Tasigna 150 Mg Kapsül Kısa Ürün BilgisiKISA URUN BILGISI1. BEŞERI TIBBI URUNUN ADITASİGNA 150 mg kapsül 2. KALITATIF VE KANTİTATIF BILEŞIMEtkin madde:Nilotinib hidroklorür monohidrat 165,45 mg (150 mg nilotinib baza eşdeğer) Yardımcı maddeler:Laktoz monohidrat 117,08 mg Yardımcı maddeler için, 6.1'e bakınız. 3. FARMASÖTIK FORMKapsül Üzerinde boylamasına siyah renkli NVR/BCR baskısı olan 1 boyutunda kırmızı opak sert jelatin kapsül içerisinde beyaz ila sarımsı toz. 4. KLİNIK ÖZELLIKLER4.1. Terapötik endikasyonlarTASİGNA, -Yeni tanı konmuş Philadelphia kromozomu pozitif kronik miyeloid lösemili (Ph+ KML) yetişkin hastalarda kronik evre tedavisinde endikedir. 4.2. Pozoloji ve uygulama şekliTedavi, KML hastalarının tedavisinde deneyimli bir hekim tarafından başlatılmalıdır. Pozoloji:Tedavi, klinik yarar gözlendiği sürece veya kabul edilemez toksisite ortaya çıkana kadar sürdürülmelidir. Bir dozun atlanması durumunda, hasta ilave bir doz almayıp, reçete edilen bir sonraki doz ile devam etmelidir. TASİGNA için önerilen doz günde iki kez 300 mg'dır. Günde bir kez 400 mg dozu için (bkz. aşağıda doz ayarlamaları), 200 mg kapsüller mevcuttur. Birinci basamak tedavi olarak TASİGNA ile tedavi edilen ve kalıcı derin moleküler yanıt elde eden kronik fazdaki yetişkin Philadelyhia kromozomu pozitif KML hastaları(MY4.5)En az 3 yıl süreyle günde iki kez 300 mg dozunda TASİGNA ile tedavi edilmiş olan kronik fazdaki uygun yetişkin Philadelphia kromozomu pozitif (Ph+) KML hastalarında,tedavinin kesilmesinden hemen önce eğer derin moleküler yanıt en az bir yıl süreylekalıcı olmuşsa, tedavinin kesilmesi düşünülebilir. TASİGNA tedavisinin kesilmesisüreci, KML hastalarının tedavisinde deneyimli bir hekim tarafından başlatılmalıdır(bkz. bölüm 4.4 ve 5.1). TASİGNA tedavisini bırakan uygun hastalar BCR-ABL transkript düzeylerini ve diferansiyelli (lökosit formülü de içeren) tam kan sayımlarını bir yıl süreyle her ay,ardından ikinci yılda 6 haftada bir ve sonrasında 12 haftada bir ölçtürmelidir. BCR-ABLtranskript düzeyleri izlemi, Uluslararası Ölçekte (IS) moleküler yanıt düzeylerini ölçmeküzere valide edilmiş olan, en az MY4.5 (BCR-ABL/ABL <%0,0032 IS) hassasiyetesahip bir kantitatif tanı testi ile gerçekleştirilmelidir. Tedavisiz faz sırasında doğrulanmış MY4 (MY4=BCR-ABL/ABL <%0,01 IS) yanıtını kaybeden fakat MMY (MMY=BCR-ABL/ABL < % 0,1 IS) yanıtını kaybetmeyenhastalarda BCR-ABL transkript düzeyleri, BCR-ABL düzeyleri MY4 ile MY4.5 arasıbir aralığa dönene kadar 2 haftada bir izlenmelidir. BCR-ABL düzeylerinin arka arkayaen az 4 ölçümde MMY ile MY4 arasında korunduğu hastalar orijinal izlem planına geridönebilir. MMY'yi kaybeden hastalar, remisyon kaybının meydana geldiğinin bilindiği tarihi izleyen 4 hafta içinde tedaviye yeniden başlamalıdır. Eğer hastada tedavi kesilmedenönce doz azaltımı yapılmışsa, TASİGNA tedavisi, günde iki kez 300 mg ya da günde birkez 400 mg şeklinde azaltılmış doz düzeyinde yeniden başlatılmalıdır. TASİGNAtedavisine yeniden başlayan hastalar, MMY tekrar elde edilene kadar ayda bir vesonrasında 12 haftada bir BCR-ABL transkript düzeylerini ölçtürmelidir (bkz.bölüm 4.4). Doz ayarlamaları ya da doz değişiklikleri:Temelde yatan lösemi ile ilişkili olmayan hematolojik toksisiteler (nötropeni, trombositopeni) nedeniyle TASİGNA tedavisinin geçici olarak kesilmesi ve/veyadozunun azaltılması gerekebilir (bkz. Tablo 1).
Eğer klinik olarak anlamlı orta dereceli veya şiddetli hematolojik olmayan toksisite gelişirse, doz uygulamasına ara verilmeli ve hastalar gerekli şekilde izlenmeli ve takipedilmelidir. Eğer önceki doz erişkin hastalarda günde iki kez 300 mg ise, toksisitekaybolduktan sonra doz uygulamasına erişkin hastalarda günde bir kez 400 mg'dandevam edilebilir. Şayet önceki doz erişkin hastalarda günde bir kez 400 mg ise, tedavikesilmelidir. Eğer klinik açıdan uygunsa, dozun erişkin hastalarda günde iki kez 300mg'a tekrar yükseltilmesi düşünülmelidir. Serum lipaz düzeyinde yükselme: Derece 3 ya da 4 lipaz yükselmeleri için, yetişkin hastalarda dozlar günde bir kez 400 mg'a indirilmelidir veya kesilmelidir. Serum lipazdüzeyleri ayda bir kez ya da klinik olarak endike olduğu şekilde takip edilmelidir (bkz.Bölüm 4.4). Bilirubin ve hepatik transaminazlarda yükselme: Erişkin hastalarda bilirubin düzeylerinde ve hepatik transminazlarda 3. veya 4. derecedeki yükselmeler için, dozlargünde bir kez 400 mg'a indirilmelidir veya kesilmelidir (bkz. Bölüm 4.8). Ayda bir kezveya klinik olarak uygun olduğu durumlarda bilirubin ve serum hepatik transaminazlarölçülmelidir. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek fonksiyonu bozulmuş hastalarda klinik çalışmalar gerçekleştirilmemiştir. Nilotinib ve metabolitleri böbrekler aracılığıyla atılmadığı için, böbrek yetmezliği olan hastalarda toplam vücut klirensinde bir azalma beklenmemektedir. Karaciğer yetmezliği:Karaciğer yetmezliği nilotinibin farmakokinetiği üzerinde hafif derecede etki gösterir. Karaciğer yetmezliği olan hastalarda doz ayarlaması gerekli görülmemekle birlikte,karaciğer yetmezliği olan hastalarda dikkatli kullanılmalıdır. (bkz. Bölüm 4.4). Kalp rahatsızlıkları:Klinik çalışmalara, kontrol altında olmayan ya da anlamlı kalp hastalığı (yakın zamanda geçirilmiş miyokard infarktüsü, konjestif kalp yetmezliği, stabil olmayan anjina ya daklinik olarak anlamlı bradikardi dahil olmak üzere) olan hastalar dahil edilmemiştir. Kalprahatsızlığı olan hastalarda dikkatli kullanılmalıdır (bkz. Bölüm 4.4). TASİGNA tedavisi ile total serum kolesterol düzeylerinde artışlar bildirilmiştir (bkz., Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Lipid profilleri TASİGNA tedavisibaşlatılmadan önce belirlenmeli ve tedavi başlatıldıktan 3 ve 6 ay sonra ve kronik tedavisırasında en az yılda bir kez değerlendirilmelidir. TASİGNA tedavisi ile kan glukozu düzeylerinde artışlar bildirilmiştir (bkz. 4.4 Özel kullanım uyarıları ve önlemleri). Kan glukoz düzeyleri TASİGNA ile tedavibaşlatılmadan önce değerlendirilmeli ve tedavi sırasında izlenmelidir. Pediyatrik popülasyon:Kronik faz Philadelphia kromozomu pozitif KML'li 2 ila <18 yaş pediyatrik hastalarda nilotinibin güvenliliği ve etkililiği belirlenmiştir (bkz. bölüm 4.8, 5.1 ve 5.2). 2 yaşınaltındaki pediyatrik hastalarda ya da akselere faz veya blast krizdeki Philadelphiakromozomu pozitif KML'li pediyatrik hastalarda deneyim bulunmamaktadır. Yeni tanıalmış 10 yaşın altındaki pediyatrik hastalarda veri mevcut değildir ve yeni tanı almışimatinibe dirençli veya imatinibi tolere edemeyen 6 yaşın altındaki hastalarda sınırlı verimevcuttur. Geriyatrik popülasyon:Klinik çalışmalara katılan deneklerin yaklaşık % 12'sini 65 yaş ve üzeri hastalar oluşturmuştur. 18 ila 65 yaş arası yetişkinlere kıyasla 65 yaş ve üzeri hastalardagüvenlilik ve etkililik açısından herhangi bir majör farklılık gözlenmemiştir. Uygulama sıklığı ve süresi:TASİGNA günde iki kez yaklaşık 12 saat aralıklarla, aç karnına alınmalıdır. Tedavi, hastaya faydalı olduğu sürece devam ettirilmelidir. Uygulama şekli:Sadece ağızdan kullanım içindir. Kapsüller suyla yutulmalıdır. İlaç alınmadan en az 2 saat öncesine kadar ve ilaç alındıktan sonra en az 1 saat herhangi bir gıdatüketilmemelidir. Kapsülleri yutamayan hastalar için her bir kapsülün içeriği bir çay kaşığı elma püresi içinde eritilebilir ve hemen sonrasında yutulmalıdır. Bir çay kaşığından fazla elma püresialınmamalı ve elma püresi dışında bir gıda kullanılmamalıdır (bkz. Bölüm 4.4 ve 5.2). 4.3. KontrendikasyonlarTASİGNA, nilotinibe ya da Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılığı olan hastalarda kullanılmamalıdır. 