Tenipra 240 Mg Gastrorezistan Sert Kapsül Kısa Ürün BilgisiKISA URUN BILGISI¡Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır?1. BEŞERI TIBBİ ÜRÜNÜN ADITENİPRA 240 mg gastrorezistan sert kapsül 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir kapsül 240 mg dimetil fumarat içerir.Yardımcı maddeler:Yardımcı maddeler için 6.1'e bakınız.3. FARMASOTİK FORMGastrorezistan sert kapsül. Opak açık yeşil gövdeli ve opak açık yeşil kapaklı sert jelatin kapsül içerisinde beyaz veya beyazımsı, enterik kaplı mini tabletler 4. KLİNİK BİLGİLER4.1 Terapötik endikasyonlarTENİPRA, relapsing-remitting multipl sklerozu (RRMS) olan yetişkin ve 13 yaş ve üzeri pediyatrik hastaların tedavisinde endikedir. 4.2 Pozoloji ve uygulama şekliTENİPRA tedavisi, multipl skleroz tedavisinde uzmanlaşmış bir doktorun denetimi altında başlatılmalıdır. Pozoloji/uygulama sıklığı ve süresiBaşlangıç dozu, günde iki kez 120 mg'dir. Yedi gün sonra, günde iki kez 240 mg'lık önerilen idame doza çıkarılmalıdır. (Bölüm 4.4'e bakınız.). Eğer hasta bir dozu kaçırırsa, çift doz alınmamalıdır. Hasta kaçırılan dozu ancak dozlar arasında 4 saat bıraktığında alabilir. Aksi takdirde, hasta bir sonraki planlanan doza kadarbeklemelidir. Dozun günde iki kez 120 mg'ye geçici olarak azaltılması kızarma ve gastrointestinal advers reaksiyonların meydana gelmesini azaltabilir. Bir ay içinde, günde iki kez 240 mg'lik önerilenidame doza yeniden başlanmalıdır. TENİPRA yiyeceklerle birlikte alınmalıdır (bkz. Bölüm 5.2). Kızarma veya gastrointestinal advers reaksiyonlar gelişen hastalarda, TENİPRA'nın yiyeceklerle birlikte alınması tolereedilebilirliğini iyileştirebilir (bkz. Bölüm 4.4, 4.5 ve 4.8). Uygulama şekliTENİPRA oral yoldan kullanım içindir. Kapsül bütün olarak yutulmalıdır. Mikrotabletlerin enterik kaplaması midede tahriş edici etkileri önlemekte olduğundan; kapsül veya içeriği ezilmemeli, bölünmemeli, çözülmemeli,emilmemeli veya çiğnenmemelidir. Özel popülasyonlara ilişkin ek bilgiler Böbrek/Karaciğer yetmezliği:TENİPRA böbrek ve karaciğer yetmezliği olan hastalarda çalışılmamıştır. Klinik farmakoloji çalışmalarına dayalı olarak, hiçbir doz ayarlaması gerekmemektedir (bkz. Bölüm 5.2). Şiddetliböbrek veya şiddetli karaciğer yetmezliği olan hastaları tedavi ederken dikkatli olunmalıdır(bkz. Bölüm 4.4). Pediyatrik popülasyon:Pozoloji yetişkinlerde ve 13 yaş ve üzerindeki pediyatrik hastalarda aynıdır. Şu anki mevcut veriler 4.4, 4.8, 5.1 (farmakodinamik ve farmakokinetik özellikler) bölümlerindesunulmaktadır. 10 ila 12 yaş arasındaki çocuklarda sınırlı veri mevcuttur. 10 yaşından küçük çocuklarda TENİPRA'nın güvenlilik ve etkililiği henüz belirlenmemiştir. Geriyatrik popülasyon:Dimetil fumarat ile yapılan klinik çalışmalar 55 yaş ve üstündeki hastalarda sınırlıdır ve bu çalışmalar, genç hastalardan farklı yanıt verip vermediğini belirlemek için yeterli sayıda 65yaş ve üzeri hasta içermemiştir (bkz. Bölüm 5.2). Etkin maddenin etki şekline dayalı olarak,yaşlılarda doz ayarlaması gerektirecek hiçbir teorik neden yoktur. 4.3 KontrendikasyonlarTENİPRA, etkin maddeye veya Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine aşırı duyarlılığı bulunan kişilerde kontrendikedir. TENİPRA, şüpheli veya doğrulanmış İlerleyici Multifokal Lökoensefalopati (PML) bulunan kişilerde kontrendikedir. 4.4 Özel kullanım uyarıları ve önlemleriŞiddetli ve uzamış lenfopeni varlığında TENİPRA kullanan hastalarda Progresif Multifokal Lökoensefalopati (PML) vakaları meydana gelmektedir. PML'ye dair ilkbelirti ve semptom görüldüğünde TENİPRA tedavisi hemen durdurulmalı ve uygundiagnostik değerlendirme yapılmalıdır (bkz. PML altbaşlığı).Kan/Laboratuvar testleriDimetil fumarat ile tedavi edilen hastalarda yapılan klinik araştırmalarda, böbrek fonksiyon testlerinde değişiklikler görülmüştür (bkz. Bölüm 4.8). Bu değişikliklerin klinik önemibilinmemektedir. Böbrek fonksiyonu (örn. kreatinin, kan üre azotu ve idrar tahlili)değerlendirmelerinin tedaviye başlamadan önce, tedavinin 3. ve 6. ayı sonunda, ardından her6 ila 12 ayda bir ve klinik olarak gerekli görüldüğünde yapılması önerilir. Karaciğer enzim artışı (>3 Üst normal limit (ULN)) ve total bilirubin seviyelerinin yükselmesi (>2 ULN) dahil ilaca bağlı karaciğer hasarı, dimetil fumarat tedavisindenkaynaklanabilmektedir. Başlangıç zamanı hemen, birkaç hafta veya daha uzun bir süre sonraolabilir. Tedavi sonrası, advers reaksiyonların çözümü gözlemlenmiştir. Klinik olarakbelirtildiği gibi, tedaviye başlamadan önce ve tedavi sırasında serum aminotransferazları (örn.alanin aminotransferaz (ALT) ,aspartat aminotransferaz (AST)) ve total bilirubin seviyelerinindeğerlendirilmesi önerilmektedir. TENİPRA ile tedavi edilen hastalarda lenfopeni gelişebilir (bkz. Bölüm 