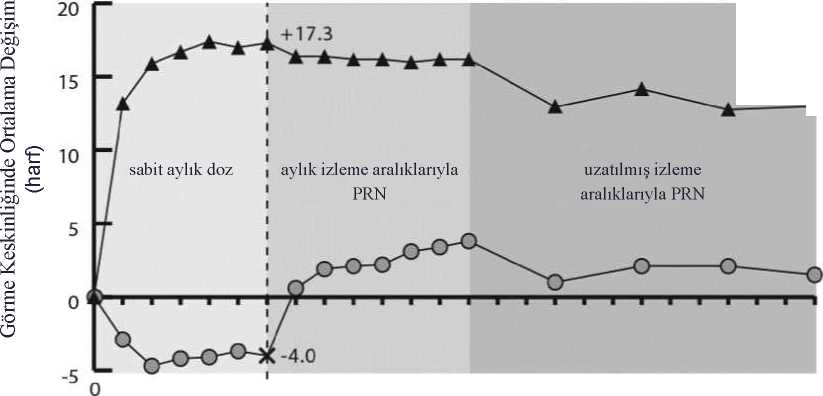
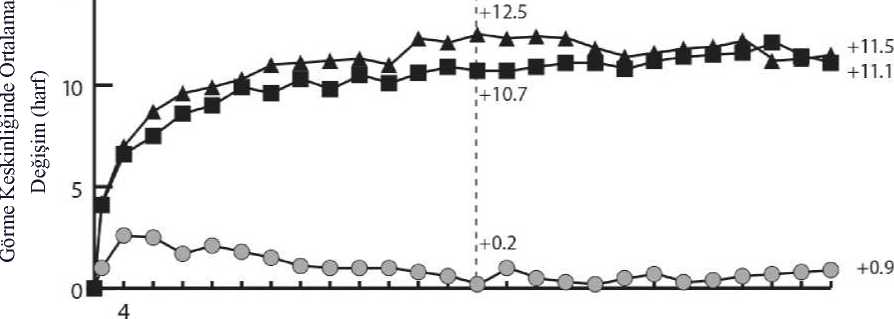
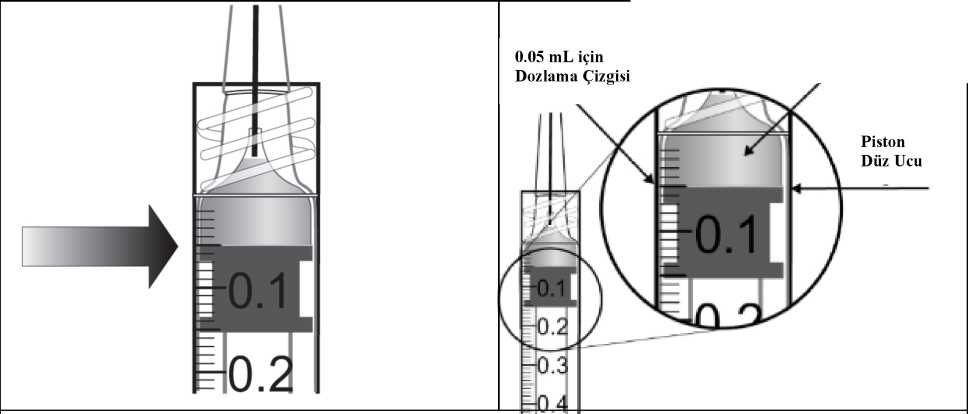
Eylea 40mg/ml İntravitreal Enjeksiyon İçin Çözelti İçeren Flakon Kısa Ürün BilgisiKISA URUN BILGISI1. BEŞERI TIBBİ ÜRÜNÜN ADIEYLEA® 40 mg/ml intravitreal enjeksiyon için çözelti içeren flakon Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:1 mL intravitreal enjeksiyon için çözeltide, Aflibersept* 40 mg Bir flakon, yaklaşık, en az 4 mg aflibersepte eşdeğer 0,1 mL'lik ekstrakte edilebilir hacim içerir. Bu, 2 mg aflibersept içeren 0,05 mL tek bir dozun sağlanabilmesi için yeterli miktar ihtiva eder. *İnsan VEGF (Vasküler Endotelyal Büyüme Faktörü) reseptörleri 1 ve 2'nin ekstraselüler bölgelerinin parçalarından meydana gelen, insan IgG1'in Fc parçası ile birleştirilmiş ve Çinhamsteri yumurtalığında (CHO) K1 hücrelerinde rekombinant DNA teknolojisi ile üretilmişfüzyon proteini. Yardımcı maddeler:1,104 mg/mL 0,537 mg/mL2,338 mg/mL Sodyum dihidrojen fosfat, monohidrat (pH ayarlaması için) Disodyum hidrojen fosfat, heptahidrat (pH ayarlaması için)Sodyum klorür Yardımcı maddelerin tam listesi için 6.1'e bakınız. 3. FARMASOTİK FORMİntravitreal enjeksiyon için çözelti Steril, berrak, renksiz ila soluk sarı arası renkte, pH'sı 6.2 olan izo-ozmotik sulu çözelti. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarEYLEA, yetişkinlerde aşağıdakilerin tedavisi için endikedir: ¦ Neovasküler (yaş tip) yaşa bağlı makula dejenerasyonu (YBMD) (bkz. bölüm 5.1), ¦ Retinal ven oklüzyonuna (retinal ven dal oklüzyonu -RVDO- ya da santral retinal venoklüzyonu -SRVO-) sekonder makula ödemine bağlı görme bozukluğu (bkz. bölüm 5.1), ¦ Diyabetik makula ödeminden (DMÖ) kaynaklanan görme bozukluğu (bkz. bölüm 5.1), ¦ Miyopik koroidal neovaskülarizasyona (miyopik KNV) bağlı görme bozukluğu (bkz.bölüm 5.1). 4.2. Pozoloji ve uygulama şekliEYLEA yalnızca intravitreal enjeksiyon içindir. EYLEA, intravitreal enjeksiyon uygulamasında deneyimli, göz hastalıkları uzmanı olan bir hekim tarafından uygulanmalıdır. Pozoloji/uygulama sıklığı ve süresi:Yaş tip YBMD (yaşa bağlı makula dejenerasyonu):Önerilen EYLEA dozu, 0,05 mL'ye eşdeğer 2 mg aflibersepttir. EYLEA tedavisinin, birbirini takip eden üç doz şeklinde ayda bir kez tek enjeksiyon olarak başlatılması ve daha sonra, tedavi aralığının iki ay olacak şekilde uzatılması önerilir. Hekimin görmeyle ilgili ve/veya anatomik sonuçlara ilişkin takdiri doğrultusunda, tedavi aralığı iki ay olarak kalabilir ya da tedavi et ve uzat dozlama rejimi kullanılarak uzatılabilir, buyaklaşımda görmeyle ilgili ve/veya anatomik sonuçların korunması için enjeksiyon aralıkları2 veya 4 haftalık kademelerle arttırılır. Görmeyle ilgili ve/veya anatomik sonuçlarda kötüleşme olması halinde, tedavi aralığı uygun şekilde kısaltılmalıdır. Enjeksiyonlar arasında izlem yapılması gerekmez. Hekimin takdirine göre, izlem vizitlerinin programı enjeksiyon vizitlerinden daha sık olabilir. Enjeksiyonlar arasında dört aydan uzun veya 4 haftadan kısa tedavi aralıkları değerlendirilmemiştir (bkz. Bölüm 5.1). RVO'ya sekonder makula ödemi (RVDO veya SRVO)Önerilen EYLEA dozu, 0,05 mL'ye eşdeğer 2 mg aflibersepttir. İlk enjeksiyondan sonra tedavinin ayda bir kez uygulanması önerilir. İki doz arasında geçen süre bir aydan kısa olmamalıdır. Anatomi ve görmeyle ilgili sonuçların hastanın tedaviden fayda görmediğine işaret etmesi halinde EYLEA tedavisi kesilmelidir. Aylık tedavinin maksimum görme keskinliği elde edilene ve/veya hastalık aktivitesine ilişkin belirti kalmayana kadar devam ettirilmesi önerilir. Ardışık üç veya daha fazla aylık enjeksiyongerekebilir. Tedavi daha sonra tedavi et ve uzat rejimiyle devam ettirilebilir, görmeyle ilgili ve/veya anatomik sonuçların stabil kalması sağlanarak tedavi aralıkları kademeli şekilde uzatılabilir,ancak bu aralıkların uzunluklarının belirlenmesi için gerekli veriler yetersizdir. Görmeyle ilgilive/veya anatomik sonuçların kötüleşmesi halinde, tedavi aralığı buna uygun şekildekısaltılmalıdır. İzlem ve tedavi programı tedavi eden hekim tarafından hastaya özgü yanıt doğrultusunda belirlenmelidir. Hastalık aktivitesinin izlemi klinik muayene, fonksiyon testi ya da görüntüleme tekniklerini (örn. optik koherens tomografisi ya da floresan anjiyografi) içerebilir. Diyabetik makula ödemiEYLEA için önerilen tedavi şekli, 0,05 mL'ye eşdeğer 2 mg afliberseptin ardışık beş doz olmak üzere ayda bir kez tek enjeksiyonudur ve takiben iki ayda bir tek enjeksiyon olarak devamettirilmesidir. Hekimin görmeyle ilgili ve/veya anatomik sonuçları değerlendirmesi doğrultusunda tedavi aralığı 2 ay olarak tutulabilir veya bireyselleştirilebilir; örneğin görmeyle ilgili ve/veyaanatomik sonuçların korunması için tedavi aralıklarının genellikle 2 haftalık kademelerleartırıldığı tedavi et ve uzat dozlama rejimi kullamlabilir. Dört aydan daha uzun tedavi aralıklarıile ilgili olarak sınırlı veri bulunmaktadır. Görmeyle ilgili ve/veya anatomik sonuçlarınkötüleşmesi halinde, tedavi aralığı buna uygun şekilde kısaltılmalıdır. 4 haftadan daha kısatedavi aralıkları değerlendirilmemiştir (bkz. Bölüm 5.1). İzlem programı tedavi eden hekim tarafından belirlenmelidir. Anatomi ve görmeyle ilgili sonuçların hastanın tedaviden fayda görmediğine işaret etmesi halinde EYLEA tedavisi kesilmelidir. Protokol T çalışma tasarımına göre, EYLEA tedavisi aşağıda belirtilen şekilde uygulanabilir: Aflibersept başlangıçta ve ardından, uygunluk eşiğinin altında bir santral alt alan kalınlığı(SAAK) ile, görme keskinliği (GK) 20/20 ya da daha iyi değilse (Yaklaşık 85 harf değerindeGK skoruna eşdeğer Snellen) ve önceki son iki enjeksiyona cevaben iyileşme ya da kötüleşmeolmamışsa 4 haftada bir enjekte edilmiştir (bkz. Bölüm 5.1). İlk yıl boyunca, takip vizitleri herdört haftada bir (±1 hafta) gerçekleşmiştir. 24. haftadaki vizitten itibaren, görme keskinliği ve SAAK ne olursa olsun, iki ardışık enjeksiyondan sonra iyileşme veya kötüleşme olmamışsa enjeksiyon durdurulmuş, ancakgörme keskinliği harf skoru veya SAAK kötüleşmişse tedavi yeniden başlatılmıştır (bkz.Bölüm 5.1) Miyopik koroidal neovaskülarizasyonEYLEA için önerilen doz 0,05 mL'ye eşdeğer 2 mg aflibersept ile tek bir intravitreal enjeksiyondur. Görmeyle ilgili ve/veya anatomik sonuçlar hastalığın persistan olduğunu düşündürdüğü takdirde ilave dozlar uygulanabilir. Rekürrensler hastalığın yeni ortaya çıkmış gibi tedaviedilmesi önerilir. İzlem programı tedavi eden hekim tarafından belirlenmelidir. İki doz arasında geçen süre bir aydan kısa olmamalıdır. Uygulama şekli:İntravitreal enjeksiyonlar tıbbi standartlar ile geçerli kılavuzlara uygun şekilde ve intravitreal enjeksiyon konusunda deneyimli bir hekim tarafından yapılmalıdır. Genel olarak, yeterlianestezi ve topikal geniş spektrumlu mikrobisit dahil aseptik koşullar (örn. perioküler deri, gözkapağı ve oküler yüzeye povidon iyodür uygulama) sağlanmalıdır. Cerrahi el dezenfenksiyonu,steril eldiven, steril örtü ve steril göz kapağı spekulumu (veya eşdeğeri) önerilmektedir. Enjeksiyon iğnesi limbusa 3.5-4.0 mm posterior konumdan vitreus boşluğuna batırılmalı, yatay meridyenden kaçınılarak kürenin merkezi hedeflenmelidir. Enjeksiyon hacmi 0.05 mLolmalı;sonraki enjeksiyonlar için diğer bir sklera bölgesi kullanılmalıdır. İntravitreal enjeksiyondan hemen sonra hastalar göz içi basınçta artış için kontrol edilmelidir. Uygun takip yöntemi optik sinir başı için perfüzyon kontrolü veya tonometri şeklinde olabilir.Gerektiğinde parasentez için uygun steril ekipman bulundurulmalıdır. İntravitreal enjeksiyondan sonra hastalar endoftalmi düşündüren her türlü semptomu gecikmeden bildirmeleri gerektiği konusunda bilgilendirilmelidir (örn. göz ağrısı, gözdekızarıklık, fotofobi, bulanık görme). Bir flakon yalnızca bir kez ve tek gözün tedavisinde kullanılmalıdır. Tek bir flakondan birden fazla dozun çekilmesi kontaminasyon ile bunu takiben enfeksiyon riskini arttıracaktır. Flakon önerilen 2 mg'lık aflibersept (0,05 mL enjeksiyonluk çözeltiye eşdeğer) dozundan fazlasını içerir. Flakondan çekilebilen hacim, enjektörden çıkabilen miktardır ve tamamıkullanılmayacaktır. EYLEA kullanıma hazır enjektörden çekilebilecek hacim en az 0,1mL'dir. Önerilen dozun enjeksiyonundan önce fazla hacim mutlaka atılmalıdır.Flakon içindeki hacmin tamamının enjekte edilmesi doz aşımına neden olabilir. Fazla tıbbi ürünle birlikte hava kabarcığının uzaklaştırılması için piston yavaşça bastırılmalı ve piston kubbesinin(kubbenin ucu değil) ile şırınga üzerindeki siyah dozlama çizgisininaynı hizaya gelmesi sağlanmalıdır(bkz. Bölüm4.9 ve 6.6).Enjeksiyondan sonra kullanılmamış olan ürün atılmalıdır. Uygulama öncesinde tıbbi ürünün hazırlanmasıyla ilgili talimatlar için, bkz. bölüm 6.6. Özel popülasyonlara ilişkin ek bilgiler:Karaciğer ve/veya böbrek yetmezliği:Karaciğer ve/veya böbrek yetmezliği olan hastalarda EY^EA ile ilgili özel bir çalışma yapılmamıştır. Mevcut veriler bu hastalarda EYLEA için doz ayarlaması gerektiğine işaretetmemektedir (bkz. bölüm 5.2). Geriyatrik popülasyon:Özel bir uygulama yapılması gerekmez. 