4.4. Özel kullanım uyarıları ve önlemleriMiyelosupresyon:TASİGNA tedavisi (Ulusal Kanser Enstitüsü Genel Toksisite Kriterleri derece 3 ve 4) trombositopeni, nötropeni ve anemi ile ilişkilendirilir. İlk 2 ay boyunca her iki haftadabir ve daha sonra ayda bir veya klinik olarak belirtildiği şekilde tam kan sayımıyapılmalıdır. Miyelosupresyon genellikle geri dönüşümlü olmuştur ve genellikleTASİGNA'ya geçici olarak ara verilerek veya doz azaltılarak yönetilmiştir (bkz. Bölüm 4.2). QT Uzaması:TASİGNA'nın konsantrasyona bağlı olarak erişkin ve pediyatrik hastalarda yüzey EKG'sinde QT aralığı ölçümü ile kardiyak ventriküler repolarizasyonu uzattığıgösterilmiştir. Faz III çalışmasında günde iki kez 300 mg TASİGNA alan, yeni tanı konmuş KML kronik evre hastalarında kararlı durumda başlangıca göre ortalama QTcF (Fridericiayöntemi ile düzeltilen QT) aralığı ortalama zamanı 6 milisaniye olmuştur. Hiçbir hastadaQTcF, 480 milisaniye üzerine çıkmamıştır. Torsades de pointes (geçici ya da uzun süreli)epizodu gözlenmemiştir. Hastalardakilere benzer maruziyetlerin gözlendiği bir sağlıklı gönüllü çalışmasında, plasebo kolunun çıkarıldığı ortalama QTcF değişimi başlangıca göre 7 milisaniye (güvenaralığı +4 milisaniye) olarak bulunmuştur. Hiçbir denekte 450 milisaniyenin üzerinde birQTcF gözlenmemiştir. İlave olarak, çalışma sırasında klinik olarak anlamlı bir aritmivakası meydana gelmemiştir. Özellikle de, herhangi bir Torsades de pointes (geçici yada uzun süreli) epizodu gözlenmemiştir. TASİGNA besinlerle ve/veya güçlü CYP3A4 inhibitörleri ve/veya QT'yi uzatma potansiyeli olduğu bilinen tıbbi ürünlerle birlikte yanlış biçimde alındığında QTaralığında anlamlı uzama meydana gelebilir (bkz. Bölüm 4.5). Hipokalemi vehipomagnezemi varlığı, bu etkiyi daha da arttırabilir. QT aralığı uzaması, hastalarıölümcül sonuç riskine maruz bırakabilir. QTc (düzeltilmiş QT) aralığında uzama görülen veya uzama riski olan aşağıdaki hastalarda TASİGNA dikkatli kullanılmalıdır: - Konjenital uzun QT sendromu olanlar, - Yakın tarihli miyokard infarktüsü, konjestif kalp yetmezliği, stabil olmayanangina veya klinik anlamlı bradikardi dahil, kontrol edilemeyen veya önemlikardiyak hastalığı olanlar, - Antiaritmik tıbbi ürünleri veya QT aralığının uzamasına neden olan diğer tıbbi ürünleri kullananlar. QTc aralığı üzerindeki bir etkinin yakından izlenmesi tavsiye edilir ve TASİGNA tedavisine başlamadan önce ve klinik olarak endike olduğu şekilde bir başlangıç EKG'siönerilir. TASİGNA uygulamasına başlanmadan önce hipokalemi veya hipomagnazemi düzeltilmeli ve tedavi sırasında periyodik olarak kontrol edilmelidir. Ani ölüm:Kalp hastalığı öyküsü veya anlamlı kardiyak risk faktörleri bulunan imatinibe dirençli veya intolere kronik ve hızlanmış evre KML hastalarında, yaygın olmayan sıklıkla(%0,1-1) ani ölüm vakaları bildirilmiştir. Bu hastalarda sıklıkla altta yatan maligniteyeek olarak komorbiditelerin de mevcut olduğu görülmüş ve eş zamanlı ilaç kullanımıtespit edilmiştir. Ventriküler repolarizasyon anomalilerinin de katkı sağlayan faktörlerolabileceği düşünülmüştür. Yeni tanı konmuş kronik evre KML hastalarında, Faz IIIçalışmasında ani ölüm vakası bildirilmemiştir. Sıvı tutulumu ve ödem:Yeni tanı konmuş KML hastalarında yürütülen bir Faz III çalışmada plevral efüzyon, pulmoner ödem ve perikard efüzyonu gibi ilaca bağlı sıvı tutulumunun şiddetli formları,yaygın olmayan sıklıkla (%0,1 ila %1) gözlenmiştir. Pazarlama sonrası bildirimlerdebenzer olaylar gözlenmiştir. Beklenmedik hızlı kilo alımı dikkatle incelenmelidir.TASİGNA ile tedavi sırasında şiddetli sıvı tutulumuna dair belirtiler ortaya çıkarsa,etiyolojisi değerlendirilmeli ve hasta bu doğrultuda tedavi edilmelidir (Hematolojikolmayan toksisitelerin yönetimine ilişkin talimatlar için bkz. Bölüm 4.2.). Kardiyovasküler olaylar:Kardiyovasküler olaylar, yeni tanı konmuş KML hastalarında yürütülen randomize bir Faz III çalışmasında bildirilmiştir ve pazarlama sonrası raporlarda gözlenmiştir. Buklinik çalışmada 60,5 ay medyan tedavi süresiyle 3 / 4. derece kardiyovasküler olaylarşunları kapsamıştır: periferik arter tıkayıcı hastalığı (günde iki kere 300 mg ve 400 mggrubunda sırasıyla %1,4 ve %1,1); iskemik kalp hastalığı (günde iki kere 300 mg ve 400mg grubunda sırasıyla %2,2 ve %6,1) ve iskemik serebrovasküler olaylar (günde iki kere300 mg ve 400 mg grubunda sırasıyla %1,1 ve %2,2). Eğer kardiyovasküler olaylarınakut belirtileri ve semptomları ortaya çıkarsa, hastalara acilen tıbbi yardım almalarıtavsiye edilmelidir. TASİGNA tedavisi sırasında hastaların kardiyovasküler durumudeğerlendirilmeli ve kardiyovasküler risk faktörleri standart kılavuzlara uygun olarakizlenmeli ve aktif olarak tedavi edilmelidir. Kardiyovasküler risk faktörlerininyönetilmesi için uygun tedavi reçete edilmelidir (Hematolojik olmayan toksisitelerinyönetimine ilişkin talimatlar için bkz. Bölüm 4.2.). Hepatit B reaktivasyonu:Hepatit B virüsü (HBV) kronik taşıyıcısı olan hastalarda, BCR-ABL tirozin kinaz inhibitörleri ile tedavi sonrası, Hepatit B reaktivasyonu ortaya çıkmıştır. Bazı vakalar,karaciğer nakli veya ölüme sebep olan akut karaciğer yetmezliği veya fulminan hepatitile sonuçlanmıştır. TASİGNA tedavisine başlanmadan önce, hastalar HBV enfeksiyonu açısından test edilmelidir. Pozitif HBV serolojisine sahip (aktif hastalığı olanlar dahil) ve tedavisırasında HBV enfeksiyonu için pozitif test sonucu veren hastalarda, tedavibaşlatılmadan önce karaciğer hastalığı ve HBV tedavisi konusunda uzman hekimleredanışılmalıdır. TASİGNA ile tedaviye ihtiyaç duyan HBV taşıyıcıları, tedavi boyuncave tedavi sonlandırıldıktan sonra birkaç ay boyunca aktif HBV enfeksiyonu bulgu vebelirtileri için yakından izlenmelidir (bkz. Bölüm 4.8). Sürekli derin moleküler yanıt elde etmiş olan kronik evredeki yetişkin Ph+ KML hastalarında özel izlemTedavinin kesilmesi için uygunlukTipik BCR-ABL transkriptleri olan e13a2/b2a2 veya e14a2/b3a2'yi eksprese ettiği doğrulanan uygun hastalarda tedavinin kesilmesi düşünülebilir. Hastalar, BCR-ABL'ninnicel tayinine, moleküler yanıtın derinlik değerlendirmesine ve TASİGNA ile tedavininkesilmesinden sonra olası bir moleküler remisyon kaybının tespitine olanak verebilecektipik BCR-ABL transkriptlerine sahip olmalıdır. Tedavisi kesilen hastaların izlemiTedavinin kesilmesine uygun hastalarda BCR-ABL transkript düzeylerinin sık izlemi, en az MY4.5 (BCR-ABL/ABL <%0,0032 IS) hassasiyet ile moleküler yanıt düzeyleriniölçeceği valide edilmiş olan kantitatif bir tanı testi ile gerçekleştirilmelidir. BCR-ABLtranskript düzeyleri, tedavi kesilmeden önce ve kesilme sürecinde değerlendirilmelidir(bkz. Bölüm 4.2 ve 5.1). Birinci veya ikinci basamak tedavi olarak nilotinib almış olan hastalarda majör moleküler yanıt kaybının (MMY=BCR-ABL/ABL <0,1 %IS) ya da ikinci basamaktedavi olarak nilotinib almış olan hastalarda doğrulanmış MY4 (MY4=BCR-ABL/ABL<0,01 %IS) kaybının (aralarında en az 4 haftanın olduğu arka arkaya iki ölçümün MY4(MY4=BCR-ABL/ABL <%0,01 IS) kaybını göstermesi), meydana geldiği bilinen tarihiizleyen 4 hafta içerisinde tedaviye yeniden başlanması gerekmektedir. Tedavisiz fazsırasında moleküler relaps meydana gelebilir ve uzun süreli veriler henüz mevcutdeğildir. Bu nedenle, olası bir remisyon kaybını belirleyebilmek için BCR-ABLtranskript düzeylerinin ve diferansiyelli tam kan sayımlarının sık izlemi kritiktir (bkz.Bölüm 4.2). Tedaviye yeniden başlandıktan sonra 3 ay içinde MMY elde edemeyenhastalarda BCR-ABL kinaz bölge mutasyon testinin yapılması gerekir. Laboratuvar testleri ve takip:Kan lipidleri:Yeni tanı konmuş KML hastalarında yürütülen bir Faz III çalışmada, günde iki kez 400 mg nilotinib ile tedavi edilen hastaların %1,1'inde total kolesterolde derece 3/4 artışgörülmüştür; öte yandan, günde iki kez 300 mg doz grubunda derece3/4 artışarastlanmamıştır (bkz. Bölüm 4.8). Lipid profillerinin TASİGNA tedavisi başlatılmadanönce belirlenmesi ve tedavi başlatıldıktan 3 ve 6 ay sonra ve kronik tedavi sırasında enaz yılda bir kez değerlendirilmesi tavsiye edilmektedir (bkz. Bölüm 4.2). HMG CoAredüktaz inhibitörüne (lipid düşürücü ajan) ihtiyaç olursa, belirli HMG CoA redüktazinhibitörleri de CYP3A4 yolağı ile metabolize edildiğinden, tedavi başlatılmadan önceBölüm 4.5'e başvurulmalıdır. Kan glukozu:Yeni tanı konmuş KML hastalarında yürütülen bir Faz III çalışmada, günde iki kez 400 mg nilotinib ile tedavi edilen hastaların %6,9'unda kan glukozunda Derece 3/4 artış vegünde iki kez 300 mg ile tedavi edilen hastaların %7,2'sinde kan glukozunda Derece 3/4artış gözlenmiştir. Glukoz düzeylerinin TASİGNA ile tedavi başlatılmadan öncedeğerlendirilmesi ve tedavi sırasında klinik olarak gerekli olduğu şekilde izlenmesiönerilmektedir (bkz. Bölüm 4.2). Test bulgularına göre tedaviye karar verilirse, hekimleryerel uygulama standartları ve tedavi kılavuzlarını izlemelidir. Diğer tıbbi ürünler ile etkileşimler:TASİGNA'nın