Azavix 100 Mg Sc Enjeksiyonluk Süspansiyon İçin Toz İçeren Flakon Kısa Ürün BilgisiKISA URUN BILGISI1. BEŞERI TIBBİ ÜRÜNÜN ADIAZAVIX 100 mg SC enjeksiyonluk süspansiyon için toz içeren flakon Steril Sitotoksik 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her flakon 100 mg azasitidin içerir.Hazırlama sonrası elde edilen süspansiyon her mL'de 25 mg azasitidin içerir. Yardımcı madde(ler):0,875 mg 1,525 mg Monosodyum Fosfat Monohidrat Disodyum Hidrojen Fosfat DihidratYardımcı maddeler için 6.1'e bakınız. 3. FARMASOTİK FORMEnjeksiyonluk süspansiyon için toz. Beyaz ila beyaza yakın liyofilize toz. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarAZAVIX hematopoietik kök hücre transplantasyonuna uygun olmayan yetişkin hastalarda: Uluslararası Prognostik Skorlama Sistemi'ne (IPSS) göre intermediate 2 ve yüksekrisk miyelodisplastik sendrom (MDS) Miyeloproliferatif bozukluk olmaksızın kemik iliği blastı %10-29 arasında olan kronikmiyelomonositer lösemi (KMML) Dünya Sağlık Örgütü (WHO) sınıflandırmasına göre %20-30 blast ve çoklu serilidisplazisi olan akut miyeloid lösemi (AML) Dünya Sağlık Örgütü (WHO) sınıflandırmasına göre %30'dan fazla kemik iliği blastıolan akut miyeloid lösemi (AML) tedavisinde endikedir.  4.2 Pozoloji ve uygulama şekliAZAVIX tedavisi kemoterapötik ajanlar konusunda tecrübeli bir hekim tarafından başlanmalı ve izlenmelidir. Hastalara, tedavi öncesinde bulantı ve kusmaya karşı anti-emetik premedikasyonuuygulanmalıdır. Pozoloji/uygulama sıklığı ve süresi:İlk tedavi siklusunda tavsiye edilen başlangıç dozu tedavi öncesi hematoloji laboratuvar değerlerinden bağımsız olarak, tüm hastalar için vücut yüzey alanına göre 75 mg/m2 dozundaolmalı, subkutan olarak 7 gün boyunca yapılan enjeksiyonları takiben 21 günlük bir araverilmelidir (28 gün süren tedavi siklusu). Hastaların en az 6 siklus tedavi alması önerilir. Hasta tedaviden fayda gördüğü sürece ya da hastalıkta ilerleme görülünceye kadar tedavi devam ettirilmelidir. Hastalar hematolojik yanıt/toksisite ve renal toksisite açısından izlenmelidir (bkz. Bölüm 4.4); bir sonraki siklusa başlarken erteleme ya da aşağıda belirtildiği şekilde doz azaltımı gerekebilir. AZAVIX ile oral azasitidin birbirinin yerine kullanılmamalıdır. Maruz kalmadaki farklılıklar nedeniyle, oral azasitidin için doz ve zamanlama önerileri enjekte edilebilir azasitidin için olanlardanfarklıdır. Sağlık profesyonellerinin tıbbi ürünün adını, dozu ve uygulama yolunu doğrulamalarıönerilir. Laboratuvar Testleri: Tedaviye başlamadan ve her tedavi siklusu öncesinde; karaciğer fonksiyon testleri, serum kreatinin ve serum bikarbonat seviyeleri ölçülmelidir. Tedaviye başlamadan ve en az her tedavisiklusundan önce, cevap ve toksisiteyi izlemek gerekli olduğu için tam kan sayımları yapılmalıdır. Hematolojik Toksisite Nedeniyle Doz Ayarlaması:Hematolojik toksisiteye bağlı olarak doz ayarlamasında; Hematolojik toksisite, trombosit sayısı <50 x 109/ L ve/veya mutlak nötrofil sayısı (MNS) <1 x 109/L ise siklus içerisinde ulaşılan"en düşük değer (en düşük) olarak tanımlanır.
İyileşme ise hematolojik toksisite gözlenen hücre serilerinde başlangıç değerleri ile en düşük değer arasındaki farkın en az yarısı kadar bir artışın olma hali olarak tanımlanır (iyileşme> Endüşük sayım + (0,5 x [Başlangıç sayım- En düşük sayım]). Tedavi öncesi başlangıca göre kan sayımı değerleri düşmemiş hastalarda (örneğin beyaz kan hücresi - BKH - > 3 x 109/L ve MNS > 1,5 x 10 9/L ve trombosit > 75 x 10 9/L)AZAVIX tedavisinebağlı olarak hematolojik toksisite ortaya çıkarsa, bir sonraki tedavi siklusu trombosit sayısı ve MNSdeğerleri düzelene kadar ertelenmelidir. 14 Gün içerisinde değerlerde iyileşme sağlanırsa herhangibir doz değişikliğine gerek yoktur. Ancak 14 gün içerisinde iyileşme sağlanamazsa bu durumdaaşağıdaki tabloya göre doz azaltılması yapılmalıdır. Doz ayarlamalarını takiben siklus 28 günedöndürülmelidir.
14 gün içerisinde değerlerde düzelme olmazsa bir sonraki siklusda verilebilecek doz miktarı (%)
% 50
% 100 * İyileşme = Sayım > En düşük sayım + (0,5 x [Başlangıç sayım - En düşük sayım]) Tedavi öncesi başlangıca göre kan sayımı değerleri düşmüş hastalarda (örneğin beyaz kan hücresiBKH -3 x 10 9/L veya MNS1,5 x 10 9/L veya trombosit75 x 10 9/L)/L)AZAVIX tedavisinitakiben BKH ya da MNS ya da trombosit sayısında, uygulama öncesine göre < %50 bir azalma yada %50'den fazla olmasına rağmen herhangi bir hücre seri farklılaşmasında iyileşme görülmesidurumunda doz ayarlamasına ya da tedavinin ertelenmesine gerek yoktur.Eğer BKH ya da MNS ya da trombosit sayısındaki azalma uygulama öncesine göre %50'den fazla ise ve herhangi bir hücre seri farklılaşmasında iyileşme görülmemesi durumunda AZAVIXtedavisinin bir sonraki siklusu, trombosit sayısı ve MNS düzelene kadar ertelenmelidir. 14 Güniçerisinde iyileşme sağlanırsa herhangi bir doz ayarlamasına gerek yoktur. Ancak 14 gün süresindebir düzelme gözlenmemesi durumunda kemik iliği hücresel yapısı değerlendirilmelidir. Eğer kemikiliği hücre düzeyi > %50 ise doz değişikliğine gerek yoktur. Kemik iliği hücre düzeyi < %50 isetedavi ertelenmeli ve doz aşağıdaki tabloya göre azaltılmalıdır: 
* İyileşme = Sayım > en düşük sayım + (0,5 x [Başlangıç sayım - En düşük sayım]) Doz ayarlamalarını takiben siklus 28 güne döndürülmelidir. Uygulama şekli:Sulandırılmış AZAVIX; üst kol bölgesi, uyluk ya da karına subkutan olarak enjekte edilmelidir. Enjeksiyon yerleri dönüşümlü olarak değiştirilmelidir. Yeni enjeksiyonlar bir öncekinden en az 2,5cm uzağa yapılmalı ve kesinlikle hassasiyet, çürük, kızarıklık ya da sertleşme olan bölgelereuygulanmamalıdır. Sulandırıldıktan sonra süspansiyon filtre edilmemelidir. AZAVIX içinsulandırma ve uygulama prosedürü için detaylı talimatlar Bölüm 6.6.da verilmiştir. Özel popülasyonlara ilişkin ek bilgiler:Böbrek Yetmezliği:Azasitidin, başlangıç doz ayarlaması olmaksızın böbrek yetmezliği olan hastalara uygulanabilir (bkz. Bölüm 5.2). Eğer serum bikarbonat düzeyinde nedeni açıklanamayan bir şekilde 20 mmol/L'nin altında azalma ortaya çıkarsa, bir sonraki siklusta doz %50 azaltılmalıdır. Eğer serum kreatininya da kan üre azot (BUN) değerleri açıklanamayan bir şekilde başlangıç değerlerinin > 2 kat üzerineve normal değerin en üst sınırı (ULN)'na çıkarsa, değerler normale ya da başlangıç düzeylerinedönene kadar bir sonraki siklus ertelenmeli ve takip eden tedavi siklusunda doz %50 azaltılmalıdır(bkz. Bölüm 4.4). Karaciğer Yetmezliği:Karaciğer yetmezliği olan hastalarda yapılmış çalışma bulunmamaktadır (bkz. Bölüm 4.4). Ciddi karaciğer yetmezliği bulunan hastalar advers olaylar için dikkatlice izlenmelidir. Tedaviyebaşlamadan önce karaciğer yetmezliği olan hastalarda başlangıç dozu için spesifik bir dozdeğişikliği önerilmemektedir; takip eden doz değişiklikleri hematolojik laboratuvar değerleriüzerinden yapılmalıdır. İleri evre malign hepatik tümorü olan hastalarda AZAVIX kontrendikedir(bkz. Bölüm 4.3 ve 4.4).