4.8). TENİPRA ile tedaviye başlamadan önce lenfositler de dahil olmak üzere mevcut tam kan sayımıyapılmalıdır. Lenfosit sayısının normal aralığın altında olduğu tespit edilirse TENİPRAtedavisine başlamadan önce olası nedenlerin kapsamlı bir değerlendirmesi tamamlanmalıdır.Dimetil fumarat önceden düşük lenfosit sayılarına sahip hastalarda çalışılmamıştır ve buhastalar tedavi edilirken dikkatli olunmalıdır. TENİPRA şiddetli lenfopeni (lenfosit sayısı <0.5 x 109/L) olan hastalarda başlatılmamalıdır. Tedaviye başladıktan sonra, her 3 ayda bir lenfosit dahil tam kan sayımı yapılmalıdır. Lenfopeni olan hastalarda ilerleyici multifokal Lökoensefalopati (PML) riskinin artması nedeniyle aşağıdakiler için son derece dikkatli olunmalıdır: * TENİPRA, 6 aydan fazla süren uzun süreli şiddetli lenfopeni (lenfosit sayısı < 0.5x 109/L) olan hastalarda kesilmelidir. * 6 aydan daha uzun süre sürekli orta derecede azalan mutlak lenfosit sayısı > 0.5 x109/L ila < 0.8 x 109/L arasında olan hastalarda, TENİPRA tedavisinin yararı/riski yenidendeğerlendirilmelidir. * Lenfosit sayısı, lokal laboratuvar referans aralığı tarafından tanımlanan normalin(LLN) alt sınırının altında olan hastalarda, mutlak lenfosit sayımlarının düzenli olarakizlenmesi önerilir. Bireysel PML riskini daha da artırabilecek ek faktörler gözönündebulundurulmalıdır (aşağıdaki PML ile ilgili alt bölüme bakınız). Lenfosit sayıları iyileşme görülünceye kadar takip edilmelidir (Bölüm 5.1'e bakınız). İyileşme üzerine ve alternatif tedavi seçeneklerinin yokluğunda, tedavinin kesilmesindensonra TENİPRA'nın yeniden başlatılıp başlatılmayacağına ilişkin kararlar klinik yargıyadayanmalıdır. Manyetik Rezonans Görüntüleme (MRI)TENİPRA ile tedaviye başlamadan önce, referans olarak bir başlangıç MRI (genellikle 3 ay içinde çektirilmiş) bulunmalıdır. İlave MRI taraması ihtiyacı, ulusal ve yerel önerilere göredeğerlendirilmelidir. MRI görüntüleme, PML riskinin arttığı düşünülen hastalarda arttırılmışizlemin bir parçası olarak düşünülebilir. Klinik olarak PML'den şüphelenilmesi halinde,acilen tanı amaçlı MRI çekilmelidir. Progresif Multifokal Lökoensefalopati (PML)Dimetil fumarat ile tedavi edilen hastalarda PML bildirilmiştir (bkz.Bölüm 4.8). PML, ölümcül olabilen veya ciddi sakatlık ile sonuçlanabilen John-Cunningham virüsünün (JCV)neden olduğu fırsatçı bir enfeksiyondur. Lenfopeni (LLN'nin altındaki lenfosit sayıları) durumunda dimetil fumarat ve fumarat içeren diğer tıbbi ürünler ile PML vakaları meydana gelmiştir. Uzun süreli orta ila şiddetlilenfopeni,Dimetil fumarat ile PML riskini arttırmaktadır, ancak hafif lenfopeni olanhastalarda risk göz ardı edilemez. Lenfopeni durumunda PML riskinin artmasına katkıda bulunabilecek ek faktörler şunlardır: * TENİPRA tedavisinin süresi. Tedavi süresi ile kesin ilişki bilinmemekle birlikte,PMLvakaları yaklaşık 1 ila 5 yıllık tedaviden sonra ortaya çıkmıştır. * CD4+ ' da ve özellikle immünolojik savunma için önemli olan CD8+ T hücresayılarındaşiddetli düşüşler (bkz. Bölüm 4.8) ve * önceki immünosupresif veya immünomodülatör tedavi (aşağıya bakınız). Doktorlar, semptomların nörolojik disfonksiyonu gösterip göstermediğini ve eğer öyleyse, bu semptomların MS için tipik olup olmadığını veya olası bir PML'yidüşündürüp düşündürmediğini belirlemek için hastalarını değerlendirmelidir. PML'i düşündüren ilk belirti ya da semptomda, TENİPRA uygulaması durdurulmalı ve kantitatif polimeraz zincir reaksiyonu (PCR) metodolojisi ile beyin omurilik sıvısında(BOS) JCV DNA'sının belirlenmesi de dahil olmak üzere uygun diagnostikdeğerlendirme yapılmalıdır. PML semptomları, MS nüksüne benzer olabilir. PML ileilişkilendirilen tipik semptomlar; çeşitli, günden haftaya ilerleyen, vücudun bir tarafındailerleyen zayıflık ya da kol ve bacaklarda hantallık, görme bozukluğu ve düşüncede,hafızada değişiklik ve konfüzyon ve kişilik değişikliklerine yol açan oryantasyondeğişiklikleridir. Doktorlar, hastanın fark edemeyeceği PML'yi düşündüren semptomlarakarşı özellikle dikkatli olmalıdır. Hastanın farkında olmadığı semptomları farkedebileceğinden, hastalara eşlerini veya bakıcılarını tedavileri hakkında bilgilendirmeleride tavsiye edilmelidir. PML sadece JCV enfeksiyonunun varlığında meydana gelebilir. Dimetil fumarat ile tedavi edilen hastalarda serum anti-JCV antikor testinin doğruluğu üzerinde lenfopenininetkisinin çalışılmadığı dikkate alınmalıdır. Ayrıca, negatif bir anti-JCV antikor testinin(normal lenfosit sayısının varlığında) sonradan JCV enfeksiyonunun meydana gelmeihtimalini ortadan kaldırmadığı göz önünde bulundurulmalıdır. Bir hastada PML gelişirse, TENİPRA kalıcı olarak kesilmelidir. İmmünosupresif ya da immünomodülatör tedavilerden önceHastalar, hastalığı modifiye eden diğer tedavilerden