75 yaşın üzerindeki DMÖ'lü hastalara ilişkin deneyim sınırlıdır. Pediyatrik popülasyon:EYLEA'nın çocuklarda ve adolesanlarda güvenlilik ve etkililiği belirlenmemiştir. EYLEA'nın yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV endikasyonları için pediyatrikpopülasyonda kullanımı söz konusu değildir. 4.3. Kontrendikasyonlar¦ Etkin madde olan aflibersepte veya bölüm 6.1'de listelenmiş olan yardımcımaddelerden herhangi birine aşırı duyarlılık, ¦ Aktif veya şüpheli oküler ya da perioküler enfeksiyon, ¦ Aktif şiddetli intraoküler inflamasyon. 4.4. Özel kullanım uyarıları ve önlemleriİzlenebilirlikBiyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası açık bir şekilde kaydedilmelidir. İntravitreal enjeksiyon ile ilişkili reaksiyonlarEYLEA ile uygulananlar dahil intravitreal enjeksiyonlar endoftalmi, intraoküler inflamasyon, regmatojen retina dekolmanı, retina yırtılması ve iatrojenik travmatik katarakt ileilişkilendirilmiştir (bkz. bölüm 4.8). EYLEA uygulanırken daima uygun aseptik enjeksiyonteknikleri kullanılmalıdır. Hastalar ayrıca, enfeksiyon oluşursa erken tedaviye olanak sağlamakiçin enjeksiyonu takip eden hafta boyunca gözlenmelidir. Hastalardan, endoftalmi ya dayukarıda bahsi geçen diğer olayları düşündüren bir semptomla karşılaşmaları halindegecikmeden durumu bildirmesi istenmelidir. Flakon, önerilen 2 mg aflibersept dozundan (0,05 mL'ye eşdeğer) fazlasını içerir. Fazla hacim uygulamadan önce dışarı atılmalıdır (bkz. bölüm 4.2 ve 6.6). EYLEA enjeksiyonu dahil intravitreal enjeksiyonlar sonrasında 60 dakika içinde göz içi basıncın arttığı görülmüştür (bkz. bölüm 4.8). Glokomun yeterli kontrol altında olmadığıhastalarda özellikle dikkatli olunmalıdır (göz içi basıncı >30 mmHg olanlara EY^EAenjeksiyonu yapılmamalıdır). Bu nedenle göz içi basınç ve optik sinir başının perfüzyonu tümolgularda izlenmeli ve uygun şekilde kontrol altına alınmalıdır. İmmünojenisiteEY^EA terapötik bir protein olduğundan, immünojenisite potansiyeli bulunmaktadır (bkz. bölüm 4.8). Hastalardan, aşırı duyarlılığa atfedilebilecek klinik bulgular olabileceği için ağrı,fotofobi ya da kızarıklık gibi intraoküler inflamasyon belirti veya semptomlarını bildirmeleriistenmelidir. Sistemik etkilerOküler olmayan hemorajiler ve arteriyel tromboembolik olaylar dahil sistemik advers olaylar, VEGF inhibitörlerinin intravitreal enjeksiyonunu takiben bildirilmiştir ve bunların VEGFinhibisyonu ile ilişkili olabileceğine yönelik teorik bir risk vardır. Son 6 ay içinde inme, geçiciiskemik atak ya da miyokard enfarktüsü öyküsü olan SRVO, RVDO, DMÖ ya da miyopikKNV'li hastaların tedavisinde güvenlilik ile ilgili veriler sınırlıdır. Bu tip hastaların tedavisindedikkatli olunmalıdır. DiğerY^MD, SRVO, RVDO, DMÖ ve miyopik KNV'nin diğer intravitreal anti-VEGF tedavilerinde olduğu gibi aşağıdaki durumlar geçerlidir: Her iki göze eşzamanlı olarak uygulanan EY^EA'nın güvenlilik ve etkililiği sistematikolarak çalışılmamıştır (bkz. bölüm 5.1). Aynı zamanda bilateral tedavi uygulanıyorsa, budurum sistemik maruziyette artışa neden olabilir ve sistemik advers olay riskiniartırabilir. Diğer anti-VEGF'lerin (vasküler endotelyal büyüme faktörü) eşzamanlı kullanımı EYLEA'nın diğer anti-VEGF tıbbi ürünlerle (sistemik ya da oküler) eşzamanlıkullanımına ilişkin veri bulunmamaktadır. Yaş tip Y^MD için yapılan anti-VEGF tedavisinden sonra retina pigment epitelinde yırtılma olmasıyla ilişkili risk faktörleri arasında retina pigment epitelinde büyük ve/veyayüksek ayrılma yer almaktadır. EYLEA tedavisine başlanırken retina pigment epitelindeyırtılmayla ilgili risk faktörleri olan hastalarda dikkatli olunmalıdır. Regmatojen retina dekolmanı veya 3. ya da 4. evre makula deliği olan hastalardatedaviden kaçınılmalıdır. Retinal yırtık olması halinde doza ara verilmeli ve tedaviye ancak yırtık onarıldıktansonra devam edilmelidir. Aşağıdaki durumlarda doza ara verilmeli ve tedaviye planlanan bir sonraki tedavizamanından önce devam edilmemelidir: o en iyi düzeltilmiş görme keskinliği (EDGK) son görme keskinliği değerlendirmesine kıyasla en az 30 harf azalma olması;o fovea merkezinin etkilendiği subretinal kanama olması veya kanama alanınıntoplam lezyon alanının %50'si kadar ya da daha fazla olması. İntraoküler ameliyat yapılması veya planlanması halinde önceki veya sonraki 28 günsüresince doza ara verilmelidir. Olası fayda fetus ile ilgili olası riske üstün gelmediği sürece gebelik sırasında EYLEAkullanılmamalıdır (bkz. bölüm 4.6). Doğurganlık çağındaki kadınlar aflibersept tedavisi sırasında ve son intravitrealenjeksiyondan sonra en az 3 ay süreyle etkili bir kontrasepsiyon yöntemi kullanmalıdır(bkz. bölüm 4.6). İskemik SRVO ve RVDO'lu hastaların tedavisine ilişkin deneyim sınırlıdır. Geridönüşümsüz iskemik görme fonksiyonu kaybına ilişkin klinik belirtilerin saptandığıhastaların tedavisi için önerilmez. Kısıtlı verilere sahip popülasyonlarTip I diyabet kaynaklı DMÖ'lü hastaların, HbA1c değeri %12'nin üzerinde olan diyabetli hastaların ya da proliferatif diyabetik retinopatili hastaların tedavisi konusunda sınırlı verimevcuttur. EYLEA, aktif sistemik enfeksiyonları bulunan hastalarda ve retina dekolmanı ya da makula deliği gibi eşzamanlı göz hastalıkları bulunan hastalarda henüz çalışılmamıştır. Ayrıcahipertansiyonu kontrol altında olmayan diyabetik hastalarda EYLEA ile tedavi deneyimiyoktur. Bu bilgi eksikliği, hastaların tedavisi sırasında hekim tarafından göz önündebulundurulmalıdır. Miyopik KNV'de, Asyalı olmayan hastaların, daha önce miyopik KNV tedavisi görmüş hastaların ve ekstrafoveal lezyonları bulunan hastaların tedavisinde EYLEA deneyimi mevcutdeğildir. Bu tıbbi ürün her dozunda 1 mmol (23 mg)'den daha az sodyum ihtiva eder; yani esasında sodyum içermediği kabul edilebilir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriHerhangi bir etkileşim çalışması yapılmamıştır. Verteporfin ile fotodinamik tedavi (PDT) ve EYLEA'nın birlikte kullanımı çalışılmamıştır; bu nedenle, güvenlilik profili belirlenmemiştir. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin herhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyonPediyatrik popülasyonlara ilişkin herhangi bir etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Doğurganlık çağındaki kadınlar aflibersept tedavisi sırasında ve son intravitreal enjeksiyondan sonra en az 3 ay süreyle etkili bir kontrasepsiyon yöntemi kullanmalıdır (bkz. bölüm 4.4). Gebelik dönemiGebelikte aflibersept kullanımıyla ilgili veri bulunmamaktadır. Hayvanlardaki çalışmalarında embriyo-fetal toksisite gösterilmiştir (bkz. bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. Oküler uygulama sonrasında sistemik maruziyet son derece düşük olmakla birlikte, olası fayda fetüs ile ilgili olası riske üstün gelmediği sürece gebelik sırasında EYLEA kullanılmamalıdır. Laktasyon dönemiAfliberseptin insanlarda anne sütüne geçip geçmediği bilinmemektedir. Anne sütü almakta olan çocuğa yönelik risk dışlanamaz. Laktasyon döneminde EYLEA kullanımı önerilmez. Emzirmeye ara verilmesi veya EYLEA kullanımından kaçınılması konusu, emzirmenin çocuğa getirdiği yarar ile tedavinin kadınagetirdiği yarar dikkate alınarak kararlaştırılmalıdır. Üreme yeteneği/FertiliteYüksek sistemik maruziyet ile hayvan çalışmalarında elde edilen bulgular afliberseptin erkek ve kadın fertilitesini etkileyebileceğini göstermektedir (bkz. bölüm 5.3). Sistemik maruziyetinson derece düşük olduğu oküler uygulamalardan sonra bu tip etkiler beklenmez. 