Pediyatrik popülasyon:Yeterli güvenlilik ve etkililik verisi bulunmadığından 18 yaş altındaki çocuklar ve adölesanlarda AZAVIX kullanımı önerilmemektedir. Halihazırda mevcut veriler Bölüm 4.8, 5.1 ve 5.2'deaçıklanmaktadır, ancak pozoloji konusunda herhangi bir tavsiyede bulunulmamaktadır. Geriyatrik popülasyon:Yaşlı hastalar için spesifik bir doz ayarlaması önerilmemektedir. Yaşlılarda böbrek fonksiyonları zaten azaldığı için böbrek fonksiyonlarının izlenmesi yararlı olabilir. 4.3 Kontrendikasyonlar- Azasitidine veya Bölüm 6.1'de listelenen herhangi bir bileşenine aşırı duyarlılığı olan hastalarda, - İlerlemiş malign karaciğer tümörü olan hastalarda (bkz. Bölüm 4.4), - Laktasyonda (bkz. Bölüm 4.6). 4.4 Özel kullanım uyarıları ve önlemleriHematolojik toksisiteAzasitidin ile tedavi esnasında, özellikle ilk 2 siklus sırasında (bkz. Bölüm 4.8), anemi, nötropeni ve trombositopeni sık gözlenmektedir. Cevap ve toksisiteyi izlemek gerekli olduğu için, en az hertedavi siklusundan önce tam kan sayımları yapılmalıdır. İlk siklus için önerilen dozunuygulanmasından sonra, en düşük sayımlara ve hematolojik cevaba dayanarak (bkz. Bölüm 4.2),daha sonraki sikluslar için doz azaltılabilir veya uygulama geciktirilebilir. Hastalara derhal febril ataklarını bildirmeleri tavsiye edilmelidir. Ayrıca hastalara ve doktorlara kanama belirtileri ve semptomları için dikkatli olmaları tavsiye edilir. Karaciğer yetmezliğiKaraciğer yetmezliği olan hastalarda herhangi bir çalışma yapılmamıştır. Metastatik hastalığa bağlı olarak büyük tümör yükü olan, özellikle albumin alt sınır değeri <30 g/L olan hastalarda, azasitidintedavisi sırasında ilerleyen karaciğer koması ve ölüm seyrek olarak rapor edilmiştir. Azasitidin,ilerlemiş malign karaciğer tümörleri olan hastalarda kontrendikedir (bkz. Bölüm 4.3). Böbrek yetmezliğiKemoterapötik ajanlarla birlikte i.v. azasitidin ile tedavi edilen hastalarda serum kreatinin düzeyi 
olarak, alkali idrar ve hipokalemi (serum potasyumu < 3mmol/L) ile birlikte serum bikarbonatlarının <20 mmol/L'ye düşmesi olarak tanımlanan renal tübüler asidoz, azasitidin ve etoposid ile tedaviedilen 5 kronik miyeloid lösemi (KML) hastasında gelişmiştir. Serum kreatinin veya BUNseviyelerinde açıklanamayan artışlar veya serum bikarbonatta azalmalar (<20 mmol/L) oluşur ise,dozaj azaltılmalı veya uygulama geciktirilmelidir (bkz. Bölüm 4.2). Hastalar, oligüri ve anüri durumunda derhal doktorlarını bilgilendirmeleri konusunda uyarılmalıdırlar. Böbrek fonksiyonu normal olan hastalar ile böbrek yetmezliği olan hastalar arasında advers etkilerin sıklığı açısından klinik bir farklılık olmamasına rağmen, azasitidin ve/veya metabolitleri esasolarak böbrekten atıldığı için böbrek yetmezliği olan hastalar yakından izlenmelidir (bkz. Bölüm4.2). Laboratuvar Testleri:Tedaviye başlamadan ve her tedavi siklusundan önce karaciğer fonksiyon testleri, serum kreatinin ve serum bikarbonat düzeyleri belirlenmelidir. Tedaviye başlamadan ve en az her tedavi siklusundan önce, cevap ve toksisiteyi izlemek gerekli olduğu için tam kan sayımları yapılmalıdır (bkz. Bölüm 4.8). Kalp ve akciğer hastalığıCiddi konjestif kalp yetmezliği, klinik olarak stabil olmayan kalp hastalığı veya akciğer hastalığı olan hastalar klinik çalışmalara (AZA PH GL 2003 CL 001 ve AZA-AML-001)alınmamıştır ve bu yüzden azasitidinin bu hastalarda güvenliliği ve etkililiği saptanamamıştır.Bilinen bir kalp veya akciğer hasta1ığı geçmişi olan hastalarda yapılan bir klinik çalışmadanalınan yeni veriler, azasitidin ile kardiyak olayların insidansında önemli bir artış olduğunugöstermiştir (bkz. Bölüm 4.8). Bu nedenle, bu hasta grubunda azasitidin kullanırken dikkatliolunması önerilir. Azasitidin ile tedavi öncesinde ve tedavi sırasında kardiyopulmonerdeğerlendirme yapılması düşünülmelidir.
Nekrotizan fasitAzasitidin ile tedavi edilen hastalarda, ölümcül vakalar da dahil olmak üzere nekrotizan fasiit rapor edilmiştir. Nekrotizan fasiit gelişen hastalarda, azasitidin tedavisi hemen durdurulmalı ve acilenuygun bir tedaviye başlanmalıdır. Tümör lizis sendromuTedavi öncesinde yüksek tömör yükü olan hastalar tümör lizis sendromu açısından risk altındadır. Bu hastalar yakın takip edilmeli ve uygun önlemler alınmalıdır. Bu tıbbi ürün her dozunda 1 mmol (23 mg)'dan daha az sodyum ihtiva eder; yani aslında sodyum içermez. Diferansiyasyon sendromuEnjekte edilebilir azasitidin alan hastalarda diferansiyasyon sendromu (retinoik asit sendromu olarak da bilinir) vakaları bildirilmiştir. Diferansiyasyon sendromu ölümcül olabilir ve semptomlarve klinik bulgular solunum sıkıntısı, pulmoner infiltratlar, ateş, döküntü, pulmoner ödem, periferiködem, hızlı kilo alımı, plevral efüzyonlar, perikardiyal efüzyonlar, hipotansiyon ve böbrekyetmezliğini içerir (bkz. Bölüm 4.8). Yüksek doz IV kortikosteroidlerle tedavi ve hemodinamikizleme, diferansiyasyon sendromunu düşündüren semptom veya bulguların ilk başlangıcındadüşünülmelidir. Semptomlar düzelene kadar enjekte edilebilir azasitidinin geçici olarak kesilmesidüşünülmeli ve devam edilirse dikkatli olunması önerilmektedir. 4.5. Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleriIn vitroin vivoetkileşim olasılığının olmadığı düşünülmektedir.Azasitidinin sitokrom P450 enzimleri üzerinde klinik olarak önemli inhibitör veya indükleyici etkisi olası değildir (bkz Bölüm 5.2). Azasitidin ile klinik ilaç etkileşme çalışmaları yapılmamıştır. Özel popülasyonlara ilişkin ek bilgilerHiçbir etkileşim çalışması yapılmamıştır.  Pediyatrik popülasyonHiçbir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyon Genel tavsiyeGebelik Kategorisi: D Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli olan kadınlar tedavi sırasında ve tedaviden sonra en az 6 ay boyunca etkili kontrasepsiyon yöntemi kullanmalıdırlar. Erkeklere tedavi sırasında çocuk sahibi olmamaları,tedavi sırasında ve tedaviden sonra en az 3 ay boyunca etkili doğum kontrol yöntemleri kullanmalarıtavsiye edilmelidir. Gebelik dönemiAzasitidinin, gebe kadınlarda kullanımına ilişkin yeterli veri yoktur. Fareler üzerinde yapılan çalışmalar üreme toksisitesi olduğunu göstermiştir (bkz. Bölüm 5.3). İnsanlar için potansiyelriski bilinmemektedir. Azasitidin, hayvan çalışmalarından elde edilen sonuçlara vemekanizmasına dayanarak gebelik sırasında, özellikle ilk trimesterde, kesinlikle gerekliolmadıkça kullanılmamalıdır. Tedavinin anne için avantajları fetus için olası risklerine karşı hervaka için tartışılarak karar verilmelidir. Laktasyon dönemiAzasitidin/ metabolitlerinin anne sütüne geçip geçmediği bilinmemektedir. Emzirilen bebekte ciddi advers reaksiyon potansiyeli nedeniyle, azasitidin tedavisi sırasında emzirme kontrendikedir. Üreme yeteneği/Fertiliteİnsanlarda azasitidinin fertilite üzerindeki etkisine dair herhangi bir veri yoktur. Hayvanlarda azasitidin kullanımının erkek fertilitesi üzerinde advers reaksiyonları görülmüştür (bkz. Bölüm 5.3).Tedaviye başlamadan önce erkek hastalara spermlerini saklamak üzere danışman aramalarıtavsiye edilmelidir.