dimetil fumarata geçirildiğinde dimetil fumaratın etkililik ve güvenliliğini değerlendiren hiçbir çalışma gerçekleştirilmemiştir. Dimetil fumarat ile tedavi edilen hastalarda PML'in gelişimine önceki immunosupresif tedavinin katkısı olasıdır. PML vakaları, daha önce natalizumab ile tedavi edilen ve PML'nin belirlenmiş bir risk olduğu hastalarda ortaya çıkmıştır. Doktorlar, natalizumabın son kesilmesinden sonraortaya çıkan PML vakalarının lenfopeniye sahip olmayabileceğini bilmelidir. Ek olarak, dimetil fumarat ile doğrulanmış PML vakalarının çoğu, daha önce immünomodülatör tedavisi olan hastalarda ortaya çıkmıştır. Hastalar, hastalığı modifiye eden diğer tedavilerden TENİPRA'ya geçirildiğinde, MS'in yeniden aktive olma riskini azaltırken ek bir immün etkiden kaçınmak için diğertedavininetki mekanizması ve yarı ömrü değerlendirilmelidir. Tam kan sayımı, TENİPRA'ya başlamadan önce ve tedavi süresince düzenli olarak önerilmektedir (Kan/laboratuar testleri bölümüne bakınız). Şiddetli böbrek ve karaciğer yetmezliğiTENİPRA şiddetli böbrek veya şiddetli karaciğer yetmezliği olan hastalarda çalışılmamıştır. Dolayısıyla, bu hastalarda dikkatli kullanılmalıdır (bkz. Bölüm 4.2). Şiddetli aktif gastrointestinal hastalıkTENİPRA şiddetli aktif gastrointestinal hastalığı olan hastalarda çalışılmamıştır. Dolayısıyla, bu hastalarda dikkatli kullanılmalıdır. KızarmaKlinik çalışmalarda, dimetil fumarat ile tedavi edilen hastaların %34'ünde kızarma meydana gelmiştir. Kızarma deneyimleyen hastaların çoğunda bu durum hafif veya orta şiddetlidir. Sağlıklı gönüllü çalışmalarından elde edilen veriler, dimetil fumarata bağlı kızarıklığın muhtemelen prostaglandin aracılı olduğunu göstermektedir. 75 mg enterik kaplı olmayanasetilsalisilik asit ile kısa süreli tedavi, tahammül edilemeyen kızarmadan etkilenen hastalardayararlı olabilir (bkz. Bölüm 4.5). İki sağlıklı gönüllü çalışmasında, dozlama periyodu boyuncakızarma oluşumu ve şiddeti azalmıştır. Klinik çalışmalarda, dimetil fumarat ile tedavi edilen toplam 2560 hastanın 3'ü aşırı duyarlılık veya anafilaktoid reaksiyonlar olması muhtemel olan şiddetli kızarma semptomlarıdeneyimlemiştir. Bu olaylar yaşamı tehdit edici değildir; ancak hospitalizasyona yol açmıştır.Hekimler ve hastalar şiddetli kızarma reaksiyonunun olması halinde bu ihtimale karşı dikkatliolmalıdırlar (bkz. Bölüm 4.2, 4.5 ve 4.8). Anafilaktik reaksiyonlarPazarlama sonrası deneyimde dimetil fumarat uygulamasını takiben anafilaksi / anafilaktoid reaksiyon vakaları bildirilmiştir. Semptomlar dispne, hipoksi, hipotansiyon, anjiyoödem,döküntü veya kaşıntıyı içerebilmektedir. Dimetil fumarat kaynaklı anafilaksinin mekanizmasıbilinmemektedir. Reaksiyonlar genellikle ilk dozdan sonra meydana gelir, ancak tedavisırasında herhangi bir zamanda da meydana gelebilir. Bu reaksiyonlar ciddi ve hayatı tehditedici olabilir. Eğer anafilaksi belirtileri veya semptomları varsa, hastalara TENİPRA'yıkullanmayı bırakmaları ve hemen tıbbi yardım almaları konusunda bilgi verilmelidir. Tedaviyeniden başlatılmamalıdır (bkz. Bölüm 4.8). EnfeksiyonlarFaz III plasebo-kontrollü çalışmalarda, dimetil fumarat veya plasebo ile tedavi edilen hastalarda enfeksiyonların insidansı (%60'a %58) ve ciddi enfeksiyonların insidansı (%2'ye%2) sırasıyla benzerdir. Ancak dimetil fumarat, immünomodülatör özellikleri nedeniyle(Bölüm 5.1'e bakınız) eğer hastada ciddi bir enfeksiyon gelişirse, TENİPRA tedavisininaskıya alınması düşünülmeli ve tedaviye yeniden başlamadan önce yararlar ve riskler yenidendeğerlendirilmelidir. TENİPRA alan hastalar, enfeksiyon semptomlarını bir doktorabildirmeleri konusunda bilgilendirilmelidir. Ciddi enfeksiyonları olan hastalar,enfeksiyon(lar) ortadan kalkana kadar TENİPRA tedavisine başlamamalıdır. Lenfosit sayıları <0,8x109/L veya 0,5x109/L olan hastalarda ciddi enfeksiyon insidansı artışı gözlenmemiştir.(bkz. Bölüm 4.8) Eğer tedaviye orta-ciddi uzamış lenfopeni varlığında devamedilirse, Progresif Multifokal Lökoensefalopati (PML) dahil fırsatçı bir enfeksiyon riski gözardı edilemez (bkz. Bölüm 4.4 alt bölüm PML). Herpes Zoster EnfeksiyonlarıDimetil fumarat ile herpes zoster vakaları meydana gelmiştir. Vakaların çoğu ciddi değildir, ancak disemine herpes zoster, herpes zoster oftalmikus, herpes zoster otikus, herpes zosternörolojik enfeksiyonu, herpes zoster meningoensefalit ve herpes zoster meningomiyelit gibiciddi vakalar bildirilmiştir. Bu vakalar tedavi sırasında herhangi bir zamanda ortayaçıkabilmektedir. TENİPRA alan hastalar, özellikle eşzamanlı lenfositopeni rapor edildiğindeherpes zoster belirtileri ve semptomları yönünden izlenmelidir. Herpes zoster oluşursa, herpeszoster için uygun tedavi uygulanmalıdır. Ciddi enfeksiyonu olan hastalarda enfeksiyonçözülene