4.7. Araç ve makine kullanımı üzerindeki etkilerEYLEA enjeksiyonu, enjeksiyon veya göz muayenesiyle ilişkili olası geçici görme bozukluğu nedeniyle araç ve makine kullanma üzerinde minör bir etkiye sahiptir. Hastalar görmefonksiyonu tamamen normale dönmedikçe araç veya makine kullanmamalıdır. 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti: Sekiz faz III çalışmanın güvenlilik popülasyonu toplam 3102 hastadan oluşmaktadır. Bu hastaların 2501'i önerilen doz olan 2 mg dozuyla tedavi edilmiştir. Enjeksiyon prosedürüne bağlı olarak çalışılan gözde meydana gelen ciddi oküler advers reaksiyonlar, EYLEA ile 1900 intravitreal enjeksiyonun 1'inden azında meydana gelmiştir(körlük, endoftalmi, retina dekolmanı, travmatik katarakt, katarakt, vitreus kanaması, vitreusdekolmanı ve göz içi basınçta artışı) (bkz. bölüm 4.4). En sık görülen advers reaksiyonlar (EYLEA ile tedavi edilen hastaların en az %5'i), konjonktival hemoraji (%25), retinal hemoraji (%11), görme keskinliğinde azalma (%11), gözağrısı (%10), katarakt (%8), göz içi basınçta artış (%8), vitreus dekolmanı (%7) ve vitreustauçuşan noktalardır (%7). Advers reaksiyonların listesi: Aşağıda açıklanan güvenlilik verileri yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV ile ilgili sekiz faz III çalışmada ortaya çıkan ve enjeksiyon prosedürü veya tıbbi ürünler makulnedensellik olasılığı bulunan tüm advers reaksiyonları içermektedir. Advers reaksiyonlar sistem organ sınıfına ve sıklığa göre, aşağıdaki şekilde listelenmiştir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1000 ila <1/100); seyrek(>1/10.000 ila <1/1000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketletahmin edilemiyor). Her sıklık grubunda, advers ilaç reaksiyonları azalan ciddiyet sıralamasında sunulmuştur. Faz III çalışmalarda (yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV endikasyonları için faz III çalışmalardan havuzlanmış veriler) veya pazarlama sonrasıaraştırmalarda hastalarda bildirilen tüm tedavi sonucu ortaya çıkan advers ilaçreaksiyonlarıBağışıklık sistemi hastalıklarıYaygın olmayan: Aşırı duyarlılık*** Göz hastalıklarıÇok yaygın: Görme keskinliğinde azalma, retinal hemoraji, konjunktival hemoraji, göz ağrısı Yaygın: Retina pigment epitelinin yırtılması*, retina pigment epitel dekolmanı*, retinal dejenerasyon, vitreus kanaması, katarakt, kortikal katarakt, nükleer katarakt, subkapsülerkatarakt, kornea erozyonu, kornea abrazyonu, göz içi basıncının yükselmesi (intraoküler basınçartışı), bulanık görme, vitreusta uçuşan noktalar, vitreus dekolmanı, enjeksiyon yerinde ağrı,gözlerde yabancı cisim hissi, lakrimasyon artışı, göz kapağında ödem, enjeksiyon yerindehemoraji, punktat keratit, konjunktival hiperemi, oküler hiperemi Yaygın olmayan: Endoftalmit**, retina dekolmanı, retina yırtılması, iritis, üveit, iridosiklit, lentiküler opasiteler, kornea epitel defekti, enjeksiyon yerinde iritasyon, gözde anormal his,göz kapağı iritasyonu, ön kamarada flare, kornea ödemi Seyrek: Körlük, travmatik katarakt, vitrit, hipopiyon * Yaş tip YBMD ile ilişkili olduğu bilinen durumlardır; sadece yaş tip YBMD çalışmalarında gözlenmiştir. ** Kültür pozitif ve kültür negatif endoftalmit *** Pazarlama sonrası dönemde, aşırı duyarlılık raporları döküntü, kaşıntı, ürtiker ve ciddi anafilaktik/anaflaktoid reaksiyon olgularının izole vakalarını içermiştir. Seçili advers reaksiyonların tanımı: Yaş tip YBMD ile ilgili faz III çalışmalarında anti-trombotik ilaç alan hastalarda konjonktival kanama insidansında bir artış olduğu saptanmıştır. Bu insidans artışı ranibizumab ve EYLEAtedavisi alan hastalar arasında benzerlik göstermektedir. Arteriyel tromboembolik olaylar (ATO'lar) sistemik VEGF inhibisyonuyla potansiyel ilişkisi olan advers olaylardır. İntravitreal VEGF inhibitörlerinin kullanımının ardından, inme vemiyokard enfarktüsü dahil arteriyel tromboembolik olay gelişmesi bakımından teorik bir risksöz konusudur. YBMD, DMÖ, RVO ve miyopik KNV'li hastalarla yürütülen EYLEA klinik çalışmalarında, arteriyel tromboembolik olaylar açısından düşük bir insidans oranı görülmüştür. Endikasyonlargenelinde, aflibersept tedavisi alan gruplar ile ilgili karşılaştırma grupları arasında kayda değerbir fark gözlenmemiştir. Tüm terapötik proteinler gibi, EYLEA'nın da immünojenisite potansiyeli bulunmaktadır. Becton Dickinson filtreli iğne EYLEA flakon ambalajında yer alır ve EYLEA'nın doğru kullanımı için gereklidir. Becton Dickinson filtreli iğne, flakon içeriğini enjektöre çekmek içinflakon tıpasına itildiğinde, parçacık oluşumunu önlemek için silikon yağı ile kayganlaştırılır. EYLEA'nın flakon formunun uygulanmasının ardından gözün vitreusunda silikon yağıyla ilişkili uçuşan nokta izole vakaları bildirilmiştir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9. Doz aşımı ve tedavisiKlinik çalışmalarda aylık aralıklarla verilen 4 mg'a kadar olan dozlar kullanılmış ve 8 mg ile izole doz aşımı olgularına rastlanmıştır. Enjeksiyon hacminin artırılmasıyla gelişen doz aşımı, göz içi basıncın artmasına neden olabilir. Bu nedenle, doz aşımı durumunda göz içi basınç izlenmeli ve tedavi eden hekimin takdirinegöre gerekli bulunduğunda, uygun tedavi uygulanmalıdır (bkz. Bölüm 6.6). 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Oftalmolojikler, Oküler vasküler hastalıklarda kullanılan ajanlar, Antineovaskülarizasyon ilaçlarıATC kodu: S01LA05 Aflibersept, insan VEGF reseptörünün 1. ve 2. ekstraselüler parçaları ile insan IgG1'inin Fc kısmının birleştirildiği rekombinant bir füzyon proteinidir. Aflibersept, rekombinant DNA teknolojisi ile Çin hamster overi (CHO) K1 hücrelerinde üretilmektedir. Aflibersept VEGF-A ile PlGF'e doğal reseptörlerinden daha yüksek afiniteyle bağlanan çözünür bir tuzak reseptör görevi görür ve böylece aynı kökenden gelen bu VEGFreseptörlerinin bağlanmasını ve aktivasyonunu inhibe edebilir. Etki mekanizması: Vasküler endotelyal büyüme faktörü-A (VEGF-A) ve plasental büyüme faktörü (PlGF), anjiyogenik faktörlerin VEGF ailesinin endotel hücreleri için potent mitojenik, kemotaktik vevasküler geçirgenlik sağlayan üyeleridir. VEGF; iki reseptör tirozin kinaz olan ve endotelhücrelerinin yüzeyinde bulunan VEGFR-1 ve VEGFR-2 üzerinden etki göstermektedir. PlGFyalnızca VEGFR-1'e bağlanmakta ve VEGFR-1, lökosit yüzeyinde de bulunmaktadır. Bureseptörlerin VEGF-A tarafından aşırı derecede aktifleştirilmesi patolojik neovaskülarizasyonve aşırı vasküler geçirgenliğe neden olabilmektedir. PlGF bu süreçlerde VEGF-A ile sinerjigösterebilir ve ayrıca lökosit infiltrasyonu ile vasküler inflamasyonu da uyardığı bilinmektedir. Farmakodinamik etkiler:Yaş tip YBMDYaş tip Y^MD patolojik koroidal neovaskülarizasyon (KNV) ile karakterizedir. KNV'den kan ve sıvı sızması retinanın kalınlaşmasına ya da ödem ve/veya sub/intra-retinal kanamaya nedenolarak görme keskinliği kaybıyla sonuçlanabilmektedir. EYLEA tedavisi alan hastalarda (art arda üç ay süresince ayda bir kez tek enjeksiyon, ardından 2 ayda bir tek enjeksiyon) santral retina kalınlığı [SRK] tedavinin başlatılmasından kısa süre sonra azalmış ve ortalama KNV lezyonu boyutunun azaldığı görülmüştür; bu durum aylık0.5 mg ranibizumab ile görülen bulgular ile uyumludur. VIEW1 çalışmasında optik koherens tomografisinde (OKT) ortalama SRK azalmaları olduğu görülmüştür (iki ayda bir 2 mg EYLEA ve ayda bir 0.5 mg ranibizumab için 52. haftadasırasıyla -130 ve -129 mikron). Yine 52 haftalık zaman noktasında, VIEW2 çalışmasında retinakalınlığı bakımından ortalama azalma olduğu OKT ile ortaya konmuştur (iki ayda bir 2 mgEYLEA ve her ay 0.5 mg ranibizumab için sırasıyla -149 ve -139 mikron). KNV boyutundakiazalma ile retina kalınlığındaki azalmanın çalışmaların ikinci yılında genel olarak