4.7. Araç ve makine kullanımı üzerindeki etkilerAzasitidinin araç ve makina kullanımına hafif ve orta derecede etkisi vardır. Azasitidin kullanımı ile yorgunluk rapor edilmiştir. Bu nedenle, araç veya makine kullanırken dikkatli olunmasıönerilmelidir. 4.8. İstenmeyen etkilerGüvenlilik profili özetiMDS, KM^L ve %20-30 kemik iliği blastlı AML'si olan yetişkinlerde:Hastaların %97'sinde AZAVIX uygulaması ile ilişkili advers reaksiyonlar oluşmuştur. Azasitidin endikasyon çalışmasında (AZA PH GL 2003 CL 001) en sık görülen ciddi advers reaksiyonlar febril nötropeni (%8) ve anemi (%2,3) olup bu çalışmayı destekleyen çalışmalarda da(CALGB 9221 ve CALGB 8921) benzer ciddi advers reaksiyonlar raporlanmıştır. Daha az sıklıktabildirilen diğer ciddi advers reaksiyonlar nötropenik sepsis (%0,8) ve bazen ölümcül sonuçlarıolabilen pnömoni (%2,5) gibi enfeksiyonları, trombositopeni (%3,5), aşırı duyarlılık reaksiyonları(% 0,25) ve kanama olaylarını [örneğin serebral kanama (%0,5), gastrointestinal kanama (%0,8) veintrakraniyal kanama (%0,5)] içermektedir. Azasitidin tedavisi ile çok yaygın görülen advers reaksiyonlar trombositopeni, nötropeni ve lökopeniyi (genellikle Derece 3-4) içeren hematolojik reaksiyonlar (% 71,4), bulantı, kusmayı(genellikle Derece 1-2) içeren gastrointestinal olaylar (%60,6) veya enjeksiyon bölgesireaksiyonlarıdır (%77,1; genellikle Derece 1-2). % 30'dan fazla kemik iliği blastı olan 65 yaş ve üstü hastalarda:Azasitidin tedavi kolunda AZA-AML-001 çalışması ile belirlenmiş olan çok yaygın ciddi advers reaksiyonlar (> % 10) arasında febril nötropeni (%25), pnömoni (%20,3) ve pireksi (%10,6)bulunmaktadır. Ayrıca daha az sıklıkla raporlanmış olan ciddi advers reaksiyonlar arasında sepsis,1), anemi (%4,2), nötropenik sepsis (% 3), idrar yolu enfeksiyonu (% 3), trombositopeni,5), nötropeni (%2,1), selülit (%2,1), baş dönmesi (% 2,1) ve dispne (%2,1) bulunmaktadır. Azasitidin tedavisi ile en sık raporlanan advers reaksiyonlar (çalışmadaki hastaların %30'unda görülen), kabızlık (%41,9), mide bulantısı (%39,8) ve ishali de içeren sindirim sistemi olayları(%36,9; genellikle Derece 1-2), pireksiyi de içeren genel bozukluklar ve uygulama bölgesine ilişkindurumlar (%37,7; genellikle Derece 1-2) ve febril nötropeni (%32,2) ve nötropeniyi (%30,1; Belge Dcgen®Hi^lie : DS¥ece 8^4)'de3içerenahiefflaiDİojiık olaylardırçaklp Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys
Advers reaksiyonların tablolaştırılmış listesi Aşağıdaki tablo azasitidin tedavisi ile ilişkili olabilecek advers reaksiyonları içermektedir. Sıklıklar, MDS ve AML üzerine yapılmış temel klinik çalışmalara ve pazarlama sonrası gözlemleredayanmaktadır. Sıklıklar şu şekilde tanımlanmıştır: çok yaygın (>1/10), yaygın (>1/100- < 1/10), yaygın olmayan (>1/1.000- <1/100), seyrek (>1/10.000- <1/1.000), çok seyrek (<1 /10.000), bilinmiyor (eldekiverilerden hareketle tahmin edilemiyor). Her bir sıklık grubu içinde advers reaksiyonlar azalanciddiyet sırasına göre verilmiştir. Advers reaksiyonlar, temel klinik çalışmaların herhangi birindegözlemlenen en yüksek sıklığa göre aşağıdaki tabloda sunulmaktadır. Tablo 1: Azasitidin ile tedavi edilen MDS veya AML hastalarında bildirilen advers reaksiyonlar (klinik çalışmalar ve pazarlama-sonrası deneyim)Sistem Organ SınıfıÇok yaygınYaygınYaygınolmayanSeyrekBilinmiyorNekrotizan fasiit* Sepsis* (Bakteriyel viral ve fungal dahil)Nötropeniksepsis* Solunum yolları enfeksiyonu (üstsolunum yollarıve bronşit dahil)İdrar yoluenfeksiyonlarıSelülitDivertiküliltihabıOral fungalenfeksiyon Pnömoni* (bakteriyel,viral vefungal dahil)Nazofarenjit EnfeksiyonlarveenfestasyonlarSinüzit Farenjit Rinit Herpes simplex Deri enfeksiyonu SeyrekkV.turkiye.gov.tr/saglilBilinmiyor-titck-ebysÇok yaygıngüvenli elYaygı^ a ile im:RSHY3S3k0SHY3akllfM0FyYnUy BelgealanmYaygınrakifolmayanSistem OrganBelge DoSl^nİf^W56Q3^

 Seçilen Advers reaksiyonların sıralanmasıHematolojik advers reaksiyonlarAzasitidin tedavisi ile ilişkili olarak çok yaygın rapor edilen (>%10) hematolojik advers reaksiyonlar, genellikle 3. veya 4. dereceden anemi, trombositopeni, nötropeni, febril nötropeni velökopenidir. Bu olayların olma riski daha çok ilk 2 siklus sırasındadır, daha sonra hematolojik fonksiyonun normale döndüğü hastalarda daha az sıklıkta oluşur. Çoğu hematolojik advers reaksiyonlar, tam kansayımlarının rutin olarak izlenmesi ve bir sonraki siklusta azasitidin uygulamasının geciktirilmesinötropeni için profilaktik antibiyotikler ve/veya büyüme faktorü desteği (ömeğin G-CSF) ve anemiveya trombositopeni için transfüzyonlar ile gerektiği gibi tedavi edilmektedir. Enfeksiyonlar Miyelosupresyon nötropeniye ve enfeksiyon riskinin artmasına neden olabilir. Azasitidin alan hastalarda nötropenik sepsisi de içeren sepsis ve pnömoni gibi ve bazıları ölümcül sonuçlara nedenolan ciddi advers reaksiyonlar rapor edilmiştir. Enfeksiyonlar, nötropeni için anti-enfektif ajanlarve büyüme faktör desteği (ömegin G-CSF) kullanımı ile kontrol altına alınabilir. Kanama Azasitidin alan hastalarda kanama görülebilir. Gastrointestinal kanama ve intrakraniyal kanama gibi ciddi advers reaksiyonlar rapor edilmiştir. Özellikle daha önceden trombositopenisi olan veyatedaviye bağlı trombositopenisi gelişen hastalar, kanama belirtileri ve semptomları için izlenmelidir. Aşırı duyarlılık Azasitidin alan hastalarda ciddi aşırı duyarlılık reaksiyonları rapor edilmiştir. Anafilaktik benzeri reaksiyon durumunda azasitidin tedavisi derhal kesilmelidir ve uygun semptomatik tedavibaşlatılmalıdır. Deri ve deri altı doku hastalıkları Deri ve deri altı advers reaksiyonlarının çoğunluğu enjeksiyon bölgesi ile ilgilidir. Bu advers reaksiyonların hiçbiri azasitidinin kesilmesine veya ana çalışmalarda azasitidin dozununazaltılmasına neden olmamıştır. Advers reaksiyonlarının çoğunluğu tedavinin ilk 2 siklusu sırasındaolmuştur ve sonraki sikluslar ile azalmaya yönelmiştir. Enjeksiyon bölgesinde Belge Dadıökünlö/ertflamasy©n/pruri4usY:döftüfffüYeri'tem veBderklezyonuhgibi'subfcutantadıvefskreaksiyonlar,
antihistaminikler, kortikosteroidler ve non-steroidal anti-enflamatuarlar (NSAII'ler) gibi ilaçların birlikte kullanımım gerektirebilir. Bu kutanöz reaksiyonlar, bazen enjeksiyon bölgesinde oluşanyumuşak doku enfeksiyonlarından ayırt edilmelidirler. Pazarlama sonrasındaki gözlemlerde;azasitidin ile birlikte nadir vakalarda, ölüme yol açan selülit ve nekrotizan fasiit gibi yumuşak dokuenfeksiyonları rapor edilmiştir. Enfeksiyöz advers reaksiyonların klinik yöntemi için Bölüm 4.8Enfeksiyonlar bölümüne bakınız. Gastrointestinal advers reaksiyonlar Azasitidin tedavisi ile çok yaygın rapor edilen advers reaksiyonlar kabızlık, ishal, bulantı ve kusmadır. Bu advers reaksiyonlar, bulantı ve kusma için anti-emetikler; ishal için anti- diyaretiklerve kabızlık için laksatif ve/veya feçes yumuşatıcıları ile semptomatik olarak tedavi edilmelidirler. Renal advers reaksiyonlar Azasitidin ile tedavi edilen hastalarda, serum kreatinin değerlerinde artış ve hematüriden renal tübüler asidoz, renal yetmezlik ve ölüme kadar giden derecelerde böbrek bozuklukları raporedilmiştir (bkz. Bölüm 4.4). Hepatik advers reaksiyonlar Azasitidin tedavisi sırasında, metastatik hastalığa bağlı olarak tümör yükü çok olan hastalarda hepatik yetmezlik, ilerleyen hepatik koma ve ölüm gözlenmiştir (bkz. Bölüm 4.4). Kardiyak olaylar Kardiyovasküler veya pulmoner hastalık geçmişi olduğu bilinen hastaların dahil edildiği bir klinik çalışmadan alınan veriler, azasitidin ile tedavi edilen yeni AML teşhisi konmuş hastalarda kardiyakolaylarda bir artış olduğunu göstermiştir (bkz. Bölüm 4.4). Yaşlı hastalar 85 yaş ve üstü hastalarda azasitidinin güvenliliği ile ilgili sınırlı bilgi bulunmaktadır (AZA-AML-001 çalışmasında tedavi edilen 85 yaş ve üstü hastalarda 14 hasta [%5,9] bulunmaktadır.) Pediyatrik yoyülasyonAZA-JMML-001 çalışmasında, 28 pediyatrik hasta (1 aydan 18 yaşına kadar) MDS (n=10) veya juvenil miyelomonositik lösemi (JMML) (n=18) için azasitidin ile tedavi edilmiştir (bkz. Bölüm
28 hastanın tümü en az 1 advers olay yaşadı ve 17'si (%60,7) en az 1 tedaviyle ilişkili olay yaşadı. Genel pediyatrik popülasyonda en sık bildirilen advers olaylar ateş, anemi, trombositopeni ve febrilnötropeni dahil hematolojik olaylar ile kabızlık ve kusma dahil gastrointestinal olaylardır. Klinik çalışmadaki üç (3) hasta, ilacın kesilmesine neden olan tedavi ilişkili olay yaşadı (ateş, hastalığın ilerlemesi ve karın ağrısı). AZA-AML-004 çalışmasında, moleküler relapslı 7 pediyatrik hasta (2-12 yaş arası), ilk tam remisyondan [CR1] azasitidin ile tedavi edilmiştir (bkz. Bölüm 5.1). 7 hastanın tümü, tedaviye bağlı en az 1 advers olay yaşamıştır. En sık bildirilen yan etkiler nötropeni, bulantı, lökopeni, trombositopeni, diyare ve alanin aminotransferaz (ALT) artışıdır. İkihasta, dozun kesilmesine yol açan (ateşli nötropeni, nötropeni) tedaviyle ilişkili bir olay yaşamıştır.Klinik çalışma sırasında azasitidin ile tedavi edilen sınırlı sayıda pediyatrik hastada yeni güvenliliksinyali tespit edilmemiştir. Genel güvenlik profili, yetişkin popülasyonun güvenlilik profiliyletutarlıdır. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesine olanak sağlar. Sağlıkmesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi(TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr[email protected]:4.9. Doz aşımı ve tedavisiKlinik çalışmalar sırasında azasitidin ile doz aşımı bir vakada rapor edilmiştir. Hasta, önerilen başlangıç dozunun neredeyse 4 katı olan, yaklaşık 290 mg/m2 tek bir i.v. dozu aldıktan sonra,hastada ishal, bulantı ve kusma görülmüştür. Doz aşımı durumunda, hasta uygun kan sayımları yapılarak izlenmeli ve gerekli olduğu şekilde destekleyici tedavi almalıdır. Azasitidinin doz aşımı bilinen spesifik bir antidot yoktur.