kadar TENİPRA tedavisinin durdurulması düşünülmelidir. ( bkz. Bölüm 4.8 ) Tedaviye başlamaKızarma ve gastrointestinal advers reaksiyonların meydana gelmesini azaltmak için TENİPRA tedavisine kademeli olarak başlanmalıdır. (bkz. Bölüm 4.2) Fanconi sendromuDiğer fumarik asit esterleri ile kombinasyon halinde dimetil fumarat içeren bir tıbbi ürün için Fanconi sendromu vakaları bildirilmiştir. Fanconi sendromunun erken teşhisi ve dimetilfumarat tedavisinin kesilmesi, sendrom genellikle geri dönüşümlü olduğu için böbrekyetmezliği ve osteomalazi başlangıcını önlemek için önemlidir. En önemli belirtiler şunlardır:proteinüri, glukozüri (normal kan şekeri düzeyleriyle), hiperaminoasidüri ve fosfatüri(muhtemelen hipofosfatemi ile eşzamanlı). Poliüri, polidipsi ve proksimal kas güçsüzlüğü gibisemptomlarda ilerleme olabilmektedir. Nadiren lokalize olmayan kemik ağrısı olanhipofosfatemik osteomalazi, serumda yüksek alkalin fosfataz ve stres kırıklarıoluşabilmektedir. Önemli olarak, Fanconi sendromu yüksek kreatinin seviyeleri veya düşükglomerüler filtrasyon hızı olmadan ortaya çıkabilmektedir. Belirsiz semptomlar olmasıdurumunda Fanconi sendromu düşünülmeli ve uygun incelemeler yapılmalıdır. Pediyatrik popülasyonPediyatrik hastalarda güvenlilik profili yetişkinlere kıyasla niteliksel olarak benzerdir ve bu nedenle uyarılar ve önlemler pediatrik hastalar için de geçerlidir. Güvenlik profilindeki nicelfarklılıklar için Bölüm 4.8'e bakınız. Pediyatrik popülasyonda TENİPRA'nın uzun vadeligüvenliliği henüz belirlenmemiştir. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriDimetil fumarat antineoplastik veya immünosüpresif tedavi kombinasyonları ile çalışılmamıştır ve dolayısıyla, eşzamanlı uygulama sırasında dikkatli olunmalıdır. Multiplskleroz klinik çalışmalarında, kısa süreli intravenöz kortikosteroidler ile nükslerin eşzamanlıtedavisi klinik açıdan anlamlı bir enfeksiyon artışı ile ilişkilendirilmemiştir. Ulusal aşılama programlarına göre inaktif aşıların TENİPRA tedavisi sırasında eş zamanlı uygulanması düşünülebilir. Relapsing remitting multipl sklerozu olan toplam 71 hastayıkapsayan bir klinik çalışmada, en az altı aydır (n=38) günde iki kere dimetil fumarat 240 mgya da en az üç aydır (n=33) pegile olmayan interferon alan hastalarda, tetanoz toksoid (recallantijen) ve konjuge meningokokal C polisakkarit aşısına (neoantigen) benzer bir bağışıklıktepkisi kurulurken (aşılama titresinden sonraya kadar >2 kat artışı olarak tanımlanır), konjugeedilmemiş 23 valanslı pnömokokal polisakkarit aşısının (T hücresinden bağımsız antijen)farklı serotiplerine karşı bağışıklık tepkisi her iki tedavi grubunda da değişmiştir. Her ikitedavi grubunda çok az hastada, üç aşıya karşı antikor titresinde en az 4 kat ve daha fazla artışolarak tanımlanan pozitif bir immün yanıt elde edilmiştir. Tetanoz toksoidine ve pnömokokserotip 3 polisakkarite karşı yanıtta küçük sayısal farklar pegile interferon lehine notedilmiştir. Dimetil fumarat alan hastalarda canlı atenüe aşıların etkililiği ve güvenliliği hakkında klinik veri bulunmamaktadır. Canlı aşılar artmış bir klinik enfeksiyon riski taşıyabilir ve istisnaidurumlarda, bireyin aşılanmamasının oluşturacağı risk bu potansiyel riske göre ağırbasmadıkça, TENİPRA ile tedavi edilen hastalara verilmemelidir. TENİPRA tedavisi sırasında, diğer fumarik asit türevlerinin (topikal veya sistemik) birlikte kullanımından kaçınılmalıdır. İnsanlarda, dimetil fumarat sistemik dolaşıma ulaşmadan önce esterazlar tarafından büyük ölçüde metabolize edilmektedir. Sonraki metabolizma, sitokrom P450 (CYP) sistemine dahilolmadan trikarboksilik asit siklusu üzerinden meydana gelmektedir. Potansiyel ilaç etkileşimriskleri, in vitroCYP-inhibisyon ve indüksiyon çalışmaları, bir p-glikoprotein çalışması ya dadimetil fumarat ve monometil fumaratın (dimetil fumaratın primer bir metaboliti) proteinebağlanma çalışmalarından belirlenmemiştir.Multipl sklerozu olan hastalarda yaygın olarak kullanılan tıbbi ürünler, intramüsküler interferon beta-1a ve glatiramer asetat, dimetil fumarat ile potansiyel etkileşimler için klinikolarak test edilmiştir ve dimetil fumaratın farmakokinetik profilini değiştirmemiştir. Sağlıklı gönüllü çalışmalarından elde edilen veriler, dimetil fumarata bağlı kızarmanın muhtemelen prostaglandin aracılı olduğunu göstermektedir. Sağlıklı gönüllülerde yapılan ikiçalışmada, dimetil fumarattan 30 dakika önce, sırası ile 4 gün ve 4 hafta boyunca dozlanan325 mg (veya eşdeğeri) enterik kaplı olmayan asetilsalisilik asitin uygulanması, dimetilfumaratın farmakokinetik profilini değiştirmemiştir. Asetilsalisilik asit tedavisi ile ilişkilipotansiyel riskler, Relapsing Remitting MS hastalarında dimetil fumarat ile eşzamanlıuygulama