korunduğusaptanmıştır. ALTAIR çalışması önceden tedavi almamış yaş tip YBMD'li Japon hastalarla yürütülmüş, başlangıçta ayda bir kez 2 mg EYLEA olarak uygulanan 3 enjeksiyonun ardından 2 aygeçtikten sonra bir enjeksiyon daha yapılmış ve sonrasında maksimum aralık 16 hafta olmaküzere önceden belirlenmiş kriterlere göre değişen tedavi aralıklarıyla (2 haftalık veya 4 haftalıkayarlamalar) tedavi et ve uzat rejimi kullanılmış ve VIEW çalışmalarına benzer sonuçlarsergilemiştir. Elli ikinci haftada, 2 haftalık ayarlama grubu ve 4 haftalık ayarlama grubu için OKT'de santral retina kalınlığı (SRK) değerlerinde sırasıyla -134.4 ve -126.1 mikronluk ortalama azalmalar olduğu belirlenmiştir. Elli ikinci haftada 2 haftalık ve 4 haftalık ayarlama gruplarında OKT'de sıvı saptanmayan hastaların oranı sırasıyla%68.3 ve %69.1 olarak kaydedilmiştir. SRK'deki azalma, ALTAIR çalışmasının ikinci yılındaher iki tedavi kolunda da genel olarak korunmuştur. ARIES çalışması, başlangıçtaki aylık 3 enjeksiyonun ve 2 aydan sonra bir ilave enjeksiyonun uygulanmasının hemen ardından başlatılan EYLEA 2 mg tedavi et ve uzat dozlama rejiminin,bir yıllık tedaviden sonra başlatılan tedavi et ve uzat dozlama rejimine eşdeğerliğini araştırmakiçin tasarlanmıştır. Çalışmanın seyri sırasında en az bir kez Q8'den daha sık dozlama gerektirenhastalar için SRK yüksek kalmakla birlikte SRK'de başlangıçtan 104. haftaya kadar olanortalama azalma, Q8 veya daha az sıklıktaki aralıklarla tedavi edilmiş hastalara benzer şekilde-160.4 mikrondur. SRVO ve RVDO'ya sekonder makula ödemiSRVO ve RVDO'da retinal iskemi gelişmekte ve VEGF salınımı için sinyal oluşmakta, buna karşılık olarak sıkı bağlantıların stabilizasyonu bozulmakta ve endotel hücre proliferasyonuuyarılmaktadır. VEGF'nin yukarı düzenlenmesi kan retina bariyerinin yıkımıyla, vaskülergeçirgenliğin artmasıyla, retinal ödemle ve neovaskülarizasyon komplikasyonlarıyla ilişkilidir. Ardışık 6 ayda aylık 2 mg EYLEA enjeksiyonlarıyla tedavi alan hastalarda tutarlı, hızlı ve sağlam morfolojik yanıt elde edildiği (ortalama SRK iyileşmeleriyle ölçüldüğü üzere)gözlenmiştir. Yirmi dördüncü haftada, SRK azalmasının üç çalışmada da kontrole kıyaslaistatistiksel açıdan üstün olduğu kaydedilmiştir (SRVO'da COPERNICUS: -457 ve -145mikron; SRVO'da GALILEO: -449 ve -169 mikron; RVDO'da VIBRANT: - 280 ve -128mikron). Başlangıçtaki SRK değerinde kaydedilen azalma her bir çalışmanın sonuna kadar,yani COPERNICUS çalışmasında 100 hafta, GALILEO çalışmasında 76 hafta ve VIBRANTçalışmasında 52 hafta süreyle korunmuştur. Diyabetik makula ödemiDiyabetik makula ödemi, diyabetik retinopatinin bir sonucu olup, vasküler geçirgenlikte artış ve retinal kapilerlerde görme keskinliğinin azalmasına yol açabilen hasarla karakterizedir.Büyük bölümü Tip II diyabet olarak sınıflandırılan ve EYLEA tedavisi alan hastalardamorfolojide (SRK, DRSS düzeyi) hızlı ve sağlam yanıt olduğu gözlenmiştir. VIVIDDMEDMEDMEDMEçalışmalarında söz konusu azalma 100. haftada 2Q8 EYLEA grupları için sırasıyla -195.8 ve -191.1 mikron olarak, kontrol gruplarında ise -85.7 ve -83.9 mikron olarak korunmuştur.VIVIDDMEDMEDMEDMEçalışmasındaki hastaların %98.3'ünde DRSS skorunundeğerlendirilebilir olduğu kaydedilmiştir. Elli ikinci haftada, EYLEA 2Q8 gruplarının %27.7 ve %29.1'i ile kontrol gruplarının %7.5 ve %14.3'ünde DRSS'nin en az 2 basamak iyileştiğisaptanmıştır. Söz konusu oranlar 100. haftada EYLEA 2Q8 grupları için %32.6 ve %37.1;kontrol grupları için %8.2 ve %15.6 olmuştur. VIOLET çalışmasında, tedavinin 5 ardışık aylık dozla başlatıldığı ve ardından her 2 ayda bir doz verildiği sabit aralıklarla en az bir yıllık tedaviden sonra DMÖ tedavisi için EYLEA2 mg'ın üç farklı doz rejimi karşılaştırılmıştır. Çalışmanın 52. haftasında ve 100. haftasında,yani tedavinin ikinci ve üçüncü yıllarında, SRK'deki ortalama değişiklikler tedavi et ve uzat(2T&U, pro re nataMiyopik koroidal neovaskülarizasyonMiyopik koroidal neovaskülarizasyon (miyopik KNV), patolojik miyopi bulunan yetişkinlerde görme kaybının sık rastlanan nedenlerinden biridir. Bruch membranındaki rüptürler sonucundabir yara iyileşme mekanizması olarak gelişir ve patolojik miyopide görmeyi en fazla tehditeden olayı temsil eder. MYRROR çalışmasında EYLEA tedavisi alan (tedavi başlangıcında yapılan bir enjeksiyon ile persistan hastalık veya rekürrens durumunda yapılan ilave enjeksiyonlar) hastalarda SRKtedavi başladıktan kısa süre sonra EYLEA lehine olmak üzere 24 haftada azalmış (EYLEA2 mg tedavi grubu ile kontrol grubu için sırasıyla -79 mikron ve -4 mikron) ve bu azalma 48 haftasüresince korunmuştur. Ayrıca, ortalama KNV lezyon boyutunun azaldığı saptanmıştır. Klinik etkililik ve güvenlilikYaş tip YBMDEYLEA'nın güvenlilik ve etkililiği yaş tip YBMD'li hastaların yer aldığı iki randomize, çok merkezli, çift maskeli, aktif kontrollü çalışmada (VIEW1 ve VIEW2) değerlendirilmiş; toplam2,412 hastanın tedavi alıp, değerlendirilebilir olduğu kaydedilmiştir (EYLEA alan 1,817 hasta).Hastalarda yaş aralığının 49 ila 99, ortalama yaşın 76 olduğu belirlenmiştir. Klinik çalışmalardaEYLEA tedavisine randomize edilen hastaların yaklaşık %89'unun (1,616/1,817) 65 yaş veüzeri grupta, yaklaşık %63'ünün de (1,139/1,817) 75 yaş ve üzeri grupta olduğu kaydedilmiştir. Her bir çalışmada, hastalar aşağıdaki 4 dozlama rejiminden 1'i için 1:1:1:1 şeklinde rastgele atanmıştır: 1) Ayda bir uygulanan ilk 3 dozdan sonra 8 haftada bir 2 mg EYLEA (EYLEA 2Q8); 2) 4 haftada bir uygulanan 2 mg EYLEA (EYLEA 2Q4); 3) 4 haftada bir uygulanan 0.5 mg EYLEA (EYLEA 0.5Q4) ve 4) 4 haftada bir uygulanan 0.5 mg ranibizumab (ranibizumab 0.5Q4). Çalışmaların ikinci yılında hastalar randomize edilmiş oldukları dozajı almaya devam etmiş ancak dozlama programı anatomik ve görmeyle ilgili sonuçlara ilişkin değerlendirmeye göremodifiye edilmiş ve dozlama aralığı protokolde tanımlandığı gibi maksimum 12 haftaolmuştur. Her iki çalışmada da, primer etkililik sonlanım noktası Protokol Kümesinde yer alan ve görme yeteneğinin korunduğu hastaların oranı olmuş; görme yeteneğinin korunması, 52. haftadagörme keskinliğinde başlangıca göre saptanan kaybın en fazla 15 harf olması şeklindetanımlanmıştır. VIEW1 çalışmasının 52. haftasında EYLEA 2Q8 tedavi grubundaki hastaların %95.1'inde görme yeteneği korunurken, ranibizumab 0.5Q4 grubunda bu oran %94.4 olmuştur. VIEW2çalışmasının 52. haftasında EYLEA 2Q8 tedavi grubundaki hastaların %95.6'sında görmeyeteneği korunurken, ranibizumab 0.5Q4 grubunda bu oran %94.4 olarak kaydedilmiştir.EYLEA tedavisinin ranibizumab 0.5Q4 grubuyla benzer etkililik sergilediği ve klinik açıdaneşdeğer olduğu gösterilmiştir. İki çalışmaya ait kombine analizde elde edilen sonuçların ayrıntıları aşağıdaki Tablo 1 ve Şekil 1'de yer almaktadır. Tablo 1: 52. haftadaki (primer analiz) ve 96. haftadaki etkililik sonuçları; VIEW1 ve VIEW2 çalışmalarından elde edilen birleştirilmiş veriler
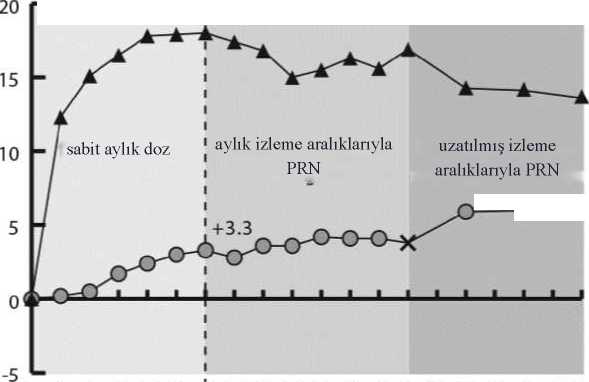
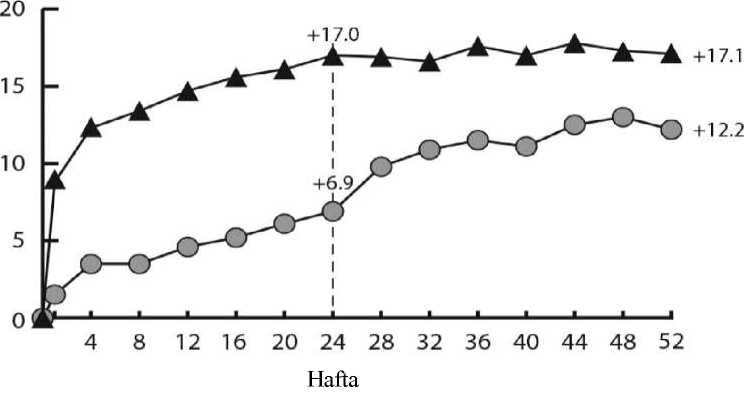
ETDRS: Erken Tedavi Diyabetik Retinopati Çalışması LS: ANCOVA'dan türetilmiş en küçük kare ortalamalarıPPS Protokol Uyarınca Seti 52. haftada PPS olan korunmuş görme keskinliği olan hastaların oranı hariç tüm analizler için Tam Analiz Seti (FAS), Yapılan Son Gözlem (LOCF) D) Normal yaklaştırmayla hesaplanan güven aralığı (CI) E) Üç aylık dozlarla tedavi başlatıldıktan sonra F) Tamamen -%10'un üzerinde bulunan güven aralığı, EYLEA'nın ranibizumab'a eşit olduğunu gösterir. Şekil 1.View1 ve View2 Çalışmalarından Birleştirilmiş Veriler için Başlangıçtan Hafta 96'ya Kadar Görme Keskinliğinde Ortalama Değişim