5. FARMAKOLOJIK ÖZELLIKLERI5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antineoplastik maddeler. Pirimidin analogları. ATC kodu: L01BC07 Etki mekanizması: Azasitidinin antineoplastik etkilerini, kemik iliğindeki anormal hematopoietik hücreler üzerinde sitotoksisite ve DNA'nın hipometilasyonu da dahil olmak üzere çoklu mekanizmalar ilegösterdiğine inanılmaktadır. Azasitidinin sitotoksik etkileri şu mekanizmalardan kaynaklanıyorolabilir: DNA, RNA ve protein sentezinin inhibisyonu, RNA ve DNA'yla birleşme ve DNA yıkımyolaklarının aktivasyonu. Non-proliferatif hücreler azasitidine göreceli olarak dirençlidir.Azasitidinin DNA'ya katılımı DNA metiltransferazlarının inaktivasyonu ve DNA'nınhipometilasyonu ile sonuçlanır. Normal hücre siklusu kontrolü, diferansiyasyonu ve ölümyolaklarında görev alan anormal derecede metillenmiş genlerin DNA hipometilasyonu, genlerinyeniden ekspresyonu ve kanser-baskılayıcı fonksiyonların tamiri ile sonuçlanabilir. DNAhipometilasyonu ile azasitidinin sitotoksik veya diğer aktivitelerinin klinik sonuçlar üzerindekigöreceli önemleri henüz bilinmemektedir. Klinik etkililik ve güvenlilik:MDS, KM^L ve kemik iliğinde %20-30 blast olan AML tamlı yetişkinlerdeAzasitidinnin etkililiği ve güvenliliği uluslararası, çok merkezli, kontrollü, açık-uçlu, randomize,paralel gruplu, Faz 3 karşılaştırmalı araştırmada (AZA PH GL 2003 CL 001) incelenmiştir.Araştırmaya Uluslararası Prognostik Skorlama Sistemine (UPSS) göre intermediate-2 ile yüksekriskli MDS ve Fransız Amerikan İngiliz (FAB) sınıflandırma sistemine göre ise RAEB, RAEB-T(%21-30 blast) ile mKMML olan MDS hastaları dahil edilmiş, sekonder MDS'si olan hastalararaştırmaya dahil edilmemiştir. Azasitidin (n=179) konvansiyonel tedavi rejimleri (n=l79) ilekarşılaştırılmıştır. Konvansiyonel tedavi rejimleri, tek başına destek tedavi (n=105), düşük dozsitarabin ve beraberinde destek tedavi (n=49) veya standart indüksiyon kemoterapi ile destektedaviden (n=25) oluşmuştur. Hastalar randomizasyondan önce doktorları tarafından 3konvansiyonel tedavi rejiminden bir tanesine seçilmişlerdir. Hasta Azasitidin grubuna randomizeolmamışsa, bu önceden seçilen rejimi almıştır. Hastanın araştırmaya dahil edilmesi için gerekenkriterlerden bir tanesi de Eastern Cooperative Oncology Group (ECOG) performansının 0-2arasında olmasıdır. Sekonder MDS'si olan hastalar araştırmaya dahil edilmemiştir. AraştırmanınBelge Dcpıpiim@ricsort^im3Mo(klisi3itoplam ısağ rfeâlfm süresidicTaAzaföitidi^s :medyaöy9ostklusk(4<-89ysiklus
aralığında) ve ortalama 10,2 siklus olacak şekilde 7 gün boyunca günlük 75 mg/m2 subkutan dozda uygulanmış ve 21 gün ara verilmiştir (28 günden oluşan tedavi siklusu). Tedavi AmaçlıPopülasyonda (ITT) yaş ortalaması 69'dur (38-88 yaş arası). 358 hasta (179 azasitidin ve 179 konvansiyonel tedavi rejimleri üzerinde yapılan ITT analizinde, azasitidin ile medyan 24,46 aylık bir sağ kalıma karşı, konvansiyonel tedavi rejimi tedavisinde 15,02aylık sağ kalım olduğu saptanmıştır. Aradaki fark 9,4 aydır. (p<0,0001). Azasitidin kullananhastalarda iki yıllık sağ kalım oranı %50,8 iken, konvansiyonel tedavi rejimi hastalarında % 26,2'dir (p<0,0001) 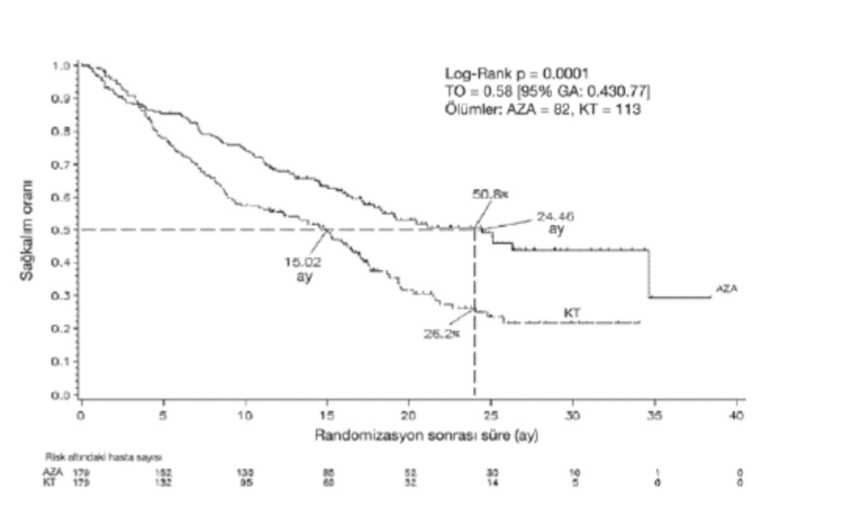 ANAHTAR: AZA= azasitidin; KT= konvansiyonel tedavi; GA= güvenlilik aralığı; TO= tehlike oranı Azasitidinin sağkalım faydaları, kontrol kolunda kullanılan konvansiyonel tedavi rejimi seçeneğinden (tek başına en iyi destek tedavi, düşük doz sitarabin ve beraberinde en iyi destek tedaviveya standart indüksiyon kemoterapisi ve beraberinde en iyi destek tedavi) bağımsız olaraktutarlıdır. itge, güvenB'felektronik imza ile imzalanmıştır.UPSS (Uluslararası Prognostik Skorlama Sistemi) sitogenetik alt grup analiz edildiğinde, tüm gruplarda (iyi, orta, kötü sitogenetikli, monozomi 7 dahil) medyan genel sağ kalım açısından benzersonuçlar. Yaş alt grupları analiz edildiğinde, tüm gruplarda medyan genel sağ kalımda bir artışgözlendi (<65 yaş, >65 yaş  Azasitidin grubunda ölüm veya AML'ye dönüşüm için geçen medyan süre 13 ay iken; bu süre konvansiyonel rejim tedavisi alan grupta 7,6 aydır. Azasitidin 5,4 aylık avantaj sağlamış olup, p-değeri 0,0025'dir. Ayrıca, azasitidin tedavisi sitopeni ve semptomlarında azalma ile birliktelikgöstermiştir. Azasitidin tedavisi, kırmızı kan hücresi (KKH) ve trombosit transfüzyonlarına olanihtiyacın azalmasına yol açmaktadır. Başlangıçta KKH transfüzyonuna bağımlı olan azasitidingrubundaki hastaların %45'i tedavi süresi boyunca KKH transfüzyonundan bağımsız hale gelirken,kombine CCR gruplarındaki ((%33,6 (%95 GA: 22,4, 44,6) istatistiksel olarak anlamlı (p < 0,0001)fark)) hastaların %11,4'ünde fark %33,6'dır. Başlangıçta KKH transfüzyonuna bağımlı olan vebağımsız hale gelen hastalarda, azasitidin grubunda KKH transfüzyon bağımsız hale gelme medyansüresi 13 aydır. Azasitidin grubunda elde edilen toplam yanıt (tam remisyon [TR] + parsiyel remisyon [PR]) %29 iken konvansiyonel tedavi rejimleri grubunda ise %12'dir (p= 0,0001). Bağımsız İncelemeKomitesi'nin AZA PH GL 2003 CL1 çalışmasında elde ettiği genel yanıt (TR + PR), azasitidingrubunda %7 (12/179) olup bu oran kombine konvansiyonel tedavi gruplarında %1 (2/179)'dur(p=0,0113). Bağımsız İnceleme Komitesi ve araştırmacı değerlendirmeleri yanıtları arasındakifarklar