güçlü CYP3A4 inhibitörü (bunlarla sınırlı olmamakla birlikte ketokonazol, itrakonazol, vorikonazol, klaritromisin, telitromisin, ritonavir) olanajanlarla bir arada uygulanmasından kaçınılmalıdır. Bu ilaçlardan herhangi biriyle tedavigerekli olduğunda, mümkünse TASİGNA tedavisine ara verilmesi önerilmektedir (bkz.Bölüm 4.5). TASİGNA tedavisine geçici olarak ara verilmesi mümkün değilse, hastanınQT aralığının uzaması açısından yakından izlenmesi gereklidir (bkz. Bölüm 4.2, 4.5 ve 5.2) . TASİGNA'nın potent CYP3A4 indükleyicileri olan tıbbi ürünlerle (fenitoin, rifampisin, karbamazepin, fenobarbital ve St. John's Wort vb.) birlikte eş zamanlı olarakkullanılması, nilotinibe maruziyeti klinik olarak anlamlı bir dereceye kadar düşürebilir.Bu nedenle, TASİGNA alan hastalarda CYP3A4 indükleme potansiyeli daha düşük olanalternatif terapötik ajanların eş zamanlı kullanımı tercih edilmelidir (bkz. Bölüm 4.5). Besin etkisi:Nilotinibin biyoyararlanımı, gıdalarla artmaktadır. TASİGNA yemekle birlikte alınmamalıdır (bkz. Bölüm 4.2 ve 4.5) ve TASİGNA, yemekten en az iki saat sonraalınmalıdır. İlaç alındıktan sonra en az 1 saat herhangi bir gıda tüketilmemelidir. Greyfurtsuyu ya da CYP3A4'ü inhibe ettiği bilinen diğer besinlerin tüketilmesindenkaçınılmalıdır. Kapsülleri yutamayan hastalar için, her bir kapsülün içeriği bir çay kaşığıelma püresi içinde eritilebilir ve hemen sonrasında yutulmalıdır. Bir çay kaşığından fazlaelma püresi alınmamalı ve elma püresi dışında bir gıda kullanılmamalıdır (bkz. Bölüm 5.2) . Karaciğer yetersizliği:Karaciğer yetersizliği, nilotinibin farmakokinetiği üzerinde hafif derecede bir etki gösterir. Tek dozluk 200 mg nilotinib uygulaması, hafif, orta ve şiddetli karaciğeryetersizliği olan hastalarda EAA değerinde, hepatik fonksiyonu normal olan bir kontrolgrubuna karşı sırasıyla %35, %35 ve %19 artışa neden olmuştur. Nilotinibin öngörülenkararlı durum Cmaks değeri, sırasıyla %29, %18 ve %22 artış göstermiştir. Klinikçalışmalara ALT ve/veya AST düzeyi normal aralığın üst sınırının 2,5 mislinden fazla(ya da hastalığa bağlı ise 5 mislinden fazla) ya da toplam bilirubin düzeyi normal aralığınüst sınırının 1,5 mislinden fazla olan hastalar dahil edilmemiştir. Nilotinib çoğunluklakaraciğer aracılığıyla metabolize edilmektedir. Bu nedenle karaciğer yetmezliği olanhastalarda nilotinib maruziyeti artabilir ve bu hastalar, dikkatle tedavi edilmelidir (bkz.Bölüm 4.2). Serum lipaz:Serum lipaz düzeylerinde yükselme gözlenmiştir. Pankreatit öyküsü olan hastalarda dikkatli olunması önerilmektedir. Abdominal semptomlarla birlikte izlenen lipazyükselmesi olgularında TASİGNA tedavisi kesilmeli ve pankreatit tanısını dışlamak içinuygun tanı yöntemlerine başvurulmalıdır. Total gastrektomi:Total gastrektomi geçiren hastalarda nilotinibin biyoyararlanımı azalabilir (bkz. Bölüm 5.2) . Bu hastaların daha sık takip edilmesi düşünülmelidir. Tümör lizis sendromu:Tümör Lizis Sendromunun (TLS) meydana gelme olasılığı nedeniyle, nilotinib tedavisine başlanmadan önce klinik açıdan anlamlı dehidratasyonun düzeltilmesi veyüksek ürik asit düzeylerinin tedavi edilmesi önerilmektedir (bkz. Bölüm 4.8). Laktoz:TASİGNA, laktoz içermektedir. Nadir kalıtımsal galaktoz intoleransı, Lapp laktaz yetmezliği ya da glukoz-galaktoz malabsorpsiyon problemi olan hastaların bu ilacıkullanmamaları gerekir. Pediyatrik popülasyonÇocuklarda, yetişkinlere göre daha yüksek sıklıkta, hafif ila orta derecede geçici aminotransferaz ve toplam bilirubin artışları yönünde laboratuvar anormalilerigözlenmiştir; bu, pediatrik popülasyonda hepatotoksisite riskinin daha yüksek olduğunugöstermektedir (bkz. Bölüm 4.8). Karaciğer fonksiyonu (bilirubin ve hepatiktransaminaz düzeyleri), aylık olarak veya klinik olarak belirtildiği şekilde izlenmelidir.Bilirubin ve hepatik transaminazlardaki yükselmeler, nilotinibe geçici olarak araverilerek, doz azaltılarak ve/veya nilotinib tedavisi durdurularak yönetilmelidir (bkz.Bölüm 4.2). KML pediatrik popülasyonunda yapılan bir çalışmada, nilotinib ile tedaviedilen hastalarda büyüme geriliği raporlanmıştır (bkz. Bölüm 4.8). Nilotinib tedavisi alanpediatrik hastalarda büyümenin yakından izlenmesi önerilir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriTASİGNA, eğer klinik endikasyon mevcut ise eritropoietin veya granülosit koloni uyarıcı faktör (G-CSF) gibi hematopoetik büyüme faktörleri ile kombinasyon halindeverilebilir. Eğer klinik olarak endike ise hidroksiüre veya anagrelid ile verilebilir. Nilotinib ağırlıklı olarak, oksidatif metabolizmaya katkı yapan ana faktör olması beklenen CYP3A4 ile karaciğerde metabolize olur. Nilotinib ayrıca çoklu ilaç dışa atımpompası P-glikoproteinin (P-gp) bir substratıdır. Bu nedenle, sistemik emilen nilotinibinabsorpsiyonu ve sonraki eliminasyonu CYP3A4 ve/veya P-gp'yi etkileyen ilaçlartarafından etkilenebilir. Nilotinibin serum konsantrasyonlarını arttırabilecek ilaçlar:Nilotinibin imatinib (P-gp ve CYP3A4'ün bir substratı ve moderatörü) ile birlikte eşzamanlı olarak kullanıldığı bir Faz I çalışmasında her iki ilaç da CYP3A4 ve/veya Pg-P üzerinde hafif bir inhibitör etki göstermiştir. İmatinibin EAA değeri %18 - %39,nilotinibin EAA değeri ise %18 - %40'a yükselmiştir. Bu değişikliklerin klinik önemiolması muhtemel değildir. Sağlıklı deneklerde güçlü CYP3A4 inhibitörü ketokonazol ile birlikte uygulandığında nilotinibin biyoyararlanımı 3 misli artmıştır. Bu nedenle güçlü CYP3A4 inhibitörleri(ketokonazol, itrakonazol, vorikonazol, ritonavir, klaritromisin ve telitromisin dahilolmakla birlikte bunlarla sınırlı değildir) ile birlikte eş zamanlı tedaviden kaçınılmalıdır(bkz. Bölüm 4.2 ve 4.4). Nilotinibe artmış maruziyet aynı zamanda orta güçte CYP3A4inhibitörleri ile de beklenebilir. CYP3A4 inhibisyonuna yol açmayan ya da minimumdüzeyde yol açan alternatif ilaçlarla birlikte eşzamanlı tedavi düşünülmelidir. Nilotinibin serum konsantrasyonlarını azaltabilecek ilaçlar:Potent bir CYP3A4 indükleyicisi olan rifampisin nilotinib Cmaks değerini %64 azaltır ve nilotinib EAA'sını %80 azaltır. Rifampisin ve nilotinib, birlikte eş zamanlı olarakkullanılmamalıdır. CYP3A4'ü indükleyen diğer ilaçların (örneğin; fenitoin, karbamazepin, fenobarbital ve St. John's Wort) birlikte eşzamanlı kullanımı da benzer şekilde, nilotinibe olanmaruziyeti klinik açıdan önemli ölçüde azaltır. CYP3A4 indükleyicilerinin endikeolduğu hastalarda, nispeten düşük bir indüksiyon potansiyeline sahip alternatif ajanlarınkullanımı düşünülmelidir. Nilotinibin çözünürlüğü, pH'a bağlıdır ve yüksek pH'da daha az çözünür. 5 gün boyunca günde bir kere 40 mg esomeprazol alan sağlıklı gönüllülerde, gastrik pH belirgin biçimdeartmış, fakat nilotinibin emilimi sadece orta düzeyde bir düşüş göstermiştir (Cmaks'ta %27düşüş ve EAAo-®'da %34 düşüş). TASİGNA ihtiyaca göre esomeprazolle veya diğerproton pompası inhibitörleri ile eş zamanlı olarak kullanılabilir. Sağlıklı gönüllülerde yapılan bir çalışmada, 400 mg'lık tekli TASİGNA dozu famotidinden 10 saat sonra ve 2 saat önce uygulandığında nilotinibin farmakokinetiğindeanlamlı bir değişiklik gözlenmemiştir. Bu nedenle, H2 blokörü ile eşzamanlı kullanımgerekli olduğunda, TASİGNA dozundan yaklaşık 10 saat önce ve yaklaşık 2 saat sonrauygulanabilir. Yukarıda bahsedilen aynı çalışmada, 400 mg'lık tekli TASİGNA dozundan 2 saat önce veya sonra bir antiasit uygulaması da (alüminyum hidroksit/magnezyumhidroksit/simetikon) nilotinib farmakokinetiğini değiştirmemiştir. Bu nedenle, gerekliolduğunda, TASİGNA dozundan yaklaşık 2 saat önce veya 2 saat sonra bir antiasituygulanabilir. Sistemik konsantrasyonları nilotinib tarafından değiştirilebilecek ilaçlar:Nilotinib CYP3A4, CYP2C8, CYP2C9, CYP2D6 ve UGTlAl'in in vitro olarak görece güçlü inhibitörü olup, en düşük Ki değeri CYP2C9 içindir (Ki=0,13 mikroM). Duyarlı bir CYP2C9 substratı olan 25 mg varfarin ile sağlıklı gönüllülerle yürütülen tek doz ilaç-ilaç etkileşimi çalışmasında 800 mg nilotinib, varfarinin farmakokinetikparametrelerinde ve protrombin zamanı (PT) ve uluslararası normalize oran (INR) ileölçüldüğünde varfarin farmakodinamiğinde herhangi bir değişikliğe yol açmamıştır.Kararlı durum verisi bulunmamaktadır. Bu çalışma, varfarin ile nilotinib arasında klinikaçıdan anlamlı ilaç-ilaç