VIEW1 ve VIEW2 çalışmalarının birleştirilmiş veri analizlerinde, EYLEA önceden tanımlı sekonder etkililik sonlanım noktası olan Ulusal Göz Sağlığı Enstitüsü Görme FonksiyonuAnketinde (NEI VFQ-25) başlangıca göre klinik açıdan anlamlı değişiklikler sağlamış,ranibizumaba kıyasla klinik açıdan anlamlı fark görülmemiştir. Bu değişikliklerin büyüklüğüyayınlanmış olan çalışmalara benzer olup, En İyi Düzeltilmiş Görme Keskinliği (EDGK)bakımından 15 harflik kazanıma denk gelmiştir. Çalışmaların ikinci yılında, etkililik genel olarak 96. haftadaki son değerlendirmeye kadar korunmuş ve hastaların %2-4'ünde tüm enjeksiyonlar aylık şekilde gerekli olmuş, üçte birindeise tedavi aralığı yalnızca bir ay olmak üzere en az bir enjeksiyon gerekmiştir. KNV alanındaki azalmaların her iki çalışmada tüm doz grupları genelinde belirgin olduğu görülmüştür. Her iki çalışmada ve kombine analizde değerlendirilebilen tüm alt gruplarda (örn. yaş, cinsiyet, ırk, başlangıçtaki görme keskinliği, lezyon tipi, lezyon boyutu) elde edilen etkililik sonuçlarıgenel popülasyonlardaki sonuçlarla tutarlılık sergilemiştir. ALTAIR, önceden tedavi almamış yaş tip Y^MD'li 247 Japon hastanın yer aldığı, tedavi et ve uzat dozlama rejiminin iki farklı ayarlama aralığı (2 haftalık ve 4 haftalık) sonrasındaEYLEA'nın etkililik ve güvenliliğinin değerlendirilmesi amacıyla tasarlanmış çok merkezli,randomize, açık etiketli 96 haftalık bir çalışmadır. Tüm hastalar 3 ay süreyle ayda bir kez 2 mg EYLEA dozları almış, ardından 2 aylık bir aralık geçtikten sonra bir enjeksiyon daha yapılmıştır. Hastalar 16. haftada iki tedavi grubuna 1:1oranında randomize edilmiştir: 1) 2 haftalık ayarlamalarla EYLEA tedavi et ve uzat 2) 4haftalık ayarlamalarla EYLEA tedavi et ve uzat. Tedavi aralığının uzatılması veya kısaltılmasıkararı, protokolde tanımlanmış görmeyle ilgili ve/veya anatomik kriterlere dayalı olarak ve heriki grup için maksimum tedavi aralığı 16 hafta olacak şekilde belirlenmiştir. Primer sonlanım noktası olarak başlangıçtan 52. haftaya kadarki ortalama EDGK değişikliği değerlendirilmiştir. Sekonder etkililik sonlanım noktaları olarak, başlangıçtan 52. haftayakadar EDGK'de >15 harf kaybı olmayan hastaların oranı ve en az 15 harf kazanımı olanhastaların oranı değerlendirilmiştir. Elli ikinci haftada, 2 haftalık ayarlamalarla tedavi et ve uzat kolunda yer alan hastalarda başlangıca göre ortalama 9 harf kazanımı olurken, 4 haftalık ayarlama grubundakiler içinortalama kazanım 8.4 harf olmuştur [harflerde LS ortalama fark (%95 GA): -0.4 (-3.8,3),ANCOVA]. On beş veya daha fazla harf kaybı olmayan hastaların oranının iki tedavi kolundabenzer olduğu görülmüştür (2 haftalık ayarlama grubunda %96.7, 4 haftalık ayarlama grubunda%95.9). Elli ikinci haftada >15 harf kazanımı olan hastaların oranı 2 haftalık ayarlamagrubunda %32.5, 4 haftalık ayarlama grubunda %30.9 olarak saptanmıştır. Tedavi aralığı 12hafta veya daha uzun olacak şekilde uzatılan hastaların oranı 2 haftalık ayarlama grubunda%42.3, 4 haftalık ayarlama grubunda %49.6 olarak kaydedilmiştir. Ayrıca, 4 haftalık ayarlamayapılan gruptaki hastaların %40.7'sinde aralık 16 hafta olacak şekilde uzatılmıştır. Elli ikincihaftaya kadar yapılan son ziyarette, 2 haftalık ve 4 haftalık ayarlama gruplarındaki hastalarınsırasıyla % 56.8 ve %57.8'inde bir sonraki enjeksiyon 12 hafta veya daha uzun aralıklarlaplanlanmıştır. Çalışmanın ikinci yılında, ortalama kazanım başlangıçtan itibaren 2 haftalık ayarlama grubu için 7.6 harf ve 4 haftalık ayarlama grubu için 6.1 harf olmakla birlikte, etkililik genellikle96. haftadaki son değerlendirmeye kadar ve bu değerlendirme de dahil olmak üzerekorunmuştur. Tedavi aralıklarını 12 haftaya veya daha ilerisine kadar uzatan hastaların oranı,2 haftalık ayarlama grubunda %56.9 ve 4 haftalık ayarlama grubunda %60.2 olmuştur. 96.haftadan önceki son vizitte, 2 haftalık ayarlama grubundaki hastaların %64.9'u ve 4 haftalıkayarlama grubundaki hastaların %61.2'si bir sonraki enjeksiyonlarını 12 hafta veya daha uzunbir aralıkta planlatmıştır. Tedavinin ikinci yılında, hem 2 haftalık hem de 4 haftalık ayarlamagrubundaki hastalara sırasıyla ortalama 3.6 ve 3.7 enjeksiyon yapılmıştır. 2 yıllık tedaviperiyodu süresince hastalara ortalama 10.4 enjeksiyon yapılmıştır. Oküler ve sistemik güvenlilik profilleri pivot VIEW1 ve VIEW2 çalışmalarında gözlenen güvenlilik ile benzerdir. ARIES çalışması daha önce tedavi almamış yaş tip YBMD'li 269 hastada yürütülen 104 haftalık, çok merkezli, randomize, açık etiketli, aktif kontrollü bir çalışmadır. Üç ardışık aylıkdozdan sonra tedavi aralığının 2 aya uzatılmasının ardından başlatılan bir tedavi et ve uzatdozlama rejiminin etkililik ve güvenlilik açısından, tedavinin birinci yılından sonra başlatılanbir tedavi et ve uzat rejimine eşdeğerliğini değerlendirmek üzere tasarlanmıştır. ARIES çalışmasında aynı zamanda araştırmacının kararı doğrultusunda her 8 haftada birden daha sık tedavi gerektiren hastaların yüzdesi de araştırılmıştır. İki yüz altmış dokuz hastadan62 hastaya çalışmanın seyri sırasında en az bir kez daha sık doz uygulanmıştır. Bu hastalarçalışmada kalmış ve araştırmacının en iyi klinik muhakemesi doğrultusunda ancak her 4haftada birden daha sık olmayacak şekilde tedavi almış ve tedavi aralıkları sonrasında tekraruzatılabilmiştir. Daha sık tedavi kararından sonraki ortalama tedavi aralığı 6.1 haftadır. 104.hafta EDGK değeri çalışmanın seyri sırasında en az bir kez daha yoğun tedavi gerektirenhastalarda gerektirmeyen hastalara kıyasla daha düşük olup, EDGK'de çalışmanın sonundabaşlangıca göre ortalama değişiklik +2.3±15.6 harftir. Daha sık tedavi edilen hastalararasında, %85.5'i görmesini korumuş, yani 15 harften az kaybetmiş ve %19.4'ü 15 harf veyadaha fazla kazanmıştır. Her 8 haftada birden daha sık tedavi edilen hastaların güvenlilik profiliVIEW 1 ve VIEW 2'deki güvenlilik verileriyle karşılaştırabilir. SRVO'ya sekonder makula ödemiEYLEA'nın güvenlilik ve etkililiği SRVO'lu hastaların yer aldığı iki randomize, çok merkezli, çift maskeli, sham kontrollü çalışmada (COPERNICUS ve GALILEO) değerlendirilmiş;toplam 358 hastanın tedavi alıp, değerlendirilebilir olduğu kaydedilmiştir (EYLEA alan 217hasta). Hastalarda yaş aralığının 22 ila 89, ortalama yaşın 64 olduğu belirlenmiştir. SRVO ileilgili çalışmalarda EYLEA tedavisine randomize edilen hastaların yaklaşık %52'sinin(112/217) 65 yaş ve üzeri grupta, yaklaşık %18'inin de (38/217) 75 yaş ve üzeri grupta olduğukaydedilmiştir. Her iki çalışmada hastalar 4 haftada bir 2 mg EYLEA (2Q4) almak üzere veya4 haftada bir toplam 6 enjeksiyon yapılan sham kontrol grubunda yer almak üzere 3:2 oranındarastgele olarak atanmıştır. Ayda bir uygulanan 6 enjeksiyondan sonra yalnızca önceden belirlenen yeniden tedavi kriterlerini karşılayan hastalara tedavi uygulanmış, GALILEO çalışmasında 52. haftaya kadarsham almaya devam eden hastalar (kontrol ile kontrol) bunun istisnası olmuştur. Bu zamannoktasından sonra tüm hastalar önceden belirlenen kriterleri karşıladıkları takdirde tedavialmıştır. Her iki çalışmada primer etkililik sonlanım noktası, 24. hafta itibarıyla EDGK'de başlangıca göre en az 15 harflik kazanımı olan hastaların oranı olarak belirlenmiştir. Sekonder etkililikdeğişkenlerinden biri olarak görme keskinliğinde 24. haftada başlangıca göre değişiklikdeğerlendirilmiştir. Tedavi grupları arasındaki farkın her iki çalışmada da EYLEA lehine olmak üzere istatistiksel açıdan anlamlı olduğu gözlenmiştir. Görme keskinliğindeki maksimum iyileşme 3. ayda eldeedilmiş, daha sonra 6. aya kadar görme keskinliği ve SRK bakımından stabilizasyonkaydedilmiştir. İstatistiksel açıdan anlamlı olan fark 52 hafta süresince korunmuştur. İki çalışmaya ait analizde elde edilen sonuçların ayrıntıları aşağıdaki Tablo 2 ve Şekil 2'de yer almaktadır. Tablo 2:COPERNICUS ve GALILEO çalışmalarında 24. Haftada, 52. Haftada ve 76/100. Haftada Etkililik Sonuçları (LOCFC) ile Tam Analiz Seti)