periferik kan sayımlarının iyileştirilmesini ve en az 56 gün bu iyileştirmenin idamesinigerektiren Uluslararası Çalışma Grubu (IWG) kriterlerinin bir sonucudur. Azasitidin tedavisinitakiben TR ve PR elde edilemeyen hastalarda da sağ kalımda avantaj gözlenmiştir. Bağımsızİnceleme Komitesinin yaptığı değerlendirmeye göre azasitidin alan hastaların %49'undahematolojik iyileşme (major veya minör) tespit edilmiş olup bu oran kombine konvansiyonel tedavirejimleri ile tedavi edilen hastalarda %29'dur (p< 0,0001). Başlangıçta bir veya daha fazla sitogenetik anormalliği olan hastalarda, major sitogenetik yanıt görülen hastaların oranı azasitidin ve kombine konvansiyonel tedavi rejimi gruplarında birbirinebenzerdir. Minör sitogenetik yanıt, kombine konvansiyonel tedavi rejimi grubu ilekarşılaştırıldığında (%10), azasitidin grubunda (%34) istatistiksel olarak anlamlı düzeyde dahayüksektir (P= 0,0015). % 30'dan fazla kemik iliği blastı olan 65 yaş ve üstü akut miyeloid lösemi (AML) hastalarıAZA-AML-001 klinik araştırmasında yer alan tedavi amaçlı hasta popülasyonuna ait sonuçlaraşağıda sunulmuştur (Bkz. 4.1- Terapötik Endikasyonlar).
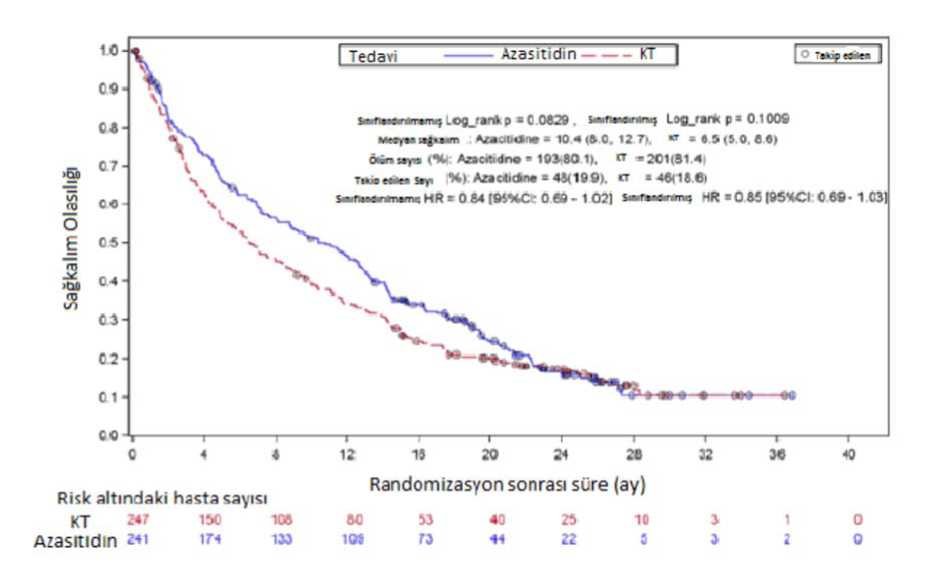
Azasitidinin etkililik ve güvenliliği hematopoietik kök hücre transplantasyonuna uygun olmayan, Dünya Sağlık Örgütü sınıflandırmasına göre 65 yaş ve üstü yeni teşhiş konmuş veya % 30'dan fazlakemik iliği blastlı ikincil AML'si olan hastalarda uluslararası, çok merkezli, kontrollü, açık-uçlu,paralel grup Faz 3 çalışması yapılmıştır. Azasitidin ile birlikte en iyi destek tedavileri (n=241)konvansiyonel tedavi rejimleri ile karşılaştırılmıştır. Konvansiyonel tedavi rejimleri, tek başınadestek tedavileri (n=45), düşük doz sitarabin ve beraberinde destek tedavileri (n=158) veya sitarabinve antrasiklin ile birlikte standart yoğunlaştırılmış kemoterapi ile beraber destek tedaviden (n=44)oluşmaktadır. Randomizasyondan önce konvansiyonel tedavi rejimi alan 3 hastadan 1'i doktorlarıtarafından seçilmişlerdir. Hastalar eğer azasitidin grubuna randomize edilmediyse önceden seçilmiştedavi rejimini almaya devam etmiştir. Çalışmaya alınma kriterleri, hastaların ECOG performansdurumlarının 0 ila 2 arasında olması ve orta dereceli veya düşük riskli sitogenetik anormalliğiolmasıydı. Çalışmanın birincil sonlanım noktası genel sağkalım olarak belirlenmiştir. Azasitidin alanlar için, 21 gün dinlenme periyodunu takiben 7 gün boyunca (28 günlük tedavi siklusu) 75 mg/m2 subkutan medyan 6 siklus (1-28 siklus) olacak şekilde uygulanırken, sadece eniyi destek tedavisi alanlarda medyan 3 siklus (1-20 siklus), düşük doz sitarabin alanlarda medyan 4siklus (1-25 siklus) ve standart yoğunlaştırılmış kemoterapi alanlarda medyan 2 siklus (1-3indüksiyon siklusu artı 1 veya 2 konsolidasyon siklusu) olacak şekilde uygulanmıştır. Bireysel başlangıç parametreleri açısından azasitidin ile konvansiyonel tedavi rejimindeki gruplar karşılaştırılabilirdir. Hastalardaki medyan yaş 75'tir (64 ile 91 yaş aralığı). % 75,2'si beyaz ırktan, %59'u erkek hastalardan oluşmaktadır. Dünya Sağlık Örgütü sınıflandırmasına göre başlangıçtahastaların % 60,7'si tek başına AML, %32,4'ü miylodisplaziye bağlı değişiklikler ile AML, %4,1'iterapiye bağlı miyeloid neoplazma ve % 2,9'u tekrar eden genetik anormallikleri ile birlikte AMLolarak kategorize edilmiştir. 488 hastanın ITT analizinde (241 hasta Azasitidin ve 247 hasta konvansiyonel tedavi rejimi ile tedavi edilmiştir.), azasitidin tedavisi alan hastalar ile konvansiyonel tedavi rejimi alan hastalar damedyan sağkalım oranı sırasıyla 10,4 ay ve 6,5 aydır. Aradaki fark 3,8 aydır (p=0,1009). Tedavietkisinin risk oranı 0,85'tir (%95 Cl=0,69, 1,03). Bir yıllık sağkalım oranları azasitidin alanhastalarda % 46,5, konvansiyonel tedavi rejimi alan hastalarda % 34,3'tür.
Önceden tanımlanmış başlangıçtaki prognostik faktörler için Cox PH modele uyarlanarak Azasitidin'in konvansiyonel tedavi rejimleri karşılaştırması için risk oranı 0,8 (%95 Cl=0,66; 0,99;p0,0355) olarak belirlenmiştir. Buna ek olarak, azasitidin ile önceden seçilmiş konvansiyonel tedavi rejimi alan hastalar karşılaştırıldığında çalışma istatistiksel olarak belirli bir fark göstermemesine rağmen, azasitidinkullanan hastaların sağkalım oranı konvansiyonel tedavi rejimi seçeneklerinden destek tedavisi vedüşük doz sitarabin artı destek tedavisi alan hastalardan daha uzundur. Yoğun kemoterapi ile destektedavisi alan hastalar ile karşılaştırıldığında ise sağkalım oranı benzerlik göstermektedir. Azasitidinin lehine toplam sağkalım yararı yönünden, bütün önceden seçilmiş alt gruplarda yaş [75 yaş altı ve 75 yaş ve üstü], cinsiyet, ırk, ECOG performans durumu [0 veya 1 ve 2], temel sitogenetikrisk (orta veya düşük), coğrafik bölge, AML'nin DSÖ sınıflandırması (miyelodisplaziye bağlıdeğişiklikler ile birlikte AML'yi de içeren), başlangıçtaki lökosit sayısı [ < 5 x109/L ve >5 x109/L],başlangıçtaki kemik iliği blastı [ % 50 ve daha az ve > % 50'den çok], önceki MDS geçmişi] bireğilim bulunmaktadır. Yalnızca çok küçük bir grupta toplam sağkalım risk oranı istatikselanlamlılığa ulaşmıştır. Bu gruplar arasında zayıf sitogenetik riski olan hastalar, miyelodisplaziyebağlı değişiklikler olan AML hastaları, 75 yaş altı hastalar, kadın hastalar ve beyaz ırktan hastalaryer almaktadır.