etkileşiminin varfarinin 25 mg'a kadarki dozlarında daha az olasıolduğunu göstermektedir. Kararlı durum verileri bulunmadığından nilotinib tedavisinebaşlandıktan sonra (en az ilk 2 hafta boyunca) varfarin farmakodinamik parametrelerinin(INR ya da PT) kontrolü önerilir. KML hastalarında 12 gün süreyle günde iki kez 400 mg dozunda uygulanan nilotinib, oral midazolamın (CYP3A4 substratı) sistemik maruziyetini (EAA ve Cmaks) sırasıyla2,6 kat ve 2 kat artırmıştır. Nilotinib orta kuvvetli bir CYP3A4 inhibitörüdür. Bununsonucu olarak, temelde CYP3A4 ile metabolize olan diğer ilaçların (örn. belirli HMG-CoA redüktaz inhibitörleri) sistemik maruziyeti, nilotinib ile bir arada uygulamadaartabilir. CYP3A4 substratları olan ve dar terapötik indekse sahip ilaçlar (bunlarla sınırlıolmamakla birlikte alfentanil, siklosporin, dihidroergotamin, ergotamin, fentanil,sirolimus ve takrolimus dahil) nilotinib ile bir arada uygulandığında uygun izlem ve dozayarlaması gerekli olabilir. Nilotinibin esas olarak CYP3A4 ile elimine edilen statinlerle kombinasyonu, rabdomiyaliz de dahil, statinle indüklenen miyopati olasılığını arttırabilir. Anti-aritmik ilaçlar ve QT uzamasına neden olabilecek diğer ilaçlar:Nilotinib; amiodaron, disopiramid, prokainamid, kinidin ve sotatol gibi anti-aritmik tıbbi ürünleri kullanan hastalar dahil QT aralığı uzaması görülmüş veya bu durumungelişebileceği hastalarda veya klorokin, halofantrin, klaritromisin, haloperidol, metadonve moksifloksasin gibi QT uzamasına yol açabilecek diğer tıbbi ürünleri kullananhastalarda dikkatli kullanılmalıdır (bkz. Bölüm 4.4). Besin etkileşimleri:TASİGNA'nın emilimi ve biyoyararlanımı, yemekle birlikte alındığında artarak, daha yüksek bir serum konsantrasyonuna yol açmaktadır (bkz. Bölüm 4.2, 4.4 ve 5.2).Greyfurt suyu ve CYP3A4'ü inhibe ettiği bilinen diğer besinlerin tüketilmesindenkaçınılmalıdır. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin hiçbir klinik etkileşim çalışması yürütülmemiştir. Pediyatrik popülasyon:Pediyatrik popülasyona ilişkin hiçbir klinik etkileşim çalışması yürütülmemiştir. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi D'dir. Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli olan kadınlar, TASİGNA tedavisi sırasında ve bitiminden 2 hafta sonrasına kadar çok etkili bir doğum kontrol yöntemi kullanmalıdır. Gebelik dönemiTASİGNA'nın gebe kadınlarda kullanımına ilişkin veri yoktur veya sınırlıdır. Hayvanlarda yapılan çalışmalar, üreme toksisitesi göstermiştir (bkz. Bölüm 5.3).TASİGNA'nın gebelik ve/ veya fetus/yeni doğan üzerinde zararlı farmakolojik etkileribulunmaktadır. TASİGNA, kadının klinik durumu nilotinib ile tedavi gerektirmedikçegebelik sırasında kullanılmamalıdır. Gebelik sırasında kullanılırsa, hasta fetüs içinpotansiyel risk konusunda bilgilendirilmelidir. Eğer nilotinib ile tedavi edilmekte olan bir hasta gebelik düşünüyorsa, Bölüm 4.2 ve 4.4'te tarif edilen tedavinin kesilmesine uygunluk kriterlerine dayalı olarak tedavininkesilmesi düşünülebilir. Tedavisiz remisyon (TFR) denenirken hastalarda gebeliklereilişkin sınırlı miktarda veri mevcuttur. Eğer TFR fazında gebelik planlanıyorsa hasta,gebelik sırasında TASİGNA tedavisine olası bir yeniden başlama gerekliliği konusundabilgilendirilmelidir (bkz. Bölüm 4.2 ve 4.4). Laktasyon dönemiNilotinibin insan sütüyle atılıp atılmadığı bilinmemektedir. Hayvanlardan elde edilen toksikolojik veriler, nilotinibin sütle atıldığını göstermektedir (bkz. Bölüm 5.3). Yenidoğanlar/bebekler için risk olasılık dışı bırakılamadığından, TASİGNA tedavisisırasında ve son dozdan sonraki 2 hafta boyunca kadınların emzirmemesi gerekir. Üreme yeteneği /FertiliteHayvan çalışmaları erkek ve dişi sıçanlarda fertilite üzerinde etki göstermemiştir (bkz. Bölüm 5.3). 4.7. Araç ve makine kullanımı üzerindeki etkilerTASİGNA'nın araç ve makine kullanma becerisi üzerinde etkisi yoktur ya da göz ardı edilebilir etkiye sahiptir. Fakat baş dönmesi, yorgunluk, görme bozukluğu ya da güvenlitaşıt kullanma ya da makine kullanma yeteneği üzerinde potansiyel bir etkiye sahip diğeristenmeyen etkiler yaşayan hastaların, bu istenmeyen etkiler devam ettiği sürece buaktiviteleri gerçekleştirmemeleri tavsiye edilir (bkz. Bölüm 4.8). 4.8. İstenmeyen etkilerGüvenlilik profilinin özetiGüvenlilik profili, onaylı endikasyonlarda 13 klinik çalışmada TASİGNA ile tedavi edilen 3.422 hastanın birleştirilmiş verilerine dayanmaktadır: yeni tanı konmuş kronikevrede Philadelphia kromozomu pozitif kronik miyelojenöz lösemi (KML) görülenyetişkin ve pediatrik hastalar (2.414 hastayla 5 klinik çalışma), kronik ve hızlanmış evrePhiladelphia kromozomu pozitif KML'si olan, imatinib dahil önceki tedaviye dirençveya intolerans gösteren yetişkin hastalar (939 hastayla 6 klinik çalışma) ve imatinibdahil önceki tedaviye direnç veya intolerans gösteren kronik evre Philadelphiakromozomu pozitif KML'si olan pediatrik hastalar (69 hastayla 2 klinik çalışma). Bubirleştirilmiş veriler, 9.039,34 hasta-maruziyet yılını temsil eder. Nilotinibin güvenlilik profili, endikasyonlar arasında tutarlıdır. Birleştirilmiş güvenlilik verilerinden en yaygın advers reaksiyonlar (insidans >%15) şunlardır: döküntü (%26,4), üst solunum yolu enfeksiyonu (farenjit, nazofarenjit, rinitdahil) (%24,8), baş ağrısı (%21,9), hiperbilirubinemi (kanda artmış bilirubin dahil)(%18,6), artralji (%15,8), yorgunluk (%15.4), bulantı (%16,8), kaşıntı (%16,7) vetrombositopeni (%16,4). Advers reaksiyonların listesi Klinik çalışmalardan ve pazarlama sonrası raporlardan advers reaksiyonlar, MedDRA sistem organ sınıfı ve sıklık kategorisine göre aşağıda listelenmiştir. Sıklık kategorilerişu kural kullanılarak tanımlanır: çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygınolmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000);bilinmiyor (mevcut verilerden hareketle tahmin edilemiyor). Enfeksiyonlar ve enfestasyonlarÇok yaygın: Üst solunum yolu enfeksiyonu (farenjit, nazofarenjit, rinit dahil) Yaygın: Folikülit, bronşit, kandidiyaz (oral kandidiyaz dahil), pnömoni, gastroenterit, idrar yolu enfeksiyonu Yaygın olmayan: Herpes virüsü enfeksiyonu, anal apse, kandidiyaz (kandida enfeksiyonu), çıban, sepsis, subkutan apse, ayak mantarı Seyrek: Hepatit B reaktivasyonu (Kist ve polipler de dahil olmak üzere) İyi huylu ve kötü huylu neoplazmalarYaygın olmayan: Deri papillomu Seyrek: Oral papillom, paraproteinemi Kan ve lenf sistemi hastalıklarıÇok yaygın: Anemi, trombositopeni Yaygın: Lökopeni, lökositoz, nötropeni, trombositemi Yaygın olmayan: Eozinofili, febril nötropeni, lenfopeni, pansitopeni Bağışıklık sistemi hastalıklarıYaygın olmayan: Hipersensitivite Endokrin hastalıklarıÇok yaygın: Büyüme geriliği Yaygın: Hipotiroidizm Yaygın olmayan: Hipertiroidizm Seyrek: Sekonder hiperparatiroidizm, tiroidit Metabolizma ve beslenme hastalıklarıYaygın: Elektrolit dengesizliği (hipomagnezemi, hiperkalemi, hipokalemi, hiponatremi, hipokalsemi, hiperkalsemi, hiperfosfatemi dahil), diabetes mellitus, hiperglisemi,hiperkolesterolemi, hiperlipidemi, hipertrigliseridemi, azalmış iştah, gut, hiperürisemi,hipofosfatemi (azalmış kan fosforu dahil) Yaygın olmayan: Dehidratasyon, artmış iştah, dislipidemi, hipoglisemi Seyrek: İştah bozukluğu, tümör lizis sendromu Psikiyatrik hastalıklarıYaygın: Depresyon, uykusuzluk, anksiyete Yaygın olmayan: Amnezi, zihin bulanıklığı durumu, oryantasyon bozukluğu Seyrek: Disfori Sinir sistemi hastalıklarıÇok yaygın: Baş ağrısı Yaygın: Baş dönmesi, hipoestezi, parestezi, migren Yaygın olmayan: Serebrovasküler olay, intrakraniyel/serebral hemoraji, iskemik inme, geçici iskemik atak, serebral enfarktüs, bilinç kaybı (senkop dahil), tremor, dikkatbozukluğu, hiperestezi, disestezi, letarji, periferik nöropati, huzursuz bacak sendromu,yüz felci Seyrek: Baziler arter stenozu, beyin ödemi, optik nörit Göz hastalıkları:Yaygın: Konjonktivit, göz kuruluğu (kseroftalmi dahil), göz iritasyonu, hiperemi (skleral, konjonktival, oküler), bulanık görme Yaygın olmayan: Görme bozukluğu, konjonktival hemoraji, azalmış görme keskinliği, göz kapağı ödemi, blefarit, fotopsi, alerjik konjonktivit, diplopi, gözde hemoraji, gözdeağrı, gözde kaşıntı, gözde şişlik, oküler yüzey hastalığı, periorbital ödem, fotofobi Seyrek: Koryoretinopati, papilloödem