel risk oluştur.
6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesi:
Mikrokristalin selüloz (Tip-101, Tip 102)
Hidroksipropil Metil Selüloz (E5)
Kolloidal Silikon Dioksit Magnezyum stearatKrospovidon XLPovidon K-30Opadry turuncu*
*Opadry turuncu;
Hipromelloz
Titanyum dioksit
Polietilen glikol/Makrogol
Demir oksit; sarı tahriş olamayan
Kırmızı demir oksit boyar maddelerini içermektedir.
6.2. Geçimsizlikler
Geçerli değildir.
6.3. Raf ömrü
24 ay
6.4. Saklamaya yönelik özel uyarılar
30 oC'nin altındaki oda sıcaklığında orijinal ambalajında saklanmalıdır.
6.5. Ambalajın niteliği ve içeriği:
PVC/PVDC-Alu blister ambalaj
NİBTU 100 mg film kaplı tablet, 120 adet, blisterde
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler
Kullanılmamış olan ürünler ya da atık materyaller 'Tıbbi Atıkların Kontrolü Yönetmeliği' ve 'Ambalaj Atıklarının Kontrolü Yönetmeliği'ne uygun olarak imha edilmelidir.
7. RUHSAT SAHİBİ
Onko İlaç Sanayi ve Ticaret A.Ş Gebze OSB2 Mah. 1700. Sk. 1703/2Çayırova/KOCAELİTelefon: 0850 250 66 56e-mail:
info@onkokocsel .com
8. RUHSAT NUMARASI
2022/773
9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİ
İlk Ruhsat Tarihi:Ruhsat Yenileme Tarihi:
10. KÜB'ÜN YENİLENME TARİHİ
İlaç Bilgileri
Nibtu 100 Mg Film Kaplı Tablet
Etken Maddesi: İmatinib Mesilat