Hematolojik ve sitogenetik cevaplar araştırmacılar ve IRC tarafından benzer sonuçlar ile değerlendirilmişlerdir. IRC tarafından tam yanıtların oranı (tam remisyon [CR] ve kan sayımıdüzelmesiz tam remisyon [CRi]) azasitidin grubu için % 28,7, ve birleştirilmiş konvansiyonel tedavirejimi için % 25,1 olarak belirlenmiştir (p=0,5384). CR ve Cri'ye ulaşan hastalarda, remisyon içinmedyan süre Azasitidin kullanan hastalarda 10,4 ay (% 95 CI =7,5, 15,2) olup, konvansiyonel tedavirejimi alan hastalarda ise 12,3 aydır (% 95 CI = 9, 17). Azasitidin ile tedavi edilen ve tam yanıtsağlanamayan hastalarda konvansiyonel tedavi rejimlerine göre sağ kalım avantajı gösterilmiştir. Azasitidin tedavisi periferik kan değerlerini iyileştirmiş ve eritrosit ve trombosit transfüzyonu ihtiyacını azaltmıştır. Eğer hasta sırasıyla 56 gün (8 hafta) boyunca veya randomizasyon öncesi birveya daha fazla eritrosit veya trombosit transfüzyonu almışsa, başlangıçta eritrosit veya trombosittransfüzyonuna bağımlı kabul edilmiştir. Eğer hasta sırasıyla tedavi süresi boyunca ve raporlamaperiyodunda ardışık gelen herhangi 56 gün boyunca eritrosit veya trombosit transfüzyonualmıyorsa, eritrosit veya trobosit transfüzyonuna bağımlı olmadığı düşünülmektedir. Başlangıçta eritrosit transfüzyonuna bağımlı olan azasitidin grubundaki hastalardan % 38,5'inin (% 95 Cl=31,1, 46,2) tedavi periyodu süresince eritrosit transfüzyonuna bağımlılığı kalmamıştır.Birleştirilmiş konvansiyonel tedavi rejimi alan hastalarda bu oran %27,6'dır (%95 GA=20,9, 35,1).Başlangıçta eritrosit transfüzyonuna bağımlı olan ve tedavi ile transfüzyona bağımsız hale gelenhastalar için, transfüzyona bağımsız hale gelmek için geçen medyan süre azasitidin grubunda 13,9ay iken konvansiyonel tedavi rejimi alan hastalarda ise bu süreye ulaşılamamıştır. Çalışma başlangıcında trombosit transfüzyonuna bağımlı olan Azasitidin grubundaki hastalardan %40,6'sının (% 95 GA=30,9, 50,8) tedavi periyodu süresince trombosit transfüzyonunabağımlılığı kalmamıştır. Birleştirilmiş konvansiyonel tedavi rejimi alan hastalarda buoran %29,3'tür (% 95 GA=19,7, 40,4). Başlangıçta trombosit transfüzyonuna bağımlı olan ve tedaviile transfüzyona bağımsız hale gelen hastalar için, transfüzyona bağımsız hale gelmek için geçenmedyan süre azasitidin grubunda 10,8 ay iken konvansiyonel tedavi rejimi alan hastalarda ise busüre 19,2 aydır. Sağlığa Bağlı Yaşam Kalitesi (HRQoL), Avrupa Organizasyonu Kanser Araştırma ve Tedavi Çekirdek Yaşam Kalitesi (EORTC QLQ-C30) anketi kullanılarak belirlenmiştir. HRQoL verileritest çalışmasındaki bütün popülasyonun alt kümesi için analiz edilebilir. Analizde bazı sınırlamalar
olmasına rağmen, elde bulunan veriler azasitidin tedavisi sırasında hastaların yaşam kalitesinde anlamlı bir kayıp yaşamadıklarını göstermektedir. Pediyatrik popülasyonAZA-JMML-001 çalışması, yeni tanı almış ileri MDS veya JMML'li pediyatrik hastalarda HSCT'den önce azasitidinin farmakokinetiğini, farmakodinamiğini, güvenliğini ve aktivitesinideğerlendirmek için gerçekleştirilmiş; Faz 2, uluslararası, çok merkezli, açık etiketli bir çalışmadır.Klinik çalışmanın birincil amacı azasitidinin 3. siklus, 28. günde yanıt oranı üzerindeki etkisinideğerlendirmektir. Hastalar (MDS, n = 10; JMML, n = 18, 3 aylık - 15 yaş arası; %71 erkek), minimum 3 siklus ve maksimum 6 siklus boyunca 28 günlük bir siklusun ilk 7 günü boyunca, günlük 75 mg/m2 intravenözazasitidin dozu ile tedavi edilmiştir. MDS koluna hasta alımı, 10 MDS hastasından sonra etkililik gözlenmemesi nedeniyle durdurulmuştur: bu 10 hastada doğrulanmış yanıt kaydedilmemiştir. JMML çalışma kolunda, 18 hasta (13 PTPN11, 3 NRAS, 1 KRAS somatik mutasyonu ve 1 nörofibromatozis tip 1 klinik tanılı [NF 1]) kaydedildi. On altı hasta 3 siklus, 5 hasta 6 siklus tedaviyitamamladı. Toplam 11 JMML hastasında, 3. siklusun 28. gününde klinik yanıt alınmıştır. Bu 11hastanın 9'unda (%50) doğrulanmış bir klinik yanıt gözlenmiştir (3 hastada cCR - doğrulanmış tamyanıt ve 6 hastada cPR - doğrulanmış kısmi yanıt). Azasitidin ile tedavi edilen hasta kohortunda, 7(%43,8) hastada sürekli trombosit yanıtı (sayımlar > 100 x 109/L) gözlenmiş ve HSCT'de 7 (%43,8)hasta transfüzyona ihtiyaç duymuştur. 18 hastadan 17'si HSCT'ye geçmiştir. Çalışma tasarımı nedeniyle (az hasta sayısı ve karışıklığa neden olan çeşitli faktörler), bu klinik çalışmadan HSCT öncesi azasitidinin JMML hastalarında sağkalımı arttırdığı veya arttırmadığısonucu çıkarılamaz. AZA-AML-004 çalışması, ilk tam remisyondan sonraki moleküler relaps gelişen AML tamlı pediyatrik hastalarda ve moleküler relapstaki AML'li çocuklarda ve genç erişkinlerde anti-kansertedavisine kıyasla azasitidinin güvenliğini, farmakodinamiğini ve etkinliğini değerlendirmek içinbir Faz 2, çok merkezli, açık etiketli bir çalışmadır.