yapılmadan önce düşünülmelidir. Asetilsalisilik asitin uzun süreli (> 4 hafta) süreklikullanımı araştırılmamıştır (bkz. Bölüm 4.4 ve 4.8). Nefrotoksik tıbbi ürünler (örn. aminoglikozidler, diüretikler, nonsteroidal anti-inflamatuar ilaçlar veya lityum), TENİPRA alan hastalarda renal advers reaksiyon (örn. proteinüri bkz.Bölüm 4.8) potansiyelini arttırabilir (bkz. Bölüm 4.4 Kan/Laboratuvar testleri). Orta miktarda alkol tüketimi dimetil fumarata maruziyeti değiştirmemiştir ve advers reaksiyonlarda bir artışla ilişkilendirilmemiştir. Alkol gastrointestinal adversreaksiyonların sıklığını arttırabileceği için, TENİPRA'yı aldıktan sonra bir saat içindeyüksek miktarlarda sert alkollü içkilerin (alkol hacmi %30'un üzerinde olan)tüketilmesinden kaçınılmalıdır. İn vitrovivoÖzel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyon:Etkileşim çalışmaları sadece yetişkinlerde yapılmıştır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)TENİPRA'nın çocuk doğurma potansiyeli bulunan kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. TENİPRA gebelik sırasında ve uygun kontrasepsiyon yöntemlerini kullanmayan çocuk doğurma potansiyeline sahip kadınlarda önerilmemektedir (bkz. Bölüm 4.5). Gebelik dönemiHayvanlar üzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. Dimetil fumaratın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. TENİPRA gebelikte yalnızca gerekli olduğunda ve beklenen yararın fetüse olan potansiyel zarardan daha fazla olması durumunda kullanılmalıdır. TENİPRA gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Laktasyon dönemiDimetil fumaratın veya metabolitlerinin insan sütü ile atılıp atılmadığı bilinmemektedir. Yeni doğanlar/infantlar için risk göz ardı edilemez. Emzirmenin durdurulup durdurulmayacağınaya da TENİPRA tedavisinin durdurulup durdurulmayacağına/tedaviden kaçınılıpkaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası ve TENİPRAtedavisinin emziren anne açısından faydası dikkate alınmalıdır. Üreme yeteneği / FertiliteDimetil fumaratın insanlarda fertilite üzerine etkilerine dair hiçbir veri bulunmamaktadır. Klinik öncesi çalışmalardan elde edilen veriler, dimetil fumaratın fertiliteyi azaltma riskininolduğunu düşündürmemektedir (bkz. Bölüm 5.3). 4.7 Araç ve makine kullanımı üzerindeki etkilerTENİPRA'nın araç ve makine kullanma yetisi üzerinde etkisi yoktur veya ihmal edilebilir düzeydedir. TENİPRA'nın araç ve makine kullanma yetisi üzerine hiçbir çalışmayürütülmemiştir ancak klinik çalışmalarda bu yetiye potansiyel etkisi olan dimetil fumarat ileilişkili hiçbir etki gözlenmemiştir. 4.8 İstenmeyen etkilerGüvenlilik profilinin özeti Dimetil fumarat ile tedavi edilen hastalarda en yaygın advers reaksiyonlar (insidans >%10); kızarma ve gastrointestinal rahatsızlıklardır (ishal, bulantı, karın ağrısı, üst karın ağrısı).Kızarma ve gastrointestinal rahatsızlıklar, tedavinin erken dönemlerinde başlama eğilimigösterir (en çok ilk bir ay içinde). Kızarma ve gastrointestinal rahatsızlıklar yaşayanhastalarda, bu olaylar TENİPRA tedavisi boyunca aralıklarla meydana gelmeye devamedebilir. Dimetil fumarat ile tedavi edilen hastalarda tedavinin kesilmesine (insidans >%1) yolaçan en yaygın olarak bildirilen advers reaksiyonlar kızarma (%3) ve gastrointestinalolaylardır (%4). Plasebo-kontrollü ve kontrolsüz klinik çalışmalarda, toplam 2513 hasta TENİPRA'yı 12 yıla kadar olan süreler boyunca kullanmıştır ve toplam maruziyet 11.318 kişi yılına eşdeğerolmuştur. Toplam 1.169 hasta TENİPRA ile en az 5 yıl ve 426 hasta en az 10 yıl tedavigörmüştür. Kontrolsüz klinik çalışmalardan elde edilen deneyimler, plasebo kontrollü klinikçalışmalardaki deneyimler ile uyumlu bulunmuştur. Klinik çalışmalardan kaynaklanan advers reaksiyonlar, ruhsatlandırma sonrası güvenlik ve spontan raporlar aşağıdaki tabloda sunulmaktadır. Advers reaksiyonlar, MedDRA Sistem Organ Sınıfı altında MedDRA tercih edilen terimlere göre belirtilmiştir. Advers reaksiyonların insidansı aşağıdaki kategorilere göre ifadeedilmektedir: - Çok yaygın (> 1/10) - Yaygın (>1/100 ila <1/10) - Yaygın olmayan (>1/1.000 ila <1/100) - Seyrek (>1/10.000 ila <1/1.000) - Çok seyrek (< 1/10. 000) - Bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
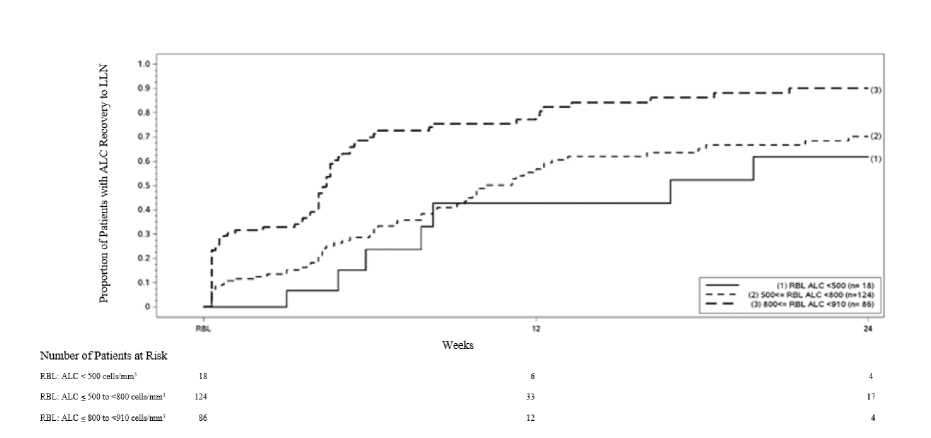