Kulak ve iç kulak hastalıklarıYaygın: Vertigo, kulak ağrısı, tinit Yaygın olmayan: Bozulmuş işitme (hipoakuzi) Kardiyak hastalıklarıYaygın: Angina pektoris, aritmi (atriyoventriküler blok, kardiyak çarpıntı, ventriküler ekstrasistol, taşikardi, atriyel fibrilasyon, bradikardi dahil), çarpıntı, uzamışelektrokardiyogram QT'si, koroner arter hastalığı Yaygın olmayan: Miyokard enfarktüsü, kardiyak üfürüm, perikardiyel efüzyon, kalp yetmezliği, diyastolik fonksiyon bozukluğu, sol dal bloku, perikardit Seyrek: Siyanoz, azalmış ejeksiyon fraksiyonu Bilinmiyor: Ventriküler fonksiyon bozukluğu Vasküler hastalıklarıYaygın: Hipertansiyon, kızarma, periferik arteriyel oklüzif hastalık Yaygın olmayan: Hipertansif kriz, intermitan kladikasyo, periferik arter stenozu, hematom, arteriyoskleroz, hipotansiyon, tromboz Seyrek: Hemorajik şok Solunum, göğüs bozuklukları ve mediastinal hastalıklarÇok yaygın: Öksürük Yaygın: Dispne, efor dispnesi, burun kanaması, orofaringeal ağrı Yaygın olmayan: Pulmoner ödem, plevral efüzyon, interstisyel akciğer hastalığı, plevratik ağrı, plörezi, boğaz tahrişi, disfoni, pulmoner hipertansiyon, hırıltı Seyrek: Faringolaringeal ağrı Gastrointestinal hastalıklarıÇok yaygın: Bulantı, üst karın ağrısı, kabızlık, ishal, kusma Yaygın: Pankreatit, karın rahatsızlığı, karın şişliği, mide gazı, karın ağrısı, dispepsi, gastrit, gastroözofageal reflü, hemoroid, stomatit Yaygın olmayan: Gastrointestinal hemoraji, melena, ağızda ülserasyon, özofageal ağrı, ağız kuruluğu, diş hassasiyeti (diş hiperestezisi), disguzi, enterokolit, gastrik ülser, dişeti iltihabı, hiatus hernisi, rektal hemoraji Seyrek: Gastrointestinal ülser perforasyonu, hematemez, özofageal ülser, ülseratif özofajit, retroperitoneal hemoraji, subileus Hepato-bilier hastalıklarıÇok yaygın: Hiperbilirubinemi (kanda artmış bilirubin dahil) Yaygın: Anormal karaciğer fonksiyonu Yaygın olmayan: Hepatotoksisite, toksik hepatit, sarılık, kolestaz, hepatomegali Deri ve deri altı doku hastalıklarıÇok yaygın: Döküntü, kaşıntı, alopesi Yaygın: Gece terlemeleri, egzama, ürtiker, hiperhidroz, kontüzyon, akne, dermatit (alerjik, eksfolyatif ve akneiform dahil), cilt kuruluğu, eritem Yaygın olmayan: Eksfolyatif döküntü, ilaç erüpsiyonu, deride ağrı, ekimoz, yüzde şişlik, kabarcık, dermal kistler, eritema nodozum, hiperkeratoz, peteşi, ışığa duyarlılık,psöriyazis, deri renginde bozukluk, deri eksfoliyasyonu, deri hiperpigmentasyonu, derihipertrofisi, deri ülseri Seyrek: Eritema multiforme, palmar-plantar eritrodizestezi sendromu, sebasöz hiperplazi, deri atrofisi Kas-iskelet bozukluklar, bağ doku ve kemik hastalıklarıÇok yaygın: Miyalji, atralji, sırt ağrısı, uzuvda ağrı Yaygın: Kas-iskelet göğüs ağrısı, kas-iskelet ağrısı, boyun ağrısı, kas güçsüzlüğü, kas spazmları, kemik ağrısı Yaygın olmayan: Kas-iskelet katılığı, eklem şişliği, artrit, yan ağrısı Böbrek ve idrar yolu hastalıklarıYaygın: Polaküri, disüri Yaygın olmayan: İdrara çıkma aciliyeti, noktüri, kromatüri, hematüri, böbrek yetmezliği, idrar inkontinansı Üreme ve meme hastalıklarıYaygın: Ereksiyon bozukluğu, menoraji Yaygın olmayan: Meme ağrısı, jinekomasti, meme ucu şişliği Seyrek: Meme sertleşmesi Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarÇok yaygın: Yorgunluk, ateş Yaygın: Göğüs ağrısı (kardiyak dışı göğüs ağrısı dahil), ağrı, göğüs rahatsızlığı, bitkinlik, yorgunluk ve periferik ödem, titremeler, grip benzeri hastalık Yaygın olmayan: Yüz ödemi, yerçekimine bağlı ödem, vücut ısısında değişiklik hissetme (sıcak hissetme, üşüme dahil), lokalize ödem Seyrek: Ani ölüm AraştırmalarÇok yaygın: Artmış alanin aminotransferaz, artmış lipaz Yaygın: Azalmış hemoglobin, kanda artmış amilaz, artmış aspartat aminotransferaz, kanda artmış alkalin fosfataz, artmış gama-glutamil transferaz, kanda artmış kreatininfosfokinaz, azalmış kilo, artmış kilo, artmış kreatinin, artmış total kolesterol Yaygın olmayan: Kanda artmış laktat dehidrogenaz, kanda artmış üre, kanda artmış konjuge olmayan bilirubin, kanda artmış paratiroid hormonu, kanda artmış trigliseritler,azalmış globülinler, artmış lipoprotein kolesterol (düşük yoğunluklu ve yüksekyoğunluklu dahil), artmış troponin Seyrek: Azalmış kan glukozu, azalmış kan insülini, artmış kan insülini, azalmış insülin C-peptid Not: Advers ilaç reaksiyonlarının tümü pediatrik çalışmalarda gözlenmemiştir. Seçili advers reaksiyonların açıklaması Ani ölümGeçmiş tıbbi kardiyak hastalık öyküsü veya önemli kardiyak risk faktörleri olan imatinibe dirençli veya intolere kronik veya hızlanmış evrede KML görülen hastalardainsani amaçlı ilaca erken erişim programlarında ve/veya TASİGNA klinikçalışmalarında yaygın olmayan (%0.1 ila 1) ani ölüm vakaları bildirilmiştir (bkz. Bölüm4.4). Hepatit B reaktivasyonuBCR-ABL TKİ'larla ilişkili olarak hepatit B reaktivasyonu bildirilmiştir. Bazı vakalar karaciğer nakline veya ölümcül bir sonuca yol açan akut karaciğer yetmezliği veyafulminan hepatit ile sonuçlanmıştır (bkz. Bölüm 4.4). Pediatrik popülasyonKronik evrede Philadelphia kromozomu pozitif KML'li pediatrik hastalarda (2 ila <18 yaşında) (n=58) nilotinibin güvenliliği, bir ana çalışmada 60 aylık bir periyottaaraştırılmıştır (bkz. Bölüm 5.1). Pediatrik hastalarda gözlenen advers reaksiyonlarınsıklığı, tipi ve şiddeti genellikle yetişkinlerde gözlenenle tutarlıdır; buna istisnalaryetişkin hastalarda daha yüksek sıklıkta bildirilmiş hiperbilirubinemi/kan bilirubini artışı(Derece 3/4: %10.3) ve transaminaz artışıdır (AST Derece 3/4: %1,7, ALT Derece 3/4:%12,1). Bilirubin ve hepatik transaminaz düzeyleri tedavi sırasında izlenmelidir (bkz.Bölüm 4.2 ve 4.4). Pediatrik popülasyonda büyüme geriliğiYeni tanı konmuş hastalarda 51,9 aylık ve imatinibe/dasatinibe dirençli veya imatinibe intolere Ph+ KML-KF hastalarında 59,9 aylık medyan maruziyet ile KML'li pediatrikpopülasyonda yürütülen bir çalışmada, sekiz hastada büyümede yavaşlama (başlangıcagöre en az iki ana yüzdelik doğruyu geçen) gözlenmiştir: beşi (%8,6) başlangıca göre ikiana yüzdelik doğruyu ve üçü (%5,2) başlangıca göre üç ana yüzdelik doğruyu geçmiştir.Büyüme geriliği ile ilişkili olaylar 3 hastada (%5,2) bildirilmiştir. Nilotinib tedavisi alanpediatrik hastalarda büyümenin yakından izlenmesi önerilir (bkz. Bölüm 4.4). Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarakizlenmesine olanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli adversreaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. ([email protected]. Doz aşımı ve tedavisiNilotinible, belirtilmeyen sayıda TASİGNA kapsülün alkol veya diğer ilaçlarla birlikte içildiği kasıtlı doz aşımına ilişkin münferit raporlar alınmıştır. Advers olaylar nötropeni,kusma ve sersemlik olmuştur. EKG değişiklikleri veya hepatoksisite bildirilmemiştir.Bildirilen sonuçlar, olayların reversibl olduğu yönündedir. Doz aşımı durumunda, hasta gözlem altında tutulmalı ve uygun destekleyici tedavi uygulanmalıdır. 5.FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antineoplastik ajanlar, protein kinaz inhibitörleri, BCR-ABL tirozin kinaz inhibitörleri ATC Kodu: L01EA03 Etki mekanizması: Nilotinib, hem hücre dizilerinde hem de Philadelphia-kromozomu pozitif primer lösemi hücrelerinde, BCR-ABL onkoproteininin ABL tirozin kinaz aktivitesinin güçlü birinhibitörüdür. İlaç, ATP bağlanma yerine son derece yüksek bir afinite ile bağlanarak,vahşi-tip BCR-ABL'yi güçlü bir şekilde inhibe etmekte ve BCR-ABL'nin imatinibekarşı dirençli 32/33 mutant formuna karşı etkinlik sağlamaktadır. Bu biyokimyasaletkinliğin bir sonucu olarak, nilotinib KML hastalarından alınan Philadelphia-kromozomu pozitif primer lösemi hücrelerinde ve hücre dizilerinde selektif olarakproliferasyonu inhibe etmekte ve apopitozu indüklemektedir. KML sıçan modellerinde,nilotinib monoterapi olarak oral uygulamayı takiben tümör yükünü azaltmakta ve sağkalımı uzatmaktadır. Farmakodinamik etkilerTASİGNA'nın, KML tedavisi için önerilen terapötik dozlarda oral uygulamayı takiben elde edilen aralıktaki konsantrasyonlarda inhibe ettiği PDGF (Platelet kaynaklı büyümefaktörü), KIT ve Efrin reseptör kinazları dışında, Src de dahil olmak üzere incelenendiğer protein kinazların çoğuna karşı herhangi bir etkisi yoktur ya da çok az bir etkisivardır (bkz. Tablo 2).