Azasitidin, 7 hastada (yaşları 2 ila 12 arasında, ortanca 6, 7 yıl ve %71,4'ü erkek olan), her 28 günlük siklusun, ilk 7 gününde 100 mg/m2 olacak şekilde en fazla 3 siklus kullanılmıştır. 84. günde, 5 hastada minimal rezidüel hastalık (MRH) değerlendirmesi yapıldı ve 4 hastada ya (n=3) moleküler stabilizasyon ya da (n = 1) moleküler iyileşme tespit edildi ve bir hastada ise klinik nüksgörüldü. Azasitidin ile tedavi edilen 7 hastanın altısına (%90 [%95 GA = 0,4; 1]) HKHN uygulandı.Bu küçük örneklem büyüklüğü nedeniyle, azasitidinin pediyatrik AML'deki etkililiği belirlenemez.Güvenlilik bilgileri için bkz. Bölüm 4.8. 5.2 Farmakokinetik özellikler Genel özelliklerEmilim:Azasitidin tek 75 mg/m2 subkutan doz uygulamasından sonra, azasitidin 0,5 saatte oluşan (ilk numune alma noktası) 750±403 ng/mL'lik doruk plazma konsantrasyonlarıyla hızla absorbeedilmiştir. Eğri altındaki alana (EAA) dayanarak subkutan uygulama sonrası azasitidinin I.V. azasitidine (tek 75 mg/m2 doz) göre biyoyararlanımı eğri altındaki alan (EAA) olarak yaklaşık %89'dur.Azasitidinin subkutan uygulamasının eğri altındaki alanı ve maksimum plazma konsantrasyonu(Cmaks) yaklaşık 25-100 mg/m2 doz aralığı içinde orantılıdır. Dağılım:IV uygulamanın ardından ortalama dağılım hacmi 76±26 L ve sistemik klirensi 147±47 L/saattir. Biyotransformasyon:İn vitroverilere göre sitokrom P450 izoenzimleri (CYPler), UDP-glukuronoziltransferazlar (UGTler), sulfotransferazlar (SULTlar) ve glutatyon transferazların (GSTler) azasitidinmetabolizmasında yer almadığı görülmektedir.Azasitidin metabolizması, sitidin deaminaz aracılığı ile oluşan deaminasyon ve spontan olarak gelişen hidroliz ile gerçekleşmektedir. İnsan karaciğeri S9 fraksiyonlarında metabolit oluşumununNADPH'dan bağımsız olduğu gözlenmiştir, bu durum metabolik basamakların sitozolik enzimlertarafından katalizlendiğine işaret etmektedir. İnsan hepatosit kültürleri üzerinde yapılan in vitroBelge Dotiptutı-tediiebilecek
konsantrasyonlardan yaklaşık 30 kat daha yüksek konsantrasyonlarda) sitokrom P450 izoenzimleri (CYP) olan 1A2, 2C19 veya 3A4 veya 3A5'i" indüklemediğini göstermektedir. 100 ^M azasitidinile inkübe edilen bir seri P450 izoenziminde (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ve 3A4)inhibisyon oluşturmamıştır. Bu nedenle klinik olarak elde edilebilir azasitidin plazmakonsantrasyonlarında enzim inhibisyonu olasılığı düşünülmemektedir. Eliminasyon:Azasitidin s.c. uygulamadan sonra 41±8 dakikalık ortalama eliminasyon yarılanma ömrü t(1/2) ile hızlı bir şekilde plazmadan atılır. Günde 1 defa 7 gün boyunca subkutan 75 mg/m2 azasitidinuygulamasından sonra herhangi bir birikme oluşmaz. Azasitidin ve/veya metabolitleri başlıca idrarla atılır. 14C-azasitidinin s.c. ve i.v. uygulamasının ardından, uygulanan radyoaktivitenin <%l'i feçes ile atılırken, % 50-85'i idrar ile atılır. Hastalardaki karakteristik özelliklerÖzel popülasyonlar:Karaciğer yetmezliğinin (bkz Bölüm 4.2), cinsiyetin, yaşın veya ırkın azasitidinin farmakokinetiği üzerine olan etkileri incelenmemiştir. Pediyatrik popülasyonAZA-JMML-001 çalışmasında, Farmakokinetik analiz, 1. siklusun 7. gününde 10 MDS ve 18 J^IML pediatrik hasta üzerinden gerçekleştirilmiştir. (Bkz. Bölüm 5.1). Ortalama yaş MDS hastalarıiçin 13,3 (yaş aralığı 1,9-15) ve JMML hastaları için de 2,1 (yaş aralığı 0,2-6,9) idi. 75 mg/ m2 'lik bir dozun intravenöz uygulanmasını takiben azasitidin, hem MDS hem de JMML popülasyonlarında 0,083 saat içinde Cmax değerine hızlı bir şekilde ulaşmıştır. MDS ve JMMLhastaları için Cmax'ın geometrik ortalaması sırasıyla 1797,5 ve 1066,3 ng/mL iken AUC0-ro'ıngeometrik ortalaması ise 606,9 ve 240,2 ng saat/mL'dir. MDS ve JMML hastalarında geometrikortalama dağılım hacmi sırasıyla 103,9 ve 61,1 L'dir. Azasitidinin toplam plazma maruziyetininMDS hastalarında daha yüksek olduğu görülmüş; bununla birlikte hem AUC hem de Cmax değerleriiçin hastalar arasında orta ila yüksek değerli değişkenlik kaydedilmiştir.
MDS ve JMML için t^'nin geometrik ortalaması sırasıyla 0,4 ve 0,3 saat ve klerenslerin geometrik ortalaması ise sırasıyla 166,4 ve 148,3 L/saat'tir. AZA-JMML-001 çalışmasından elde edilen farmakokinetik veriler birlikte toplanmış ve AZA-2002-BA-002 çalışmasında intravenöz yolla 75 mg/m2 dozluk azasitidin uygulanan MDS'li 6 yetişkin hastadan alınan farmakokinetik verilerle karşılaştırılmıştır. Azasitidinin Cmax ve AUC0-t'nin ortalaması, intravenöz uygulamadan sonra yetişkin hastalar ve pediatrik hastalar arasındabenzerdir (sırasıyla, 2.750 ng/mL'ye karşı 2.841 ng/mL ve 1.025 ng^saat/mL'ye karşılık 882,1ng^saat/mL). AZA-AML-004 Çalışmasında farmakokinetik analiz, yedi hastanın doz sonrası en az bir ölçülebilir farmakokinetik konsantrasyon saptanabilmiş altısının verileriyle yapılmıştır. (bkz. Bölüm 5.1).AML hastalarının medyan yaşı 6,7 ve yaş aralığı ise 2-12 idi. 100 mg/m2 'lik çoklu dozun bir çok kez verilmesi sonrasında 1.siklusun 7. günü Cmax ve AUC0-tau geometrik ortalamaları sırasıyla 1.557 ng/mL ve 899,6 ng-saat/mL olmuştur. Hastalar arası Cmax ve AUC0-tau değerlerinde yüksek değişkenlik olduğu gözlenmiştir (CV yüzdesi Cmax veAUC0-tau için sırasıyla %201,6 ve %87,8 olmuştur). Azasitidin, intravenöz uygulamadan sonraortalama 0,09 saatlik bir medyan sürede hızla Cmax'a ulaşmış ve 0,38 saatlik bir geometrik ortalamayarılanma ömrü (t1/2) ile azalmıştır. Klirens ve dağılma hacmi için geometrik ortalama sırasıyla127,2 L/sa ve 70,2 L'dir. AML'li çocuklarda ilk tam remisyon (CR1)'dan sonra moleküler relapsda gözlenen farmakokinetik (azasitidin) maruziyet, MDS'li 10 çocuk ve JMML'li 18 çocuğun havuzlanmış verilerinden eldeedilen maruziyet ile karşılaştırılabilir ve ayrıca MDS'li yetişkinlerdeki azasitidin maruziyeti ilekarşılaştırılabilir seviyededir. Böbrek yetmezliğiBöbrek yetmezliğinin, tek ve çoklu subkutan uygulamalardan sonra azasitidinin farmakokinetik maruziyetinde herhangi bir önemli etkisi yoktur. Tek 75 mg/m2 subkutan doz uygulamasından sonra,normal böbrek fonksiyonu olan hastalara kıyasla hafif, orta ve ciddi böbrek yetmezliği olanhastaların ortalama maruziyet değerleri (EAA ve Cmaks), sırasıyla %11-21, %15-27 ve %41-66oranında artmıştır. Bununla birlikte, maruziyet, normal böbrek fonksiyonu olan hastalar için Belge Dtgözlerieriıayay geaetfflar&zi^et'aralfğındldtr. Azasi^idjffve/veya:metab^ttleri esasoiatafeıböb^ekten
atıldığı için böbrek yetmezliği olan hastaların yakından izlenmesi koşulu ile, azasitidin, başlangıç doz ayarlaması olmaksızın böbrek yetmezliği olan hastalara uygulanabilir. Farmakogenomikler:Azasitidin metabolizması üzerinde bilinen sitidin deaminaz polimorfizmlerinin etkisi incelenmemiştir. 5.3. Klinik öncesi güvenlilik verileriAzasitidin in vitrobakteriyel ve memeli hücre sistemlerinde hem gen mutasyonlarını hem de kromozomal anomalileri indükler. Azasitidinin potansiyel karsinojenitesi farelerde ve sıçanlardaincelenmiştir. Azasitidin 52 hafta boyunca haftada 3 defa intraperitonal (i.p.) uygulandığında, dişifarelerde hematopoetik sistem tümörlerini indüklemiştir. 50 hafta süreyle i.p. olarak uygulananazasitidin ile tedavi edilen farelerde lenforetiküler sistem, akciğer, süt bezi ve deri tümörlerinininsidansının arttığı görülmüştür. Sıçanlarda bir tümör oluşturma çalışmasında testiküler tümörlerininsidansı artmıştır.Farelerde yapılan ilk embriyotoksisite çalışmalarında, organogenezis sırasında azasitidinin tek bir i.p. enjeksiyonundan sonra, intrauterin embriyonal ölüm %44 sıklıkta (artan rezorpsiyon) görülmüştür. Azasitidin verilen farelerde, sert damağın kapanması sırasında veya kapanmasından önce beyinde gelişimsel anormallikler görülmüştür. Sıçanlara preimplantasyon sürecinde verildiğinde, azasitidinherhangi bir advers etki göstermemiştir; fakat organogenezis sırasında verildiğinde açıkçaembriyotoksiktir. Organogenezis sırasında sıçanlarda meydana gelen fetal anomaliler şunlardır:MSS anomalileri (eksensefali, ensefalosel), kol-bacak anomalileri (mikromeli, yumru ayak,sindaktili, oligodaktili) ve diğerleri (mikroftalmi, mikognazi. Gastroşizis, ödem ve kaburgaanormallikleri). Azasitidinin, tedavi edilmemiş dişi fare ile çiftleşmeden önce erkek fareye uygulanması, fertilite azalması ve embriyonik ve postnatal gelişim sırasında yavrunun kaybı ile sonuçlanmıştır. Erkeksıçanlara verilmesi, testis ve epididimislerin ağırlığının azalması, sperm sayısının azalması, gebelikoranlarının azalması, çiftleşen dişilerde embriyoların kaybı ve anormal embriyo artışı ilesonuçlanmıştır (bkz. Bölüm 4.4).