Seçilen advers reaksiyonların tanımlanması KızarmaPlasebo-kontrollü çalışmalarda, dimetil fumarat tedavisi alan hastalarda plaseboya kıyasla kızarmanın (%34'e karşı %4) ve ateş basmasının (%7'ye karşı %2) insidansı artmıştır.Kızarma, çoğunlukla kızarma veya ateş basması olarak tanımlanır; ancak başka olayları daiçerebilir (örn. sıcaklık, kızarma, kaşıntı ve yanma hissi). Kızarma olayı tedavinin erkendönemlerinde başlama eğilimi gösterir (en çok ilk bir ay içinde) ve kızarma deneyimleyenhastalarda, bu durum dimetil fumarat tedavisi boyunca aralıklarla meydana gelmeye devamedebilir. Kızarma yaşayan hastaların çoğunluğu kızarma olayını hafif veya orta şiddetteyaşamıştır. Genel olarak, dimetil fumarat ile tedavi edilen hastaların %3'ü kızarma nedeniyletedaviyi sonlandırmıştır. Yaygın eritem, döküntü ve/veya kaşıntı ile karakterize olan ciddikızarma insidansı, dimetil fumarat ile tedavi edilen hastaların %1'inden daha azındagörülmüştür (bkz. Bölüm 4.2, 4.4 ve 4.5). GastrointestinalGastrointestinal olayların insidansı, (örn. ishal [%14'e karşı %10], bulantı [%12'ye karşı %9], üst karın ağrısı [%10'a karşı %6], karın ağrısı [%9'a karşı %4], kusma [%8'e karşı %5] vedispepsi [%5'e karşı %3]) sırasıyla plaseboya kıyasla dimetil fumarat ile tedavi edilenhastalarda artmıştır. Gastrointestinal olaylar tedavinin erken dönemlerinde başlama eğilimigösterir (en çok ilk bir ay içinde) ve gastrointestinal olayları deneyimleyen hastalarda buolaylar dimetil fumarat tedavisi boyunca aralıklarla meydana gelmeye devam edebilir.Gastrointestinal olayları deneyimleyen hastaların çoğunluğunda bunlar genellikle hafif veyaorta şiddettedir. Dimetil fumarat ile tedavi edilen hastaların %4'ü, gastrointestinal olaylarabağlı olarak tedaviyi sonlandırmıştır. Gastroenterit ve gastrit de dahil olmak üzere, ciddigastrointestinal olayların insidansı dimetil fumarat ile tedavi edilen hastaların %1'indegörülmüştür (bkz. Bölüm 4.2). Karaciğer fonksiyonlarıPlasebo-kontrollü çalışmalardan elde edilen verilere dayanarak, yükselmeler gözlenen hastaların çoğunda, hepatik transaminazlar üst normal limitinin (ULN) 3 katından daha azolarak saptanmıştır. Dimetil fumarat ile tedavi edilen hastalarda, hepatik transaminazyükselmelerinin plaseboya göre artmış insidansı, en çok tedavinin ilk 6 ayında gözlenmiştir.Alanin aminotransferaz ve aspartat aminotransferazın üst normal limitin 3 katından fazlayükselmesi, sırasıyla plasebo ile tedavi edilen hastaların %5 ve %2'sinde ve dimetil fumaratile tedavi edilen hastaların %6 ve %2'sinde görülmüştür. Yükselen karaciğertransaminazlarına bağlı olarak ilacın kesilmesi %1'den düşüktür ve dimetil fumarat veyaplasebo ile tedavi gören hastalarda benzerdir. Plasebo kontrollü çalışmalarda transaminazlardaULN'nin > 3 katı yükselmeler ile birlikte total bilirubinde ULN'nin > 2 katı yükselmelergözlenmemiştir. Karaciğer enzimlerinin yükselmesi ve ilaca bağlı karaciğer hasarı vakaları (toplam bilirubinin üst normal limitin (ULN) >2 katı kadar yükselme ile eş zamanlı, üst normal limitin (ULN) >3katı transaminaz yükselmesi), pazarlama sonrası deneyimde dimetil fumarat uygulamasınıtakiben bildirilmiştir ve tedavisinin kesilmesi ile çözümlenmiştir. LenfopeniPlasebo kontrollü çalışmalarda, çoğu hasta (>%98) tedaviye başlamadan önce normal lenfosit değerlerine sahiptir. Dimetil fumarat ile tedavi edildiğinde ortalama lenfosit sayıları sonradanmeydana gelen bir plato düzeyi ile ilk bir yıl azalmıştır. Ortalama olarak, lenfosit sayıları,başlangıç değerinin yaklaşık %30'u azalmıştır. Ortalama ve medyan lenfosit sayıları normalsınırlar dahilinde kalmıştır. Plasebo ile tedavi edilen hastaların <%1'inde ve dimetil fumaratile tedavi edilen hastaların %6'sında, lenfosit sayısı 0,5x109/L'den daha az gözlenmiştir.Plasebo ile tedavi edilen hastaların hiçbirinde 0,2x109/L'den daha az lenfosit sayısıgözlenmezken, dimetil fumarat ile tedavi edilen 1 hastada gözlenmiştir. Klinik çalışmalarda (hem kontrollü hem de kontrolsüz), dimetil fumarat ile tedavi edilen hastaların %41'inde lenfopeni mevcuttur (bu çalışmalarda <0.91x109/L olaraktanımlanmıştır). Hastaların %28'inde hafif lenfopeni (>0.8x109/L ve <0.91 x109/Larasında) gözlenmiştir; hastaların %11'inde en az altı ay süren orta lenfopeni(>0.5x109/L ve <0.8x109/L arasında); hastaların %2'sinde en az 6 ay süren şiddetlilenfopeni (<0.5x109/L) gözlenmiştir. Şiddetli lenfopenisi olan grupta, sürekli tedaviile lenfosit sayımlarının çoğu <0.5x109/L olarak kalmıştır. Ek olarak, kontrolsüz, ileriye dönük, pazarlama sonrası bir çalışmada, dimetil fumarat ile tedavinin 48.haftasında (n=185) CD4+ T hücreleri, hastaların %6'sı veya %37sine kadarorta derecede (>0.2x109/L'den <0.4x109/L'ye kadar sayılan) veya ciddi (<0.2x109/L)derecedeazalırken, sırasıyla, CD8 + T hücreleri <0.2x109 / L sayımlarında hastaların %59'una kadar ve <0.1x109 sayımlarda hastaların % 25'ine kadar daha sıklıkla azalmıştır. Kontrollü ve kontrolsüz klinik çalışmalarda, lenfosit sayısı normalin alt sınırının (LLN) altındayken TENİPRA tedavisini bırakan hastalar, lenfosit sayısının LLN'ye geri dönüşüaçısından izlenmiştir (Bkz. Bölüm 5.1). PML ve fırsatçı enfeksiyonlar dahil olmak üzere enfeksiyonlarDimetil fumarat ile ilerleyici multifokal Lökoensefalopatiye (PML) neden olan John Cunningham virüsü (JCV) ile enfeksiyon vakaları bildirilmiştir (bkz.Bölüm 4.4). PMLölümcül olabilir veya ciddi sakatlığa neden olabilir. Klinik çalışmalardan birinde, dimetilfumarat alan bir hasta, ölümcül bir sonuçla uzun süreli şiddetli lenfopeni (lenfosit sayısıağırlıklı olarak <0.5x109/l 3.5 yıl) durumunda PML geliştirmiştir. Pazarlama sonrası ,PML orta ve hafif lenfopeni varlığında da meydana gelmiştir (yerel laboratuvar referansaralığı tarafından tanımlandığı gibi>0.5x109/L ila <LLN). PML tanısı sırasında T hücre alt kümelerinin belirlenmesi ile birkaç PML vakasında, CD8 + T hücre sayımlarının <0.1x109/L'ye düştüğü, CD4+ T hücre sayımlarındakiazalmaların değişken olduğu (<0.05 ila 0.5x109/L arasında değişen) ve lenfopeninin genelşiddeti ile daha fazla ilişkili olduğu bulunmuştur (<0.5 x109/L ila <LLN). Sonuç olarak, bu hastalarda CD4+/CD8+ oranı artmıştır. Uzun süreli orta ila şiddetli lenfopeni, TENİPRA ile PML riskini arttırmaktadır, ancak hafif lenfopeni olan hastalarda PML de ortaya çıkmıştır. Ayrıca, pazarlama sonrasındaPML vakaların büyük çoğunluğu >50 yaş hastalarda meydana gelmiştir. Dimetil fumarat kullanımı ile herpes zoster enfeksiyonları bildirilmiştir. 1736 MS hastasının dimetil fumarat ile tedavi edildiği devam eden uzun süreli bir uzatmaçalışmasında, yaklaşık % 5'inde, çoğunluğu hafif ila orta şiddette olan bir veya daha fazlaherpes zoster olayı yaşanmıştır. Ciddi herpes zoster enfeksiyonu yaşayanlar da dahilolmak üzere çoğu denekte lenfosit sayımı normalin alt sınırının üzerinde olmuştur.LLN'nin altında eşzamanlı lenfosit sayısı olan deneklerin çoğunda, lenfopeni orta veyaşiddetli olarak değerlendirilmiştir. Pazarlama sonrası ortamda, herpes zoster enfeksiyonuvakalarının çoğu ciddi değildir ve tedavi ile çözülmüştür. Pazarlama sonrasıdüzenlemelerde herpes zoster enfeksiyonu olan hastalarda mutlak lenfosit sayısı (ALC)hakkında sınırlı veri mevcuttur. Bununla birlikte, rapor edildiğinde, çoğu hastada orta(<0,8x109/L ila 0,5x109/L) veya ciddi (<0,5^109/L ila 0,2^109/L) lenfopeni görülmüştür(bkz. Bölüm 4.4). Laboratuvar anomalileriPlasebo-kontrollü çalışmalarda, üriner keton ölçümü (1+ veya daha yüksek) plaseboya (%10) kıyasla dimetil fumarat ile tedavi edilen hastalarda (%45) daha yüksektir. Klinik çalışmalardahiçbir istenmeyen klinik sonuç gözlenmemiştir. Plaseboya kıyasla dimetil fumarat ile tedavi edilen hastalarda 1,25-dihidroksivitamin D düzeyleri azalmıştır (2 yılda başlangıca göre medyan yüzde azalma; sırasıyla %25 ve %15) veplaseboya kıyasla dimetil fumarat ile tedavi edilen hastalarda paratiroid hormon (PTH)düzeyleri artmıştır (2 yılda başlangıca göre medyan yüzde artış; sırasıyla %29 ve %15). Heriki parametre için ortalama değerler normal aralık dahilinde kalmıştır. Tedavinin ilk 2 ayında ortalama eozinofil sayılarında geçici bir artış görülmüştür. Pediyatrik popülasyon10 ila 18 yaş arası RRMS'li pediyatrik hastalarda (7 gün boyunca günde iki kez 120 mg, ardından tedavinin geri kalanı için günde iki kez 240 mg) 96 haftalık açık etiketli, randomize,aktif kontrollü bir çalışmada; çalışma popülasyonu, n= 78), pediyatrik hastalardaki güvenlilikprofili, daha önce yetişkin hastalarda gözlenene benzer görünmüştür. Pediyatrik klinik deney tasarımı, yetişkin plasebo kontrollü klinik deneylerden farklı idi. Bu nedenle, klinik araştırma tasarımının, pediyatrik ve yetişkin popülasyonları arasındaki adversreaksiyonlardaki sayısal farklılıklara katkısı göz ardı edilemez. Pediyatrik popülasyonda aşağıdaki yan etkiler yetişkin popülasyona göre daha sık (>%10) rapor edilmiştir: TENİPRA ile tedavi edilen hastaların %28'inde, interferon beta-1a ile tedavi edilen hastaların ise %36'sında baş ağrısı bildirilmiştir. TENİPRA ile tedavi edilen hastaların %74'ünde, interferon beta-1a ile tedavi edilen hastalarda ise %31'inde gastrointestinal bozukluklar bildirilmiştir. Bunlar arasında karın ağrısı ve kusmaTENİPRA ile en sık bildirilenler olmuştur. TENİPRA ile tedavi edilen hastaların %32'sinde, interferon beta-1a ile tedavi edilen hastaların ise %11'inde solunum, göğüs ve mediastinal bozukluklar bildirilmiştir. Bunlar arasındaorofaringeal ağrı ve öksürük TENİPRA ile en sık bildirilenler olmuştur. Dismenore, TENİPRA ile tedavi edilen hastaların %17'sinde ve interferon beta 1a ile tedavi edilen hastaların %7'sinde rapor edilmiştir. 13 ila 17 yaşları arasındaki RRMS'li pediyatrik hastalarda (7 gün boyunca günde iki kez 120 mg, ardından tedavinin geri kalanı için günde iki kez 240 mg; güvenlik popülasyonu, n=22)yapılan 24 haftalık küçük, açık etiketli, kontrolsüz bir çalışmada, 96 haftalık bir uzatmaçalışmasının ardından (günde iki kez 240 mg; güvenlik popülasyonu n=20), güvenlik profiliyetişkin hastalarda gözlenene benzer görünmüştür. 