A) Fark, EYLEA 2 mg Q4 hafta eksi kontrol'dür. B) Fark ve güven aralığı (CI), bölge için ayarlanan Cochran-Mantel-Haenszel (CMH) testi(COPERNICUS için dünyanın geri kalanına karşı Amerika ve GALILEO için Asya/Pasifik'e karşıAvrupa) ve başlangıç BCVA kategorisi (> 20/200 ve < 20/200) kullanılarak hesaplanır. C) BCVA: En İyi Düzeltilmiş Görme Keskinliği ETDRS: Erken Tedavi Diyabetik Retinopati ÇalışmasıLOCF: Yapılan Son Gözlem SD: Standart sapma LS: ANCOVA'dan türetilmiş en küçük kareortalamaları D) Tedavi grubu, bölge (COPERNICUS için dünyanın geri kalanına karşı Amerika ve GALILEO içinAsya/Pasifik'e karşı Avrupa) ve başlangıç BCVA kategorisi (> 20/200 ve < 20/200) faktörlerini içerenbir ANCOVA modeline göre LS ortalama fark ve güven aralığı E) COPERNICUS çalışmasında kontrol grubu hastaları, 24. haftadan 52. haftaya kadar ihtiyaçduyulduğunda her 4 haftada bir kadar sık EYLEA alabilmişlerdir; hastaların her 4 haftada bir vizitleriolmuştur. F) COPERNICUS çalışmasında hem kontrol grubu hem de EYLEA 2 mg hastaları 52. haftadanbaşlamak üzere 96. haftaya kadar ihtiyaç duyulduğunda her 4 haftada bir kadar sık EYLEA 2 mg almıştır;hastaların üç ayda bir zorunlu vizitleri bulunmaktadır ancak gerektiğinde 4 haftada bir kadar sıkgörülmeleri muhtemeldir. G) GALILEO çalışmasında, hem kontrol grubu hem de EYLEA 2 mg hastaları, 52. haftadan başlayarak68. haftaya kadar ihtiyaç duyulduğunda her 8 haftada bir EYLEA 2 mg almıştır; hastaların her 8 haftada bir zorunlu vizitleri olmuştur. Şekil2:COPERNICUS
. EYLEA 2mgKontrol Grubu
-o 5^ Kontrol grubunun EYLEA 2 mg ile PRN tedavisine geçirilmesini göstermektedirGALILEO çalışmasında EYLEA grubunun %86.4'ü (n=89) ve sham grubunun %79.4'ü (n=54) için başlangıçta perfüze SRVO kaydedilmiştir. Yirmi dördüncü haftada bu oranEYLEA grubu için %91.8 (n=89), sham grubu için %85.5 (n=47) olmuştur. Bu oranlar 76.haftada korunmuş, EYLEA grubu için %84.3 (n=75), sham grubu için %84 (n=42) olarakbelirlenmiştir. COPERNICUS çalışmasında EYLEA grubunun %67.5'i (n = 77) ve sham grubunun %68.5'i (n = 50) için başlangıçta perfüze SRVO kaydedilmiştir. Yirmi dördüncü haftada bu oranEYLEA grubu için %87.4 (n=90), sham grubu için %58.6 (n=34) olmuştur. Bu oranlar 100.haftada korunmuş, EYLEA grubu için %76.8 (n = 76), sham grubu için %78 (n = 39) olarakbelirlenmiştir. Sham tedavi kolundaki hastalara 24. haftadan itibaren EYLEA uygulanmasınaizin verilmiştir. EYLEA tedavisinin görme fonksiyonu üzerindeki faydalı etkisi başlangıçta perfüze olan ve olmayan hastaların oluşturduğu alt gruplarda benzerlik göstermiştir. Her iki çalışmadadeğerlendirilebilen diğer alt gruplarda (örn. yaş, cinsiyet, ırk, başlangıçtaki görme keskinliği,SRVO süresi) gözlenen tedavi etkileri genel popülasyonlardaki sonuçlarla tutarlılıksergilemiştir. GALILEO ve COPERNICUS çalışmalarında elde edilen kombine verilere göre, EYLEA önceden tanımlı sekonder etkililik sonlanım noktası olan Ulusal Göz Sağlığı Enstitüsü GörmeFonksiyonu Anketinde (NEI VFQ-25) başlangıca göre değişiklik bakımından klinik açıdananlamlı değişiklikler sağlamıştır. Bu değişikliklerin büyüklüğü yayınlanmış olan çalışmalarabenzer olup, En İyi Düzeltilmiş Görme Keskinliği (EDGK) bakımından 15 harflik kazanımadenk gelmiştir. RVDO'ya sekonder makula ödemiEYLEA'nın güvenliliği ve etkililiği hemi-retinal ven oklüzyonu dahil RVDO'ya sekonder makula ödemi bulunan hastaların yer aldığı randomize, çok merkezli, çift maskeli, aktifkontrollü bir çalışmada (VIBRANT) değerlendirilmiştir. Toplam 181 hasta tedavi almış veetkililik için değerlendirilebilir olarak nitelenmiştir (EYLEA alan 91 hasta). Hastalarda yaşaralığının 42 ila 94, ortalama yaşın 65 olduğu belirlenmiştir. SRVO ile ilgili çalışmalardaEYLEA tedavisine randomize edilen hastaların yaklaşık %58'inin (53/91) 65 yaş ve üzerigrupta, yaklaşık %23'ünün de (21/91) 75 yaş ve üzeri grupta olduğu kaydedilmiştir. Çalışmada,hastalar ayda bir uygulanan ilk 6 enjeksiyondan sonra 8 haftada bir 2 mg EYLEA veyabaşlangıçta uygulanan lazer fotokoagülasyon (lazer kontrol grubu) için 1:1 oranında rastgeleatanmıştır. Lazer grubundaki hastaların 12. haftadan itibaren minimum 12 haftalık aralıklarlaolmak üzere ilave lazer fotokoagülasyon (kurtarma amaçlı lazer tedavisi') almalarına izinverilmiştir. Önceden belirlenen kriterlere göre, lazer grubundaki hastaların 24. haftadanitibaren 3 ay süreyle 4 haftada bir ve ardından 8 haftada bir 2 mg EYLEA ile kurtarma tedavisialmaları mümkün olmuştur. VIBRANT çalışmasında primer etkililik sonlanım noktası, 24. hafta itibarıyla EDGK'de başlangıca göre en az 15 harflik kazanımı olan hastaların oranı olarak belirlenmiş ve EYLEAgrubunun lazer kontrolden üstün olduğu görülmüştür. Sekonder sonlanım noktalarından biri olarak 24. haftada başlangıca göre görme keskinliğinde kaydedilen değişiklik değerlendirilmiş ve VIBRANT çalışmasında EYLEA lehine istatistikselolarak anlamlı fark saptanmıştır. Görmeyle ilgili iyileşmenin seyri hızlı olmuş ve 3. ayda pikyapmış, bu iyileşme 12. aya kadar korunmuştur. Lazer grubunda 67 hasta 24. haftadan başlayarak EYLEA ile kurtarma tedavisi almış (Aktif Kontrol/EYLEA 2 mg grubu), bu durum görme keskinliğinde 24. haftadan 52. haftaya yaklaşık5 harflik iyileşmeyle sonuçlanmıştır. VIBRANT çalışmasına ait analizde elde edilen sonuçların ayrıntıları aşağıdaki Tablo 3 ve Şekil 3'de yer almaktadır. Tablo 3:VIBRANT çalışmasında 24. haftada ve 52. haftada Etkililik Sonuçları (LOCF ile Tam
A) Fark, EYLEA 2 mg Q4 hafta eksi Lazer Kontrol'dür. B) Fark ve %95 CI, bölge (Japonya'ya karşı Kuzey Amerika) ve başlangıç BCVA kategorisi(> 20/200 ve < 20/200) için ayarlanmış Mantel-Haenszel ağırlıklandırma şeması kullanılarakhesaplanmıştır. F) Nominal p değeri cS
H 'â.BS 'rS)5b^ o OJ:0 Ü Başlangıçta, EYLEA ve lazer gruplarındaki perfüze hastaların oranı sırasıyla %60 ve %68 olarak kaydedilmiştir. Bu oranlar 24. haftada sırasıyla %80 ve %67 olmuştur. EYLEAgrubundaki perfüze hastaların oranı 52 hafta süreyle korunmuştur. Hastaların 24 haftadanitibaren EYLEA kurtarma tedavisi için uygun olarak değerlendirildiği lazer grubunda perfüzehastaların oranı 52. haftaya kadar %78'e yükselmiştir. Diyabetik makula ödemiEYLEA'nın güvenliliği ve etkililiği DMÖ tanılı hastaların yer aldığı iki randomize, çok
DME
ve VISTADME)
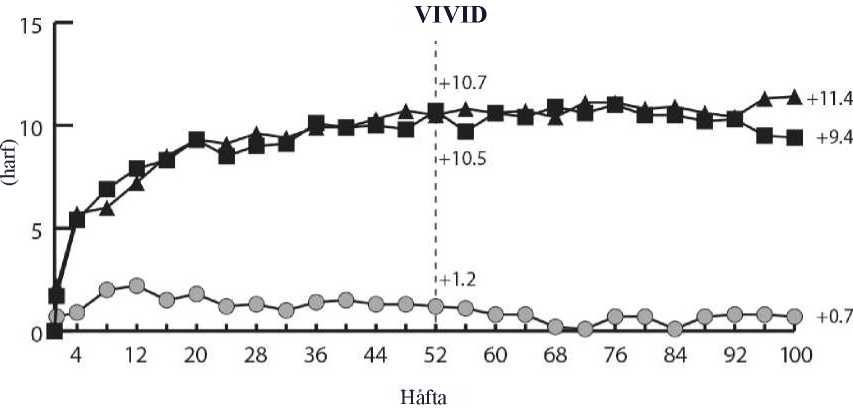
merkezli, çift maskeli, aktif kontrollü çalışmada (VIVID değerlendirilmiştir. Toplam 862 hasta tedavi almış ve etkililik için değerlendirilebilir olarak nitelenmiş, 576 hasta EYLEA almıştır. Hastalarda yaş aralığının 23 ila 87, ortalama yaşın 63olduğu belirlenmiştir. DMÖ ile ilgili çalışmalarda EYLEA tedavisine randomize edilenhastaların yaklaşık %47'sinin (268/576) 65 yaş ve üzeri grupta, yaklaşık %9'unun da (52/576)75 yaş ve üzeri grupta olduğu kaydedilmiştir. Her iki çalışmada da hastaların çoğunda Tip IIdiyabet olduğu belirlenmiştir. İki çalışmada da, hastalar 3 doz rejiminden birine 1:1:1 oranında rastgele atanmıştır: 1) Başlangıçta ayda bir kez 5 enjeksiyondan sonra 8 haftada bir 2 mg EYLEA (EYLEA2Q8); 2) 4 haftada bir 2 mg EYLEA uygulaması (EYLEA 2Q4) ve 3) Maküler lazer fotokoagülasyonu (aktif kontrol). 24. haftadan başlayarak, önceden belirlenen görme kaybı eşiğini karşılayan hastalar ek tedavi için uygun bulunmuş, böylece EYLEA gruplarındaki hastalar lazer, kontrol grubundakihastalar ise EYLEA alabilmiştir. Her iki çalışmada, primer etkililik sonlanım noktası olarak 52 haftada başlangıca göre ortalama EDGK değişikliği değerlendirilmiş ve hem EYLEA 2Q8 hem de EYLEA 2Q4 gruplarında kontrol grubuna kıyasla istatistiksel olarak anlamlı üstünlük kaydedilmiştir. Bu yarar 100. haftaya kadar korunmuştur. VIVIDDMEDMEçalışmalarında ait analizde elde edilen sonuçların ayrıntıları aşağıdaki Tablo 4 ve Şekil 4'te yer almaktadır.Tablo 4:VIVIDDMEDMEçalışmasında 52. haftada ve 100. haftada Etkililik Sonuçları
A 5 aylık enjeksiyonlarla tedavi başlatıldıktan sonra B Tedavi grubu için bir eş değişken ve bir faktör olarak başlangıç BCVA ölçümünü içeren bir ANCOVA modeline göre LS ortalaması ve CI. Ek olarak, VIVIDDME için bir faktör olarak bölge (Japonya'ya karşıAvrupa/Avustralya) ve VISTADME için bir faktör olarak MI ve/veya CVA öyküsü dahil edilmiştir. C Fark, EYLEA grubu eksi aktif kontrol (lazer) grubudur D Güven aralığı (CI) ve istatistiksel test ile fark, VIVIDDME için bölgeye (Japonya'ya karşı Avrupa/Avustralya) göre ve VISTADME için MI veya CVA medikal öyküsüne göre ayarlanan Mantel-Haenszel ağırlıklandırmaşeması kullanılarak hesaplanırE BCVA: En İyi Düzeltilmiş Görme Keskinliği ETDRS: Erken T edavi Diyabetik Retinopati Çalışması LOCF: Y apılan Son Gözlem LS: ANCOVA'dan türetilmiş en küçük kare ortalamaları CI: Güven aralığı