6. FARMASOTIK ÖZELLİKLER6.1. Yardımcı maddelerin listesiSükroz Monosodyum Fosfat Monohidrat Disodyum Hidrojen Fosfat DihidratEnjeksiyonluk su 6.2. GeçimsizliklerBu ürün, Bölüm 6.6'da6.3. Raf ömrüAçılmamış toz flakonu:24 ayHazırlandıktan sonra: AZAVIX buzdolabında saklanmayan enjeksiyonluk su ile hazırlandığında, hazırlanan tıbbi ürün 25°C'de 45 dakika ve 2-8°C'de 8 saat süre ile kimyasal ve fiziksel stabilitesinikorur. Hazırlanan tıbbi ürünün raf ömrü buzdolabında (2-8°C) saklanan enjeksiyonluk su ile uzatılabilir. AZAVIX buzdolabında (2-8°C) saklanan enjeksiyonluk su ile hazırlandığında, hazırlanan tıbbi ürün2-8°C'de 22 saat süre ile kimyasal ve fiziksel stabilitesini korur. Mikrobiyolojik açıdan hazırlanan ürün derhal kullanılmalıdır. Hemen kullanılmayacak ise, kullanım öncesi saklama süresi ve koşulları kullanıcının sorumluluğundadır ve buzdolabında saklanmayanenjeksiyonluk su ile hazırlandığında 2-8°C'de 8 saatten fazla ve buzdolabında (2-8°C) saklananenjeksiyonluk su ile hazırlandığında 2-8°C'de 22 saatten fazla olmamalıdır. 6.4. Saklamaya yönelik özel tedbirler25°C'nin altındaki oda sıcaklığında saklayınız. Hazırlanan tıbbi ürünün saklama koşulları için Bölüm 6.3'e bakınız 6.5. Ambalajın niteliği ve içeriğiPolipropilen başlıklı alüminyum kapakla kapatılmış ve kauçuk liyo-tıpalı 40ml'lik renksiz cam flakon (tip I). Ambalaj büyüklüğü: 1 flakon içinde 100 mg azasitidin
6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanma talimatıGüvenlik için öneriler:AZAVIX sitotoksik bir ilaçtır ve diğer potansiyel toksik bileşiklerde olduğu gibi, azasitidin süspansiyonlarım hazırlarken ve taşırken dikkatli olunmalıdır.Antikanser ilaçların imhası ve doğru şekilde tutulma prosedürleri uygulanmalıdır. Hazırlanan azasitidin süspansiyonu cilt ile temas ederse, derhal ve iyice su ve sabun ile yıkanmalıdır. Mukus membranlarla temas ederse, su ile iyice yıkanmalıdır. Hazırlama prosedürü:1. Aşağıdaki malzemeler hazırlanmalıdır: Azasitidin flakonu: enjeksiyonluk su flakonu(ları); steril olmayan cerrahi eldiven; Alkollü bezler; 5 mL'lik, iğneli enjeksiyon şırmgası(ları). 2. Şırıngaya 4 mL enjeksiyonluk su çekilmeli, şırıngada hiç hava olmamalıdır. 3. 4 mL enjeksiyonluk su içeren şırınganın iğnesi plastik kapaklı azasitidin flakonuna batırılmalı veenjeksiyonluk su flakona enjekte edilmelidir. 4. İğne ve şırınga, azasitidin flakonundan çıkarıldıktan sonra azasitidin flakonu kuvvetleçalkalanarak bulanık, homojen bir süspansiyon elde edilmelidir. Bu noktada süspansiyonun hermL'sinde 25 mg azasitidin (100 mg/4 mL) bulunur. Oluşan ilaç homojen, bulanık birsüspansiyondur, herhangi bir topak içermemelidir. Eğer büyük partikül veya topak mevcutsa ürünatılmalıdır. Etkin maddeyi uzaklaştırabileceği için süspansiyonu filtre etmeyiniz. Bazı adaptörlerde,şırıngalarda ve doz sistemlerinde filtrelerin bulunduğu dikkate alınmalıdır. Bu nedenle, bu tipsistemler ilaç hazırlandıktan sonra uygulama için kullanılmamalıdır.5. Azasitidin flakonunun plastik kapağı temizlenmeli ve yeni bir şırınga batırılmalıdır. Flakon tersdöndürülmeli, iğne ucunun sıvı seviyesinin altında olduğundan emin olunmalıdır. Şırınganınpistonu çekilerek doz için gerekli miktarda ilaç çekilmeli ve şırıngada hava olmamasına dikkatedilmelidir. Daha sonra şırınga ve iğnesi flakondan çıkarılmalı ve şıranganın iğnesi atılmaldır. 6. Şırıngaya yeni bir subkutan iğne ucu (25 ölçek önerilmektedir) takılır. Enjeksiyon bölgesindelokal reaksiyon insidansını azaltmak için iğne ucu enjeksiyondan önce temizlenmemelidir. 7. 1 flakondan fazla gerektiği zaman yukarıdaki basamaklar takip edilerek yeni ilaç süspansiyonuhazırlanır. 1 flakondan fazla gereken dozlarda doz eşit bölünmelidir (örneğin doz 150 mg =6 mLise 2 şırınganın her biri 3 mL süspansiyon içermelidir). Flakon ve iğne içindeki gecikmeden dolayı,flakondan bütün süspansiyonu çekmek mümkün olmayabilir. 8. Dozlama yapılan şırınganın içerikleri hastaya uygulanmadan önce tekrar çalkalanmalıdır. Belge DcErijefesiyteri %'ırasi®9aısispa»si^bnûn'i§ısp'yaklaşıkB20°C-25°©io^alidır.»$üspartsiyob*buanık bir
görünüm elde edilene kadar iki el arasında kuvvetle yuvarlanarak çalkalanır. Büyük partikül veya topak mevcutsa ürün atılmalıdır.AZAVIX süspansiyonu kullanılmadan hemen önce hazırlanmalı, oluşan süspansiyon 45 dakika içinde kullanılmalıdır. Süspansiyonun hazırlanmasından sonra 45 dakikadan daha uzun süregeçmesi halinde ilaç uygun şekilde atılmalı ve yeni bir doz hazırlanmalıdır. Alternatif olaraksüspansiyonun hastaya uygulanmadan önce hazırlanması gerektiği durumlarda hazır ilaç,hazırlandıktan hemen sonra buzdolabına (2-8°C) konulmalıdır. Süspansiyon bu şekildebuzdolabında maksimum 8 saat bekleyebilir. İlacın buzdolabında 8 saatten uzun süre kalmasıdurumunda süspansiyon uygun şekilde atılmalı ve yeni bir doz hazırlanmalıdır. Buzdolabında (2-8°C) saklanan enjeksiyonluk su ile hazırlandığında, hazırlandıktan sonra hemen buzdolabına (2-8°C) konulmalıdır. Süspansiyon buzdolabında en fazla 22 saat bekleyebilir. İlacınbuzdolabında 22 saatten uzun süre kalması durumunda süspansiyon uygun şekilde atılmalı ve yenibir doz hazırlanmalıdır. Süspansiyonu içeren şırınga hastaya uygulanmadan önce 30 dakikaya varan sürelerde buzdolabı dışında bekletilerek ısısının yaklaşık 20-25°C'ye ulaşması sağlanmalıdır. Eğer buzdolabı dışındageçen bu süre 30 dakikayı geçerse süspansiyon uygun şekilde atılmalı ve yeni bir dozhazırlanmalıdır. Tek dozun hesaplanmasıVücut yüzey alanına (VYA) göre toplam doz aşağıdaki şekilde hesaplanabilir: Toplam doz (mg)= Doz (mg/m2) x VYA (m2) Aşağıda 1,8 m2'1ik ortalama VYA değerine göre azasitidin dozlarının nasıl olması gerektiğine dair örnek bir tablo verilmiştir.
Uygulama şekli:Süspansiyonu hazırladıktan sonra filtre etmeyiniz!
Hazırlanan AZAVIX subkutan olarak üst kola, uyluğa veya karna 25 ölçekli iğne kullanarak enjekte edilmelidir (45-90° açı ile iğneyi sokunuz). 4mL'den büyük dozlar iki ayrı bölgeye enjekte edilmelidir. Enjeksiyon yapılan alan değiştirilmelidir. Yeni enjeksiyonlar, eski enjeksiyon bölgesine en az 2,5 cm uzaklıkta yapılmalıdır ve asla yumuşak, morarmış, kırmızı ve sert olan yerlere enjeksiyonyapılmamalıdır. Kullanılmamış olan ürünler ya da atık materyaller "Tıbbi Atıkların Kontrolü Yönetmeliği" ve "Ambalaj Atıklarının Kontrolü Yönetmelikleri"ne uygun olarak imha edilmelidir. Sitotoksik ve sitostatik beşeri tıbbi ürünlerin kullanımları sonucu boşalan iç ambalajlarının atıkları TEHLİKELİ ATIKTIR ve bu atıkların yönetimi 2/4/2015 tarihli ve 29314 sayılı Resmi Gazetedeyayımlanan Atık Yönetimi Yönetmeliğine göre yapılır. 7. RUHSAT SAHİBİTeva İlaçları San. ve Tic. A.Ş. Ümraniye/ İstanbul 8. RUHSAT NUMARASI2017/198 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 06.04.2017 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ
|
İlaç BilgileriAzavix 100 Mg Sc Enjeksiyonluk Süspansiyon İçin Toz İçeren FlakonEtken Maddesi: Azasitidin Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
















Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.