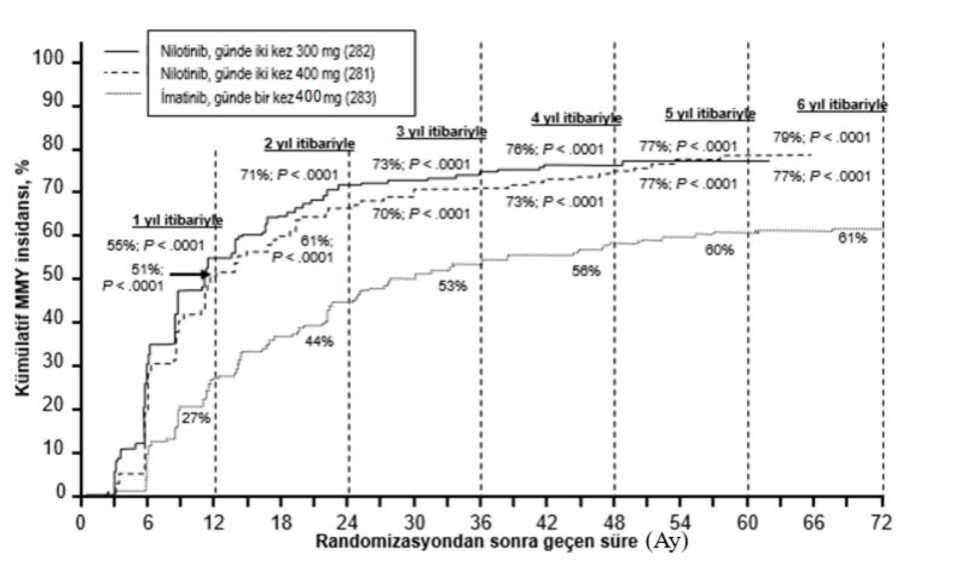
Klinik etkililik: Yeni tanı konmuş kronik evre KML 'de klinik çalışmalarSitogenetik olarak doğrulanmış yeni tanı konmuş yetişkin Ph+ KML kronik evre hastalarında nilotinib ve imatinib etkinliklerinin karşılaştırılması amacıyla 846 yetişkinhastada açık etiketli, çok merkezli, randomize bir Faz III çalışması yürütülmüştür.Hastalar altı ay önce tanı almıştır ve hidroksiüre ve/veya anagrelid hariç önceden tedavigörmemişlerdir. Hastalar günde iki kez nilotinib 300 mg (n=282), günde iki kez nilotinib400 mg (n=281) veya günde bir kez imatinib 400 mg (n=283) almak üzere 1:1:1 oranındarandomize edilmiştir. Randomizasyon, tanı anındaki Sokal risk skoruna görekatmanlandırılmıştır. Başlangıç karakteristikleri üç tedavi kolu arasında dengeli olmuştur. Medyan yaş her iki nilotinib kolunda 47 yıl ve imatinib kolunda 46 yıl olmuş, günde iki kez nilotinib 300mg, günde iki kez nilotinib 400 mg ve günde bir kez imatinib 400 mg kollarındahastaların sırasıyla %12,8, %10 ve %12,4'ünün >65 yaşında olduğu belirlenmiştir. Erkekhastaların sayısı kadın hastalardan biraz daha fazla olmuştur (günde iki kez nilotinib 300mg, günde iki kez nilotinib 400 mg ve günde bir kez imatinib 400 mg kollarında sırasıyla%56, %62,3 ve %55,8). Hastaların %60'ından fazlası beyazdır ve tüm hastaların %25'iAsyalıdır. Birincil veri analizi zaman noktası 846 hastanın tümünün 12 aylık tedaviyi tamamladığı (ya da daha önce ayrıldığı) zaman olmuştur. Müteakip analizler hastaların 24, 36, 48, 60ve 72 aylık tedaviyi tamamladığı (ya da daha önce ayrıldığı) zamanları yansıtmaktadır.Tedavide geçen medyan süre nilotinib tedavi kollarında yaklaşık 70 ay ve imatinibgrubunda 64 aydır. Kullanılan medyan doz yoğunluğu günde iki kez 300 mg nilotinibiçin 593 mg/gün, günde iki kez 400 mg nilotinib için 772 mg/gün ve günde bir kez 400mg imatinib için 400 mg/gün şeklindedir. Bu çalışma devam etmektedir. Birincil sonlanım noktası 12. ayda majör moleküler yanıt (MMY) olmuştur. MMY, RQ PCR ile ölçülen uluslararası ölçeğe (IS) göre <%0,1 BCR-ABL/ABL% şeklindetanımlanmış olup standardize başlangıç değerinden >3 log BCR-ABL transkriptazalmasına karşılık gelmektedir. 12. ayda MMY oranı, günde iki kez 300 mg nilotinibgrubunda, günde bir kez 400 mg imatinib grubu ile karşılaştırıldığında istatistiksel olarakanlamlı düzeyde daha yüksek olmuştur (%44,3 karşısında %22,3, p<0,0001). 12. ayda MMY oranı günde iki kez 400 mg nilotinib grubunda da, günde bir kez 400 mg imatinib grubu ile karşılaştırıldığında, istatistiksel olarak anlamlı düzeyde daha yüksekbulunmuştur (%42,7 karşısında %22,3, p<0,0001). 3, 6, 9 ve 12. aylarda MMY oranları günde iki kez 300 mg nilotinib grubu için sırasıyla %8,9, %33, %43,3 ve %44,3, günde iki kez 400 mg nilotinib grubu için sırasıyla %5,%29,5%, %38,1 ve %42,7 ve günde bir kez 400 mg imatinib için sırasıyla %0,7, %12,%18 ve %22,3 olmuştur. 12, 24, 36, 48, 60 ve 72. aylardaki MMY oranları Tablo 3'te gösterilmektedir. Tablo 3 MMY oranı
5 Sadece belirli bir zaman noktasında MMY olan hastalar o zaman noktası için yanıt veren hastalar olarak dahil edilmiştir. Tüm hastaların toplamda 395'i (%46,7) (günde iki kez nilotinib 300 mg grubunda 130, günde iki keznilotinib 400 mg grubunda 110 ve imatinib grubunda 155 hasta); eksik/değerlendirilebilir olmayan PCR ölçümleri(n=25), tedavi başlangıcında atipik transkriptler (n=8) veya 72 aylık zaman noktasından önce ayrılma (n=362)nedeniyle 72. ayda MMY için değerlendirilebilir olmamıştır.Farklı zaman noktalarına göre MMY oranları (o zaman noktasında veya öncesinde yanıt veren hasta olarak MMY'ye ulaşan hastalar dahil), kümülatif MMY insidansındasunulmaktadır (Bkz. Şekil 1).
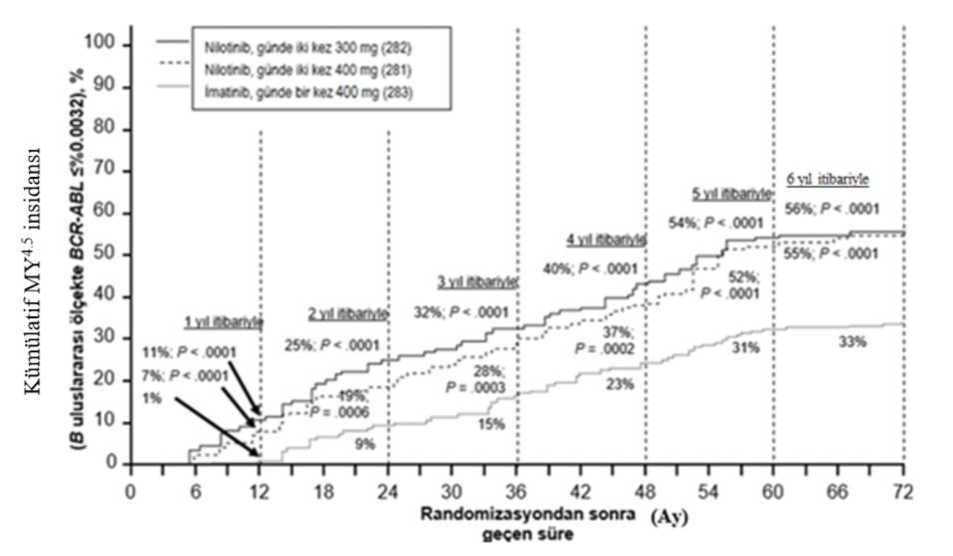
Tüm Sokal risk gruplarında tüm zaman noktalarındaki MMY oranı iki nilotinib grubunda da imatinib grubundan daha yüksek olmuştur. Retrospektif bir analizde, günde iki kez 300 mg nilotinib kullanan hastaların %91'i (234/258) tedavinin 3. ayında <%10 BCR-ABL düzeyleri elde ederken, bu oran gündebir kez imatinib 400 mg kullanan hastalarda %67'dir (176/264). Tedavinin 3. ayında<%10 BCR-ABL düzeylerine sahip hastalar, bu moleküler yanıt düzeyini eldeetmeyenlere kıyasla 72. ayda daha fazla genel sağkalım göstermiştir (sırasıyla %94,5karşısında %77,1 [p=0,0005]). İlk MMY'ye kadar geçen sürenin Kaplan-Meier analizine göre, farklı zaman noktalarında MMY elde etme olasılığı, hem günde iki kez 300 mg hem de günde iki kez400 mg nilotinib için, günde bir kez 400 mg imatinib ile karşılaştırıldığında, daha yüksekolmuştur (günde iki kez 300 mg nilotinib ile günde bir kez 400 mg imatinib arasındaHR=2,17 ve katmanlandırılmış log sıra p<0,0001, günde iki kez 400 mg nilotinib ilegünde bir kez 400 mg imatinib arasında HR=1,88 ve katmanlandırılmış log sıra p<0,0001). Farklı zaman noktalarında IS ile %<0,01 ve <%0,0032 düzeylerinde moleküler yanıtı olan hastaların oranı Tablo 4'te sunulmakta ve farklı zaman noktalarına göre IS ile%<0,01 ve <%0,0032 değerlerinde moleküler yanıtı olan hastaların oranı Şekil 2 ve 3'tegösterilmektedir. IS ile <%0,01 ve <%0,0032 moleküler yanıt oranları, standardize bir başlangıç değerinden sırasıyla >4 ve >4.5 log BCR-ABL transkriptler azalmasına karşılık gelmektedir. Tablo 4 %<0,01 (4 log azalma) ve <%0,0032 (4.5 log azalma) düzeyindemoleküler yanıtı olan hastaların oranları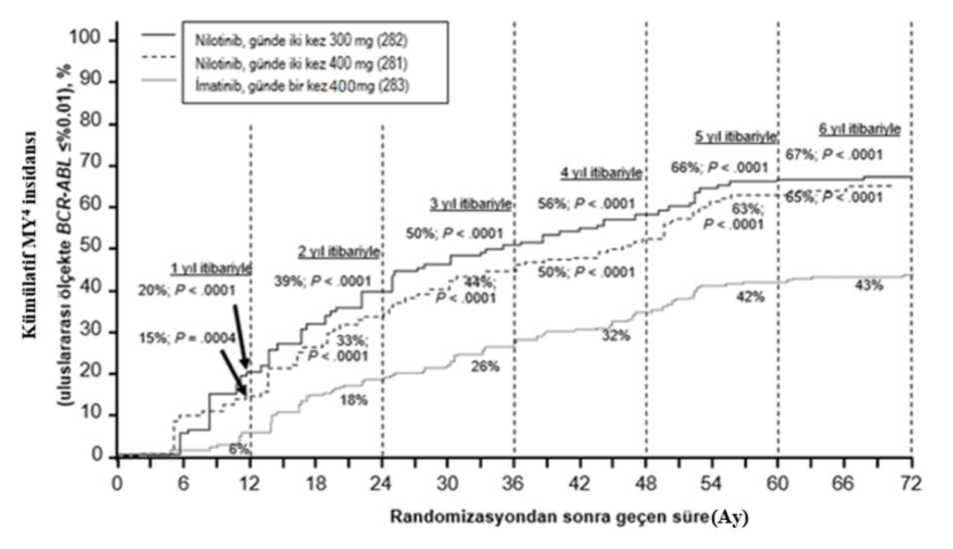
İlk MMY'nin süresine ilişkin Kaplan Meier tahminlerine dayalı olarak, MMY'ye ulaşan hastalar arasında yanıtını 72 ay sürdüren hastaların oranları günde iki kez 300 mgnilotinib grubunda %92,5 (%95 GA: %88,6 %96,4), günde iki kez 400 mg nilotinibgrubunda %92,2 (%95 GA: %88,5 %95,9) ve günde bir kez 400 mg imatinib grubunda%88 (%95 GA: %83 %93,1) olmuştur. Tam sitogenetik yanıt (TSY), değerlendirilen minimum 20 metafaza dayalı olarak kemik iliğinde %0 Ph+ metafazı şeklinde tanımlanır. 12 ay itibariyle en iyi TSY oranı (yanıtveren hastalar olarak 12. ay zaman noktasında veya öncesinde TSY'ye ulaşan hastalarıiçerir), hem günde iki kez nilotinib 300 mg hem de günde iki kez 400 mg nilotinib içingünde bir kez 400 mg imatinibe kıyasla istatistiksel olarak daha yüksektir (bkz. Tablo 5). 24 ay itibariyle TSY oranı (yanıt veren hastalar olarak 24. ay zaman noktasında veya öncesinde TSY'ye ulaşan hastaları içerir), hem günde iki kez 300 mg nilotinib hem degünde iki kez 400 mg nilotinib için günde bir kez 400 mg imatinibe kıyasla istatistikselolarak daha yüksektir.