Her çalışmada ve kombine analizde değerlendirilebilir alt gruplardaki (ör. yaş, cinsiyet, ırk, başlangıç HbA1c, başlangıçtaki görme keskinliği, önceki anti-VEGF tedavisi) tedavi etkilerigenel popülasyondaki sonuçlarla genel olarak tutarlı bulunmuştur. VIVIDDMEDMEçalışmalarında, sırasıyla 36 (%9) ve 197 (%43) hasta, 3 ay veya daha uzun arınma periyodu ile birlikte daha önce anti-VEGF tedavisi almıştır. Çalışmaya katılmadanönce VEGF inhibitörü ile tedavi edilen hasta alt grubundaki tedavi etkileri, çalışmayakatılmadan önce VEGF inhibitörü ile tedavi edilmeyen hastalarda görülen etkilere benzerolmuştur.Hekim gerekli gördüğü takdirde, bilateral hastalık bulunan hastalar diğer gözleri için anti-VEGF tedavisi almaya uygun olarak değerlendirilmiştir. VISTADME çalışmasında 217 (%70.7) EYLEA hastasına 100 haftaya kadar bilateral EYLEA enjeksiyonları yapılmış; VIVID DMEçalışmasında ise 97 (%35.8) EYLEA hastasının diğer gözüne farklı bir anti-VEGF tedavisiuygulanmıştır.Bağımsız bir karşılaştırmalı çalışmada (DRCR.net Protokol T) katı OKT ve görmeyle ilgili yeniden tedavi kriterlerine dayalı esnek bir dozlama rejimi kullanılmıştır. Aflibersept tedavigrubunda (n = 224) tedavi rejimi 52. hafta itibarıyla hastalara ortalama 9,2 enjeksiyonyapılmasıyla sonuçlanmış ve bu durumun VIVID DMEDMEDMEçalışmalarındaki EYLEA2Q8 grubuna benzer olduğu kaydedilmiştir. Protokol T'de görme bakımından başlangıca göreen az 15 harf kazanımı olan hastaların oranı %42, ortalama harf kazanımı ise 13.3 olmuştur.Güvenlilik sonuçları, oküler ve oküler olmayan advers olayların (ATE'ler dahil) genelinsidansının, çalışmaların her birinde ve çalışmalar arasında tüm tedavi grupları arasındakarşılaştırılabilir olduğunu göstermiştir.Protokol T çalışmasının tedavi protokolüne göre, çalışma ilaçları çalışma gözlerine başlangıçta ve ardından, uygunluk eşiğinin* altında bir santral alt alan kalınlığı ile, görme keskinliği 20/20ya da daha iyi değilse (Yaklaşık 85 harf değerinde GK skoruna eşdeğer Snellen) ve önceki soniki enjeksiyona cevaben iyileşme ya da kötüleşme olmamışsa 4 haftada bir enjekte edilmiştir.İyileşme görme keskinliği puanında 5 harf veya daha fazla artış (yaklaşık 1 Snellen satırı) veyasantral alt alan kalınlığında %10 veya daha fazla azalma olarak kabul edilmiştir; kötüleşme isegörme keskinliği puanında 5 harf veya daha fazla azalma veya santral alt alan kalınlığında %10veya daha fazla artış olarak kabul edilmiştir. *Uygunluk eşiği Zeiss Stratus'ta >250 mikron; Heidelberg Spectralis'te erkekler için >320 mikron veya kadınlar için >305 mikron; Zeiss Cirrus'ta erkekler için >305 mikron veyakadınlar için >290 mikron olarak tanımlanır. DMÖ'li hastalarda 100 haftalık çok merkezli, randomize, açık etiketli, aktif kontrollü bir çalışma olan VIOLET çalışmasında, tedavinin 5 ardışık aylık dozla başlatıldığı ve ardından her2 ayda bir doz verildiği sabit aralıklarla en az bir yıllık tedaviden sonra DMÖ tedavisi için üçfarklı EYLEA 2 mg doz rejimi karşılaştırılmıştır. Çalışmada, tedavi et ve uzat rejimine göre(enjeksiyon aralıklarının minimum 8 hafta tutulduğu ve klinik ve anatomik sonuçlara görekademeli olarak uzatıldığı 2T&U) 2 mg EYLEA dozunun ve gerektiğinde verilen 2 mg EYLEAdozunun (hastaların 4 haftada bir gözlemlendiği ve klinik ve anatomik sonuçlara göregerektiğinde enjekte edildiği 2PRN), tedavinin ikinci ve üçüncü yılında her 8 haftada bir (2Q8)uygulanan EYLEA'ya kıyasla eşit etkililiği değerlendirilmiştir. Primer etkililik sonlanım noktası (başlangıçtan 52. haftaya kadar EDGK'deki değişiklik) 2T&U grubunda 0,5 ± 6,7 harf ve 2PRN grubunda 1,7 ± 6,8 harf iken, 2Q8 grubunda 0,4 ± 6,7harf olarak istatistiksel olarak eşit etkililik kriterine ulaşmıştır (her iki karşılaştırma için p<0,0001; NI marjı 4 harf). EDGK'de başlangıçtan 100. haftaya kadar olan değişiklikler 52.hafta sonuçlarıyla tutarlı olmuştur: 2Q8 grubunda 0,4 ± 6,7 harfe kıyasla 2T&U grubunda -0,1± 9,1 harf ve 2PRN grubunda 1,8 ± 9 harf. 100 hafta boyunca ortalama enjeksiyon sayısı2Q8fix, 2T&U ve 2PRN için sırasıyla 12,3, 10 ve 11,5 olmuştur. Üç tedavi grubunun tümünde oküler ve sistemik güvenlilik profilleri, VIVID ve VISTA pivotal çalışmalarında gözlemlenenlere benzer olmuştur. 2T&U grubunda, enjeksiyon aralıkları için artışlar ve azalmalar araştırmacının kararına bırakılmıştır; çalışmada 2 haftalık artışlar önerilmiştir. Miyopik koroidal neovaskülarizasyonEYLEA'nın güvenliliği ve etkililiği tedavi almamış miyopik KNV'li Asyalı hastaların yer aldığı randomize, çok merkezli, çift maskeli, sham kontrollü bir çalışmada değerlendirilmiştir.Toplam 121 hasta tedavi almış ve etkililik için değerlendirilebilir olarak nitelenmiştir (EYLEAalan 90 hasta). Hastalarda yaş aralığının 27 ila 83, ortalama yaşın 58 olduğu belirlenmiştir.SRVO ile ilgili çalışmalarda EYLEA tedavisine randomize edilen hastaların yaklaşık%36'smın (33/91) 65 yaş ve üzeri grupta, yaklaşık %10'unun da (9/91) 75 yaş ve üzeri gruptaolduğu kaydedilmiştir. Hastalar çalışma başlangıcında bir kez ve primer sonlanım noktasının değerlendirileceği 24. haftaya kadar persistan hastalık veya rekürrens görüldüğü takdirde aylık ilave enjeksiyonlarşeklinde intravitreal yoldan 2 mg EYLEA veya sham enjeksiyonu almak üzere 3:1 oranındarastgele atanmıştır. Başlangıçta sham almak üzere randomize edilen hastalar 24. haftada ilkEYLEA dozunu almak için uygun olarak değerlendirilmiştir. Bunun ardından, her iki gruptakihastaların persistan hastalık veya rekürrens durumunda ilave enjeksiyon almalarına izinverilmiştir. Tedavi grupları arasındaki fark 24. haftada primer sonlanım noktası (EDGK değişikliği) için ve doğrulayıcı sekonder sonlanım noktası (EDGK'de 15 harf kazanımı olan hastaların oranı)için başlangıca kıyasla EYLEA lehine istatistiksel olarak anlamlı bulunmuştur. Her ikisonlanım noktasına ilişkin farklar 48 hafta süreyle korunmuştur. MYRROR çalışmasına ait analizde elde edilen sonuçların ayrıntıları aşağıdaki Tablo 5 ve Şekil 5'te yer almaktadır. Tablo 5:Aile Tam Analiz Seti)
F) Fark ve %95 CI ülke (ülke tanımlamaları) için ayarlanan Cochran-Mantel-Haenszel (CMH) testi kullanılarak hesaplanmıştır. Şekil 5MYRROR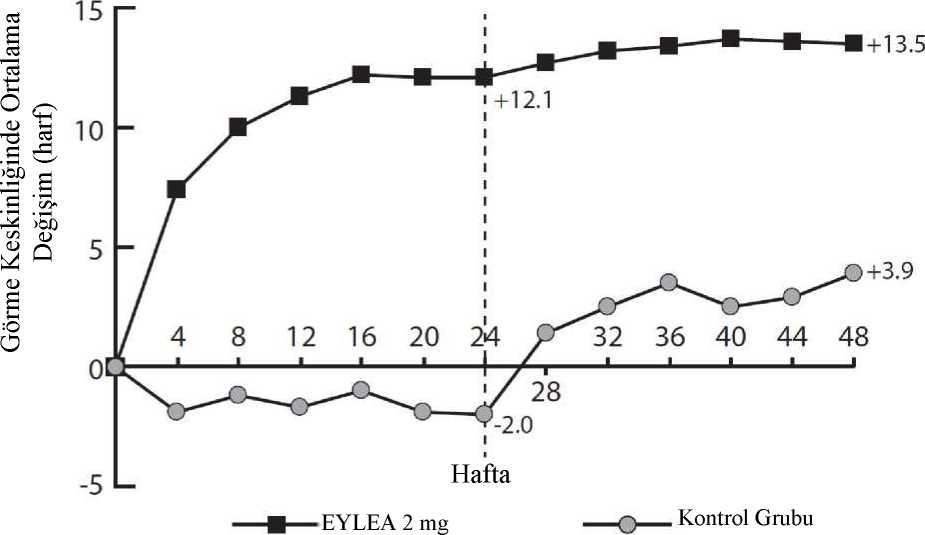
Pediyatrik popülasyonEYLEA'nın çocuklarda ve adolesanlarda güvenlilik ve etkililiği belirlenmemiştir. EYLEA'nın yaş tip YBMD, SRVO, RVDO, DMÖ ve miyopik KNV endikasyonları için pediyatrikpopülasyonda kullanımı söz konusu değildir. 5.2. Farmakokinetik özelliklerGenel ÖzelliklerEYLEA, gözde lokal etkiler oluşturmak üzere doğrudan vitreus içine uygulanır. Emilim:İntravitreal uygulamadan sonra afliberseptin gözden sistemik dolaşıma geçişi yavaştır ve sistemik dolaşımda ağırlıklı olarak VEGF ile inaktif, stabil bir kompleks halinde olduğugörülmektedir; bununla birlikte, yalnızca serbest afliberseptin endojen VEGF'ye bağlanmayeteneği vardır. Dağılım:Sık örnekleme yapılan 6 neovasküler yaş tip YBMD'li hastanın yer aldığı farmakokinetik bir alt çalışmada, plazmadaki maksimum serbest aflibersept konsantrasyonunun (sistemik Cmaks)düşük olup, 2 mg'lık intravitreal enjeksiyondan 1 ila 3 gün sonra ortalama yaklaşık 0.02 mikro(aralık: 0 ila 0.054) ve dozdan iki hafta sonra neredeyse hastaların tamamında saptanamayacakdüzeyde olduğu saptanmıştır. İntravitreal yoldan 4 haftada bir uygulanan aflibersept plazmadabirikmez. Plazmadaki ortalama maksimum serbest aflibersept konsantrasyonu hayvan modellerinde sistemik VEGF'nin biyolojik aktivitesini %50 oranında inhibe etmek için gereken aflibersept konsantrasyonuna kıyasla 50 ila 500 kat düşüktür ve bu modellerde dolaşımdaki serbestaflibersept düzeyi yaklaşık 10 mikrogram/mL olduktan sonra kan basıncında değişiklik olduğuve düzeyin 1 mikrogram/mL altına inmesiyle bunun normale döndüğü gözlenmiştir. Hastalara2 mg intravitreal uygulama yapıldıktan sonra serbest afliberseptin ortalama maksimum plazmakonsantrasyonu değerinin, sağlıklı gönüllülerle yürütülen bir çalışmada afliberseptin sistemikVEGF'ye (2.91 mikrogram/mL) yarı maksimal düzeyde bağlanması için gerekenkonsantrasyondan en az 100 kat düşük olduğu tahmin edilmektedir. Bu nedenle kan basıncıgibi sistemik farmakodinamik