Kaplan-Meier tahminlerine dayalı olarak, TSY'ye ulaşan hastalar arasında yanıtını 72 ay süreyle sürdüren hastaların oranları, günde iki kez 300 mg nilotinib grubunda %99,1(%95 GA: %97,9-%100), günde iki kez 400 mg nilotinib grubunda %98,7 (%95 GA:%97,1-%100) ve günde bir kez imatinib 400 mg grubunda %97 (%95 GA: %94,7-%99,4) şeklindedir. Tedavi sırasında hızlanmış evreye (AF) veya blast krize (BK) progresyon, randomizasyon tarihinden itibaren hızlanmış evreye veya blast krize ilk belgelenenhastalık progresyonuna ya da KML ilişkili ölüme kadar geçen süre şeklindetanımlanmıştır. Tedavi sırasında hızlanmış evreye veya blast krize progresyon toplam 17hastada gözlenmiştir: günde iki kez nilotinib 300 mg'da 2 hasta, günde iki kez nilotinib400 mg'da 3 hasta ve günde bir kez imatinib 400 mg'da 12 hasta. 72. ayda hızlanmışevreye veya blast krize progresyonu olmayan hastaların tahmini oranları sırasıyla %99,3,%98,7 ve %95,2 olmuştur (HR=0,1599 ve katmanlandırılmış log sıra günde iki keznilotinib 300 mg ile günde bir kez imatinib arasında p=0,0059, HR=0,2457 vekatmanlandırılmış log sıra günde iki kez nilotinib 400 mg ile günde bir kez imatinibarasında p=0,0185). 2 yıllık analizden sonra tedavide herhangi bir yeni AF/BK'yeprogresyon olayı bildirilmemiştir. Progresyon kriteri olarak klonal evrilme dahil, veri kesme tarihi itibariyle toplam 25 hasta tedavideyken hızlanmış evreye veya blast krize progrese olmuştur (günde iki keznilotinib 300 mg grubunda 3, günde iki kez nilotinib 400 mg grubunda 5 ve günde birkez imatinib 400 mg grubunda 17). 72. ayda klonal evrilme dahil hızlanmış evreye veyablast krize progresyonu olmayan hastaların tahmini oranları sırasıyla %98,7, %97,9 ve%93,2 olmuştur (HR=0,1626 ve katmanlandırılmış log sıra günde iki kez nilotinib 300mg ile günde bir kez imatinib arasında p=0,0009, HR=0,2848 ve katmanlandırılmış logsıra günde iki kez nilotinib 400 mg ile günde bir kez imatinib arasında p=0,0085). Toplam 55 hasta tedavi sırasında ya da tedavinin kesilmesinden sonrasındaki takip sırasında yaşamını kaybetmiştir (günde iki kez 300 mg nilotinib grubunda 21, günde ikikez 400 mg nilotinib grubunda 11 ve günde bir kez 400 mg imatinib grubunda 23). Bu55 ölümün 26'sı KML ile ilişkili olmuştur (günde iki kez 300 mg nilotinib grubunda 6,günde iki kez 400 mg nilotinib grubunda 4 ve günde bir kez 400 mg imatinib grubunda16). 72. ayda hayatta kalan hastaların tahmini oranı sırasıyla %91,6, %95,8 ve %91,4'tür(HR=0,8934 ve katmanlandırılmış log sıra günde iki kez nilotinib 300 mg ile imatinibarasında p=0,7085, HR=0,4632 ve katmanlandırılmış log sıra günde iki kez nilotinib 400mg ile imatinib arasında p=0,0314). Olay olarak sadece KML ile ilişkili ölümler gözönünde bulundurulduğunda, 72. ayda tahmini genel sağkalım oranları sırasıyla %97,7,%98,5 ve %93,9 olmuştur (HR=0,3694 ve katmanlandırılmış log sıra günde iki keznilotinib 300 mg ile imatinib arasında p=0,0302, HR=0,2433 ve katmanlandırılmış logsıra günde iki kez nilotinib 400 mg ile imatinib arasında p=0,0061). Birinci basamak tedavi olarak TASİGNA ile tedavi edilmiş ve sürekli derin moleküler yanıt elde etmiş olan kronik çevredeki yetişkin Ph+ KML hastalarında tedavinin kesilmesiAçık etiketli, tek kollu bir çalışmada, birinci basamakta > 2 yıl süreyle nilotinib ile tedavi edilip MolecularMD MRDx BCR-ABL testi ile ölçüldüğünde MY4.5 elde etmiş olankronik evredeki 215 yetişkin Ph+ KML hastası, 52 hafta daha nilotinibe devamedecekleri faza alınmıştır (nilotinib konsolidasyon fazı). 215 hastanın 190'ı (%88,4),konsolidasyon fazı sırasında aşağıdaki kriterler ile tanımlanan sürekli derin moleküleryanıt elde ettikten sonra Tedavisiz Faza (TFR) girmiştir: - Üç ayda bir yapılan (12 haftada bir alınan) değerlendirmelerin en az 4'ü en az MY4(BCR-ABL/ABL <%0,01 IS) olmuş ve bir yıl korunmuştur. - Son değerlendirme MY4.5'tir (BCR-ABL/ABL <%0,0032 IS). - İki değerlendirmeden fazlası MY4 - MY4.5 arası (%0,0032 IS < BCR-ABL/ABL<%0,01 IS) değildir. Birincil sonlanım noktası, TFR fazına başladıktan sonra 48 haftada MMY'de olan hastaların yüzdesi olarak belirlenmiştir (tedavinin yeniden başlatılması gereken hastalaryanıt vermeyen hasta kabul edilerek).
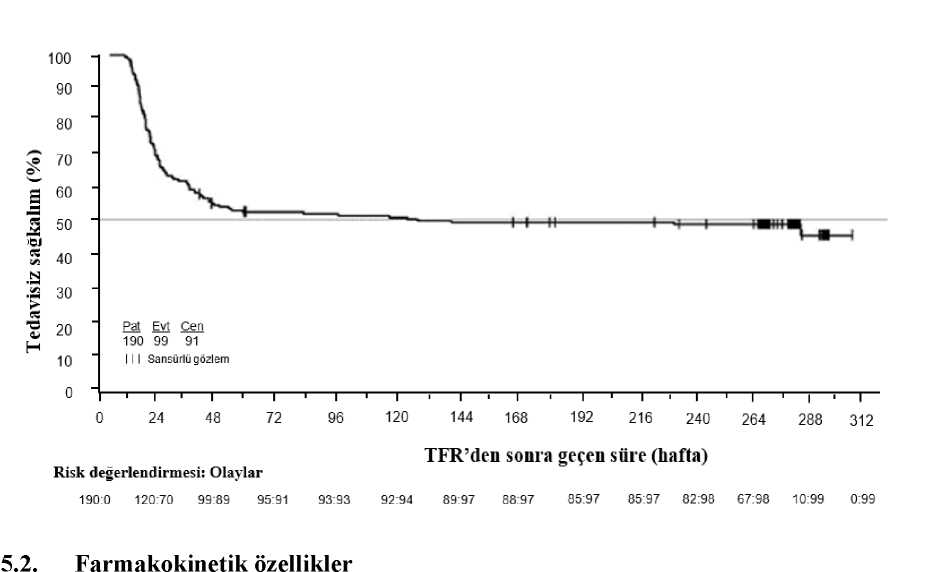
Yeniden tedavi edilen tüm hastaların %50'sinin MMY ve MY4.5'i geri kazanma süresi sırasıyla 7 ve 12,9 hafta olmuştur. Tedavinin yeniden başlamasından 24 hafta sonrayeniden kazanılan MMY kümülatif oranı %97,8 (89/91 hasta), 48 haftada geri kazanılanMY4.5 ise %91,2 (83/91 hasta) olmuştur. Ortanca tedavisiz sağkalımın (TFS) Kaplan Meier tahmini 120,1 hafta olmuştur (%95 GA: 36,9, tahmin edilemez [NE]) (Şekil 4); 190 hastanın 91'inde (%47,9) TFS olayıolmamıştır.
Emilim:Oral uygulamadan 3 saat sonra nilotinibin doruk konsantrasyonlarına ulaşılmaktadır. Oral uygulamayı takiben nilotinibin emilimi yaklaşık %30 olarak belirlenmiştir.Nilotinibin mutlak biyoyararlanımı henüz belirlenmemiştir. Bir oral çözeltiye kıyasla(pH 1,2 ila 1,3), nilotinib kapsülün bağıl biyoyararlanımı yaklaşık % 50'dir. Sağlıklıgönüllülerde TASİGNA yemekle birlikte verildiğinde, nilotinibin Cmaks değeri vekonsantrasyon-zaman eğrisinin altında kalan alanı (EAA), aç karnına alınmasına kıyaslasırasıyla %112 ve %82 oranında artmıştır. TASİGNA'nın yemekten 30 dakika ya da 2saat sonra alınması nilotinibin biyoyararlanımını sırasıyla %29 ve %15 oranındaartırmıştır (bkz. Bölüm 4.2, 4.4 ve 4.5). Nilotinibin emilimi (bağıl biyoyararlanım)sırasıyla total gastrektomi ve kısmi gastrektomi geçirmiş hastalarda yaklaşık %48 ve%22 azalabilir. Dağılım:Nilotinibin kan-plazma oranı 0,71 'dir. Plazma proteinlerine bağlanma, in vitrodeneylere dayalı olarak yaklaşık %98 oranındadır.Biyotransformasyon:Sağlıklı deneklerde tanımlanan ana metabolik yollar, oksidasyon ve hidroksilasyondur. Nilotinib, serumdaki başlıca dolaşan bileşendir. Metabolitlerin hiçbiri nilotinibinfarmakolojik aktivitesine anlamlı bir katkıda bulunmamaktadır. Nilotinib CYP2C8'denolası minör bir katkı ile başlıca CYP3A4 ile metabolize edilir. Eliminasyon:Sağlıklı gönüllülerde radyoaktif madde ile işaretlenmiş nilotinibin tek bir dozunun uygulanmasından sonra, dozun %90'ından fazlası 7 gün içerisinde çoğunlukla feçes ile(dozun %94'ü) atılmıştır. Değişmemiş ana ilaç, dozun %69'unu teşkil etmiştir. Günlük doz uygulaması ile çoklu doz farmakokinetiğinden hesaplanmış belirgin eliminasyon yarılanma ömrü yaklaşık 17 saattir. Nilotinib farmakokinetiğinde hastalararasında gözlenen değişkenlik orta düzeyde veya yüksektir. Doğrusallık / doğrusal olmayan durum:Kararlı durumda nilotinib maruziyetinin, günde bir kez 400 mg'ın üzerindeki doz düzeylerinde sistemik maruziyette doz ile orantılı artıştan daha az bir artış olmak üzere,doza bağımlı olduğu gözlenmiştir. Kararlı durumda, günde iki kez 400 mg'lık dozdauygulanan nilotinibe olan günlük sistemik maruziyetin, günde bir kez 800 mg'lık dozakıyasla %35 daha yüksek olduğu görülmüştür. Günde iki kez nilotinib 400 mg ile kararlıdurumdaki sistemik maruziyet (EAA) günde iki kez 300 mg doza göre yaklaşık %13,4daha yüksek bulunmuştur. 12 ayda nilotinib vadi ve tepe ortalama konsantrasyonu gündeiki kez 400 mg ile günde iki kez 300 mg doza göre sırasıyla %15,7 ve %14,8 daha yüksekbulunmuştur. Doz, günde iki kez 400 mg'dan günde iki kez 600 mg'a çıkarıldığında,nilotinib maruziyetinde anlamlı bir artış olmamıştır. Kararlı durum koşullarına 8. gün itibariyle erişilmiştir. İlk doz ve kararlı durum arasında nilotinibe sistemik maruziyette bir artış, günlük doz uygulama için yaklaşık 2 katken,günde iki kez doz uygulama için 3,8 kattır. Biyoeşdeğerlik/biyoyararlanım çalışmaları:Her bir kapsül içeriğinin bir tatlı kaşığı elma sosunda dağıtıldığı 200 mg'lık 2 kapsül ile 400 mg'lık tekli nilotinib doz uygulamasının, 200 mg'lık açılmamış 2 kapsül ile teklidoz uygulamasına biyoeşdeğer olduğu gösterilmiştir. Hastalardaki karakteristik özelliklerPediyatrik hastalar:Pediyatrik hastalarda en yakın 50 mg doza yuvarlanan günde iki kez 230 mg/m2 dozunda (maksimum tek doz 400 mg) nilotinib uygulanmasının ardından nilotinibin kararlı durummaruziyeti ve klirensinin, günde iki kez 400 mg ile tedavi edilen erişkinler ile benzerolduğu bulunmuştur (2 kat içinde). Tek ve çoklu dozlar sonrasında nilotinibinfarmakokinetik maruziyetinin 2 ila <10 yaş hastalar ile >10 ila <18 yaş hastalar arasındakarşılaştırılabilir olduğu görülmüştür. 5.3.Klinik öncesi güvenlilik verileriNilotinib güvenlilik farmakolojisi, tekrarlanan doz toksisitesi, genotoksisite, üreme sistemi toksisitesi, fototoksisite ve karsinojenisite (sıçan ve fare) çalışmalarındadeğerlendirilmiştir. Güvenlilik farmakolojisi çalışmalarıNilotinibin MSS ya da solunum fonksiyonları üzerinde herhangi bir etkisi olmamıştır. İn vitrokardiyak güvenlilik çalışmaları, nilotinib ile izole tavşan kalplerinde aksiyonpotansiyeli süresinde uzama ve hERG dalgalarının bloke edilmesi temelinde QT uzamasıiçin klinik öncesi bir sinyal göstermiştir. Köpeklerde özel bir telemetri çalışmasında veya39 haftaya kadar tedavi edilen köpeklerde veya maymunlarda EKG ölçümlerinde hiçbiretki görülmemiştir. Tekrarlı doz toksisitesi çalışmaları4 haftalık bir süreye kadar köpeklerde ve 9 aylık bir süreye kadar sinomolog maymunlarında gerçekleştirilen tekrarlanan doz toksisite çalışmaları karaciğerin,nilotinibin toksisitesi için başlıca hedef organ olduğunu göstermiştir. Değişimler artmışalanin aminotransferaz ve alkalin fosfataz aktivitesini ve histopatoloji bulgularını(çoğunlukla sinüzoidal hücre ya da Kupffer hücresi hiperplazisi/hipertrofisi, safra kanalıhiperplazisi ve periportal fibrozit) kapsamıştır. Genelde, klinik kimyadaki değişiklikler,4 haftalık bir iyileşme döneminden sonra tamamen düzelirken, histolojik değişiklikler,yalnızca kısmi bir tersinirlik ortaya koymuştur. Karaciğer etkilerinin görüldüğü en düşükdoz düzeylerindeki maruziyetlerin, insanlarda 800 mg/gün'lük bir dozdaki maruziyettendaha düşük olduğu gözlenmiştir. 26 haftaya kadar tedavi edilen farelerde ya da sıçanlardayalnızca küçük karaciğer değişiklikleri gözlenmiştir. Sıçan, köpek ve maymunlardakolesterol düzeylerinde çoğunlukla geri dönüşümlü artışlar gözlenmiştir. Genotoksisite çalışmalarıBakteriyel in vitroin vitroin vivomemeli sistemlerinde metabolik aktivasyonla ve metabolik aktivasyon olmaksızın gerçekleştirilen genotoksisiteçalışmaları, nilotinibin mutajenik potansiyeline ilişkin herhangi bir kanıt ortayakoymamıştır.Karsinojenisite çalışmalarıSıçanlar üzerinde yapılan 2 yıllık karsinojenisite çalışmasında neoplastik olmayan lezyonlar için başlıca hedef organın uterus olduğu görülmüştür (dilatasyon, vaskülerektazi, endotelyal hücre hiperplazisi, enflamasyon ve/veya epitelyal hiperplazi). 5, 15 ve 40 mg/kg/gün dozlarında nilotinib uygulanmasını takiben karsinojenisite kanıtınarastlanmamıştır. En yüksek doz düzeyindeki maruziyetler (EAA cinsinden) insanlarda800 mg/gün dozda nilotinibe günlük kararlı durum maruziyetinin (EAA'ya dayalı)yaklaşık 2 ila 3 katını temsil etmiştir. Nilotinibin 30, 100 ve 300 mg/kg/gün dozunda uygulandığı 26 haftalık Tg.rasH2 fare karsinojenisite çalışmasında, 800 mg/kg'lık maksimum onaylı dozda (günde iki kez 400mg olarak uygulanan) insan maruziyetinin yaklaşık 30 ila 40 katını (EAA cinsinden)temsil eden 300 mg/kg'da deri papillomu/karsinomları tespit edilmiştir. Deri neoplastiklezyonları için Etki Gözlenmeyen Düzey, 800 mg/kg'lık maksimum onaylı dozda (gündeiki kez 400 mg olarak uygulanan) insan maruziyetinin yaklaşık 10 ila 20 katını temsileden 100 mg/kg'dır. Neoplastik olmayan lezyonlar için başlıca hedef organlar deri(epidermal hiperplazi), gelişmekte olan dişler (üst kesici dişlerde enamel organın (dişminesi) dejenerasyonu/atrofisi ve üst kesici dişlerin diş etleri odontojenik epitelyumundaenflamasyon) ve timus (lenfosit azalması insidansında ve/veya şiddetinde artış)olmuştur. Üreme toksisitesi ve fertilite çalışmalarıNilotinib teratojeniteyi indüklememiş, fakat maternal toksisiteye de yol açan dozlarda embriyo- ve fetotoksisiteye yol açmıştır. Hem erkeklerin hem de kadınların tedaviedildiği fertilite çalışmasında da, kadınların tedavi edildiği embriyotoksisiteçalışmasında da artmış bir postimplantasyon kaybı gözlenmiştir. Embriyotoksisiteçalışmalarında, sıçanlarda embriyo-letalite ve fötal etkiler (çoğunlukla azalmış fetusağırlıkları, yüz kemiklerinin (kaynaşmış maksilla/zigomatik) visseral ve iskeletsel değişimlerinin prematüre füzyonu) ve tavşanlarda fetüs rezorpsiyonunda artış ve iskeletsel değişimler mevcuttur. Sıçanlarda yapılan bir doğum öncesi ve doğum sonrasıgelişim çalışmasında, annenin nilotinibe maruziyeti, fiziksel gelişim parametrelerindeilişkili değişikliklerle birlikte yavruların vücut ağırlığında azalmaya ve yavrulardaçiftleşme ve doğurganlık indekslerinde azalmaya neden olmuştur. Dişilerde Advers EtkiGözlenmeyen Düzeylerdeki nilotinib maruziyetinin genellikle, insanlarda 800 mg/günile elde edilenden daha düşük ya da buna eşit olduğu gözlenmiştir. İnsanlar için önerilen dozun yaklaşık 5 katı olan test edilen en yüksek doza kadar, erkek ve dişi sıçanlarda sperm sayısı/motilitesi ya da fertilite üzerinde herhangi bir etkigözlenmemiştir. Juvenil hayvan çalışmalarıBir çalışmada, juvenil sıçanlara oral gavaj yoluyla doğumun ilk haftasından genç erişkinlik dönemine kadar (doğumdan sonra 70. güne kadar) 2, 6 ve 20 mg/kg/gündozlarında nilotinib uygulanmıştır. Standart çalışma parametrelerinin yanı sıra,gelişimsel işaretler, MSS etkileri, çiftleşme ve fertilite için değerlendirmeler yapılmıştır.Her iki cinsiyette vücut ağırlığındaki azalmaya ve erkeklerde prepusyal ayrılmanıngecikmesine (ağırlıktaki azalmayla ilişkili olabilir) dayalı olarak, jüvenil sıçanlarda EtkiGözlenmeyen Düzey 6 mg/kg/gün olarak kabul edilmiştir. Jüvenil hayvanlar,yetişkinlere göre nilotinibe karşı artan bir duyarlılık sergilememiştir. Ek olarak, jüvenilsıçanlardaki toksisite profili, yetişkin sıçanlarda gözlemlenenle benzerdir. Fototoksisite çalışmalarıNilotinibin UV-B ve UV-A aralığında ışığı absorbe ettiği ve deriye dağılarak in vitroin vivoortamda fototoksisite görülmemiştir. Bu nedenle, nilotinibin hastalardafotosensitizasyona yol açma riski çok düşük kabul edilmektedir.6.FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiKapsül içeriğiLaktoz monohidrat 117,08 mg (inek kaynaklı) Krospovidon Poloksamer 188 Susuz koloidal silika/Koloidal silikon dioksit Magnezyum stearat Boş kapsül bileşimiJelatin (sığır kaynaklı)Titanyum dioksit (E171) Demir oksit, sarı (E172) Demir oksit, kırmızı (E172) Baskı mürekkebiŞellak (E904) (böcek kaynaklı) Demir oksit, siyah (E172) N-bütil alkol Propilen glikolDehidrate etanolİzopropil alkolAmonyum hidroksitSaf su 6.2. GeçimsizliklerYeterli veri yoktur. 6.3. Raf ömrü36 aydır. 6.4. Saklamaya yönelik özel tedbirler30°C'nin altındaki oda sıcaklığında ve orijinal ambalajında saklayınız. 6.5. Ambalajın niteliği ve içeriğiPVC/PVDC blisterler. 28 ve 112 kapsül içeren blister ambalaj. 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliği'ne uygun olarak imhaedilmelidir. 7. RUHSAT SAHİBİNovartis Sağlık, Gıda ve Tarım Ürünleri San. ve Tic. A.Ş. Kavacık/Beykoz/İstanbul 8. RUHSAT NUMARASI132/25 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 04.11.2011 Ruhsat yenileme tarihi: 07.11.2017 10. KÜB'ÜN YENİLENME TARİHİ1Yanıt oranı için Cochran Mantel Haenszel (CMH) testi p-değeri (İmatinib 400 mg'a karşı) <0,00012Sadece belli bir zaman noktasında MMY'de olan hastalar o zaman noktası için yanıt verenler olarak dahil edilir. Tüm hastaların %35,2 (n=199) si 36. ayda MMY için değerlendirilebilir değildi (300 mg nilotinib kolunda 87 hasta,imatinib kolunda 112 hasta). Bu hastaların 175'i 36. ay öncesinde tedaviyi bırakmıştı, 17 hastanın PCR sonucueksik/değerlendirilemez idi, 7 hastada ise başlangıçta atipik transkriptler saptanmıştı.3Sadece belli bir zaman noktasında MMY'da olan hastalar, o zaman noktasında yanıt verenler olarak dahil edilir. Tüm hastaların toplamda 305'i (%36,1) (Günde iki kez nilotinib 300 mg grubunda 98, günde iki kez nilotinib 400 mggrubunda 88 ve imatinib grubunda 119); eksik/değerlendirilemez PCR değerlendirmeleri (n=18), başlangıçta atipiktranskriptler (n=8) veya 48. ay zaman noktasından önce çalışmayı bırakma (n=279) nedeniyle 48. ayda MMYaçısından değerlendirilememiştir.4Sadece belli bir zaman noktasında MMY'deki hastalar o zaman noktası için yanıt verenler olarak dahil edilir.5Tüm hastaların toplamda 322'si (%38,1) (Günde iki kez nilotinin 300 mg grubunda 99,günde iki kez nilotinib 400 mg grubunda 93 ve imatinib grubunda 130); eksik/değerlendirilemez PCR değerlendirmeleri (n=9), başlangıçta atipiktranskriptler (n=8) veya 60. ay zaman noktasından önce çalışmayı bırakma (n=305) nedeniyle 60. ayda MMY içindeğerlendirilebilir bulunmamıştır.6 Bir hasta 48. haftada MMY'yi kaybetmemiş fakat TFR fazını bırakmıştır. 2 hasta için, PCR değerlendirmesi 264. haftada mevcut değildir, bu nedenle, 264. haftaveri kesme analizi için yanıtları dikkate alınmamıştır. |
İlaç BilgileriTasigna 150 Mg KapsülEtken Maddesi: Nilotinib Hidroklorür Monohidrat
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.