etkilerin ortaya çıkması olası değildir. SRVO, RVDO, DMÖ ya da miyopik KNV'li hastalarla yürütülen farmakokinetik alt çalışmalarda, plazmadaki ortalama serbest aflibersept Cmaks değeri 0.03 ila 0.05 mikrogram/mLaralığındaki değerler ve 0.14 mikrogram/mL'yi aşmayan bireysel değerler ile benzerdir. Dahasonra, serbest aflibersept plazma konsantrasyonları, genellikle bir hafta içinde alt niceliksınırıdeğerlerinin altına ya da bu sınıra yakın değerlere düşer. Tüm hastalarda 4 haftada bir sonrakiuygulamadan önce saptanamayan konsantrasyonlara ulaşılır. Biyotransformasyon:EYLEA, protein bazlı bir terapötik olduğundan, herhangi bir metabolizma çalışması yürütülmemiştir. Eliminasyon:Serbest aflibersept VEGF'ye bağlanarak stabil ve durağan bir kompleks oluşturmaktadır. Diğer büyük proteinler gibi, serbest ve bağlı afliberseptin proteolitik katabolizma ile uzaklaştırılmasıbeklenir. Hastalardaki karakteristik özelliklerBöbrek yetmezliği olan hastalar:Böbrek yetmezliği olan hastalarda EY^EA ile ilgili özel bir çalışma yapılmamıştır. VIEW2 çalışmasında yer alan ve %40'ında böbrek yetmezliği olan (%24 hafif, %15 orta dereceli, %1 şiddetli) hastalara ilişkin farmakokinetik analiz, 4 veya 8 haftada bir intravitrealuygulama sonrasında aktif ilacın plazma konsantrasyonu bakımından fark olmadığınıgöstermiştir. Benzer sonuçlar, GALILEO çalışmasında SRVO'lu hastalarda, VIVID DME5.3. Klinik öncesi güvenlilik verileriTekrarlanan doz toksisitesiyle ilgili klinik dışı çalışmalarda etkilerin yalnızca önerilen klinik dozun intravitreal yoldan uygulanmasıyla elde edilen maksimum insan maruziyetinden önemlioranda yüksek olan aşırı sistemik maruziyet ile ortaya çıktığının gözlenmesi, klinik kullanımbakımından anlamlı bir etki olmadığına işaret etmektedir. İntravitreal aflibersept tedavisi uygulanan maymunlarda maksimum insan maruziyetinden yüksek sistemik maruziyet ile nazal türbinatlardaki solunum yolu epitelinde erozyon veülserasyon geliştiği gözlenmiştir. Serbest afliberseptin Cmaks ve EAA değerleri doğrultusundasistemik maruziyeti insanlarda intravitreal 2 mg'lık doz ile gözlenen değerlere kıyasla sırasıyla200 ve 700 kat yüksek bulunmuştur. Maymunlarda Cmaks ve EAA değerleri doğrultusunda0.5 mg/göz olan Advers Etki Gözlenmeyen Düzey (NOAEL) ile sistemik maruziyetin sırasıyla42 ve 56 kat yüksek olduğu kaydedilmiştir. Afliberseptin mutajenik veya karsinojenik potansiyeliyle ilgili çalışma yapılmamıştır. İntravenöz (3 ila 60 mg/kg) ve subkutan (0.1 ila 1 mg/kg) uygulama yapılan gebe tavşanlarda yapılan embriyo-fetal gelişim çalışmalarında afliberseptin intrauterin gelişmeyi etkilediğigösterilmiştir. Maternal NOAEL, sırasıyla 3 mg/kg veya 1 mg/kg olmuştur. Gelişimle ilgiliNOAEL belirlenmemiştir. Serbest afliberseptin 0.1 mg/kg doz düzeyindeki Cmaks ve kümülatifEAA değerleri doğrultusunda sistemik maruziyeti insanlarda intravitreal 2 mg'lık doz ilegözlenen değerlere kıyasla sırasıyla 17 ve 10 kat yüksek bulunmuştur. Maymunlarda 3 ila 30 mg/kg aralığındaki dozlarda intravenöz aflibersept uygulaması yapılan 6 aylık bir çalışma kapsamında erkek ve dişi fertilitesi üzerindeki etkiler değerlendirilmiştir.Tüm doz düzeylerinde, dişilerde üremeyle ilgili hormon seviyelerinde değişiklik olmasınedeniyle menstrüasyon olmadığı veya menstrüasyonun düzensiz olduğu gözlenirken,erkeklerde sperm morfolojisi ve motilitesiyle ilgili değişiklikler olduğu saptanmıştır. Serbestaflibersept için 3 mg/kg intravenöz dozda gözlenen Cmaks ve EAA doğrultusunda, sistemikmaruziyetlerin insanlarda intravitreal 2 mg'lık dozdan sonra gözlenen maruziyete kıyaslasırasıyla yaklaşık 4,.900 kat ve 1,.500 kat yüksek olduğu belirlenmiştir. Tüm değişiklikleringeri dönüşümlü olduğu kaydedilmiştir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiPolisorbat 20 Sodyum dihidrojen fosfat, monohidrat (pH ayarlaması için) Disodyum hidrojen fosfat, heptahidrat (pH ayarlaması için) Sodyum klorür Sukroz Enjeksiyonluk su 6.2. GeçimsizliklerGeçimsizlik çalışması yapılmamış olduğundan, bu tıbbi ürün diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3. Raf ömrü24 ay 6.4. Saklamaya yönelik özel tedbirler2°C - 8°C arasında (buzdolabında) saklayınız. Dondurmayınız. Açılmamış flakon buzdolabının dışında 25°C'nin altında 24 saate kadar saklanabilir. Flakon açıldıktan sonra aseptik koşullar altında gerekli işlemlere devam edilmelidir. Işıktan korumak için orijinal ambalajında saklayınız. 6.5. Ambalajın niteliği ve içeriğiHer bir kutuda 278 mikrolitre dolum hacimli intravitreal enjeksiyon için çözelti içeren, elastomerik kauçuk tıpalı bir adet tip I cam flakon ve bir adet 18 G filtreli iğne bulunur. Herbir kullanıma hazır enjektör en az 0,1 mL çekilebilir hacim içerir. Ambalaj boyutu 1 flakon +1 filtreli iğne 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. Flakon sadece tek bir göz için tek kullanımlıktır. Flakon, önerilen 2 mg aflibersept dozundan (0,05 mL'ye eşdeğer) fazlasını içerir. Fazla hacim uygulamadan önce dışarı atılmalıdır. Uygulama öncesinde enjeksiyonluk çözelti görsel olarak incelenmelidir. Eğer çözeltide gözle görülebilen partiküller, bulanıklık ya da renk bozukluğu varsa flakon kullanılmamalıdır. Filtreli iğne: BD Küt Uçlu Filtre (Dolum) İğnesi, deriye enjeksiyon için değildir. BD Küt Uçlu Filtre (Dolum) İğnesi otoklavlanmamalıdır. Filtre iğnesi non-pirojeniktir. Ambalajı zarar görmüşse kullanılmamalıdır. Kullanılmış BD Küt Uçlu Filtre (Dolum) İğnesi onaylı keskin ve delici alet atık kutusuna atılmalıdır. Dikkat: İğne filtresinin tekrar kullanılması enfeksiyona ya da diğer hastalıklara/yaralanmalara neden olabilir. İntravitreal enjeksiyon için bir adet 30 G x ^ inç oftalmik amaçla kalifiye edilmiş enjeksiyon iğnesi kullanılması önerilmektedir. Vitreusta uçuşan noktalar riskini azaltmak için intravitrealenjeksiyonda silikon içermeyen enjektör ve enjeksiyon iğnesi kullanılması önerilmektedir. Flakonun kullanımı için talimatlar:1. Plastik kapağı çıkarınız ve flakonun kauçuk tıpasının dış kısmını dezenfekte ediniz. 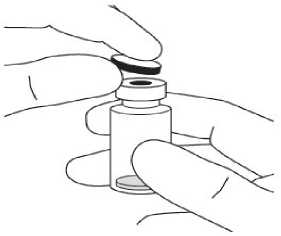 2. Kutu içinde bulunan 18 G, 5 mikron filtrelik iğneyi, 1 mL steril, Luer kilitli enjektöretakınız. 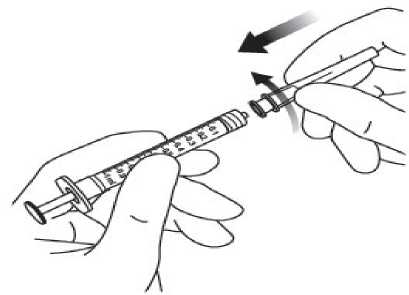 3. Filtre iğnesi flakona tamamen takılana ve uç flakonun tabanına veya alt kenarına temas edene kadar iğneyi flakon tapasının ortasına doğru itiniz._4. Aseptik teknik kullanarak, EYLEA flakonunun tüm içeriğini flakonu dik konumda tutarak enjektöre çekiniz; içeriğin tamamının çekilmesini kolaylaştırmak için flakonu hafifçe yanaeğiniz. Hava girmesini önlemek için filtre iğnesi ucunun sıvıya batırıldığından emin olunuz. İçeriği çekerken, filtre iğnesi ucunu sıvının içinde tutarak flakonu eğmeye devam ediniz.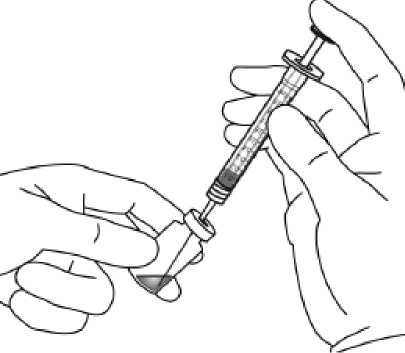 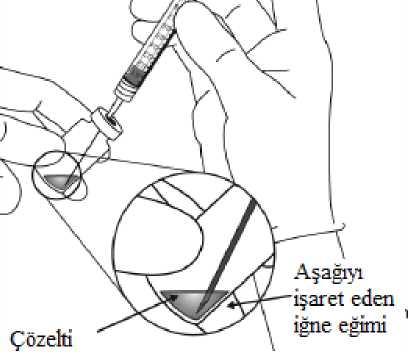 5. Flakonu boşaltırken filtreli iğneyi tamamen boşaltmak için piston çubuğunun yeteri kadar geri çekildiğinden emin olunuz._6. Filtreli iğneyi çıkartınız ve gerektiği şekilde imha ediniz. Not: Filtreli iğne, intravitreal enjeksiyon için kullanılmaz.7. Aseptik teknik kullanarak 30 G x ^ inç enjeksiyon iğnesini Luer kilitli enjektörucuna çevirerek sıkıca takınız. 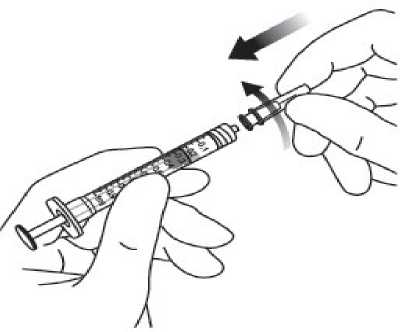 8. İğne yukarı bakacak şekilde enjektörü tutarak enjektörde hava kabarcığı olup olmadığınıkontrol ediniz. Eğer hava kabarcığı varsa,nazikçe yerinden oynatınız ve havakabarcığını çıkartınız. 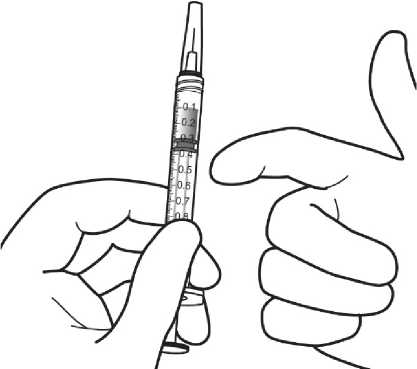 9. Tüm hava kabarcıklarım ve tıbbi ürünün fazlasını uzaklaştırmak için, düz piston kenarı ile enjektördeki 0.05 mL'yi gösteren çizgi aynı hizaya gelecek şekilde pistona hafifçe bastırınız.
7. RUHSAT SAHIBIBayer Türk Kimya San. Ltd. Şti. Fatih Sultan Mehmet Mah. Balkan Cad. No: 53 34770 Ümraniye-İstanbulTel: 0216 528 36 00Faks: 0216 645 39 50 8. RUHSAT NUMARASI2014/709 9. ILK RUHSAT TARIHI/RUHSAT YENILEME TARIHIİlk ruhsat tarihi: 19.09.2014 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENILENME TARIHI |
İlaç BilgileriEylea 40mg/ml İntravitreal Enjeksiyon İçin Çözelti İçeren FlakonEtken Maddesi: Aflibersept Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.