10 ila 12 yaş arasındaki çocuklarda sınırlı veri mevcuttur. TENİPRA'nın 10 yaşından küçük çocuklarda güvenliliği ve etkililiği henüz belirlenmemiştir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonların raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu Türkiye Farmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9 Doz aşımı ve tedavisiDimetil fumarat ile doz aşımı vakaları bildirilmiştir. Bu vakalarda tanımlanan semptomlar, TENİPRA'nın bilinen advers reaksiyon profili ile uyumludur. TENİPRA'nın eliminasyonunuarttırmak için bilinen terapötik bir müdahale yoktur ve bilinen bir antidot bulunmamaktadır.Doz aşımı durumunda, klinik olarak belirtildiği gibi semptomatik destekleyici tedaviyebaşlanması önerilmektedir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: Diğer immünosüpresanlar ATC kodu: L04AX07 Etki mekanizmasıMultipl sklerozda, dimetil fumarat tarafından sergilenen terapötik etki mekanizması tam olarak anlaşılamamıştır. Klinik öncesi çalışmalar, dimetil fumaratın farmakodinamikyanıtlarının esas olarak Nükleer faktör (eritroid-derived 2)-like 2 (Nrf2) transkripsiyonyolağının aktivasyonu aracılığıyla gerçekleştiğini göstermektedir. Dimetil fumaratın,hastalarda Nrf2-bağımlı antioksidan genlerini regüle ettiği gösterilmiştir (örn. NAD(P)Hdehidrojenaz, kinon 1; [NQO1]). Farmakodinamik etkilerİmmün sistem üzerine etkilerKlinik öncesi ve klinik çalışmalarda, dimetil fumarat anti-inflamatuar ve immünomodülatör özellikler göstermiştir. Dimetil fumarat ve dimetil fumaratın primer metaboliti monometilfumarat, klinik öncesi modellerde inflamatuvar uyaranlara yanıt olarak immün hücreaktivasyonunu ve sonrasında meydana gelen pro-inflamatuvar sitokinlerin salınımınıazaltmıştır. Psöriyazisi olan hastalarda yapılan klinik çalışmalarda, dimetil fumarat pro-inflamatuvar sitokin profillerinin (T HHH2) eğilim göstermiştir. Dimetil fumarat, multipleinflamatuvar ve nöroinflamatuvar hasar modelinde terapötik aktivite göstermiştir. MShastalarda, Faz 3 çalışmalarda, (DEFINE, CONFIRM ve ENDORSE), Dimetil fumarat iletedavi, ortalama lenfosit sayımlarından sonra ortalama bir plato göstermiştir; bu, ilk yılboyunca başlangıç değerlerinin yaklaşık %30'u kadar azalmış ve ardından bir plato izlemiştir.Bu çalışmalarda, lenfosit sayıları normalin alt smmmn (LLN, 910 hücre/mm3) altındayken TENİPRA tedavisini bırakan hastalar, lenfosit sayılarının LLN'ye geri kazanılması içinizlenmiştir. Şekil 1, uzamış şiddetli lenfopeni olmaksızın Kaplan-Meier yöntemine dayalı olarak LLN'ye ulaştığı tahmin edilen hastaların oranını göstermektedir. İyileşme başlangıç çizgisi (RBL),TENİPRA'nın kesilmesinden önceki tedavideki son ALC olarak tanımlandı. RBL'de hafif,orta veya şiddetli lenfopenisi olan 12. Hafta ve 24. Haftada LLN'ye (ALC > 0.9 x 109//L)iyileşen hastaların tahmini oranı, %95 noktasal güven aralıklarıyla Tablo 1, Tablo 2 ve Tablo3'te sunulmaktadır. Hayatta kalma fonksiyonunun Kaplan-Meier tahmin edicisinin standarthatası Greenwood formülü kullanılarak hesaplanır. Şekil 1: Kaplan-Meier Metodu; İyileşme Başlangıç Noktasından (RBL) > 910 hücre/mm3 LLN'ye İyileşen Hastaların Oranı Tablo 1: Kaplan-Meier Yöntemi; Uzamış şiddetli lenfopenisi olan hastalar hariç, LLN'ye ulaştığı tahmin edilen hastaların oranı, iyileşme başlangıcında (RBL) hafif lenfopeni
aUzun süreli şiddetli lenfopenisi olan hastalar hariç olmak üzere, RBL'de ALC < 910 ve > 800 hücre/mm3 olan hastalar. Tablo 2: Kaplan-Meier Yöntemi; Uzamış şiddetli lenfopenisi olan hastalar hariç, LLN'ye ulaştığı tahmin edilen hastaların oranı, iyileşme başlangıcında (RBL) orta derecede lenfopeni
Tablo 3: Kaplan-Meier Yöntemi; Uzamış şiddetli lenfopenisi olan hastalar hariç, LLN'ye ulaştığı tahmin edilen hastaların oranı, iyileşme başlangıcında (RBL) şiddetli lenfopeni
Klinik etkililik ve güvenlilikRelapsing-remitting multipl sklerozlu (RRMS) hastalar ile iki adet, 2 yıllık, randomize, çift kör, plasebo kontrollü çalışma [1234 hasta ile DEFINE ve 1417 hasta ile CONFIRM]yapılmıştır. MS'in ilerleyici formları olan hastalar bu çalışmalara dahil edilmemiştir. Etkililik(bkz. aşağıdaki tablo) ve güvenlilik, randomizasyondan önceki bir yıllık süreçte en az 1 nüksyaşayan hastalarda veya randomizasyondan önce altı haftada en az bir gadolinyum-tutan(Gd+) lezyon geliştirerek bir beyin Manyetik Rezonans Görüntülemesi (MRG) yapılanhastaları kapsayan 0'dan 5'e değişen Genişletilmiş Engellilik Durum Ölçeği (EDSS) skorluhastalarda kanıtlanmıştır. CONFIRM, glatiramer asetatın referans karşılaştırıcısı olarakdeğerlendirici bir kör (örn. tedavi çalışmasının sonuçlarını değerlendiren çalışmahekimi/araştırmacı körleştirilmiştir) içermiştir. DEFINE'da, hastalar aşağıdaki medyan başlangıç özelliklerine sahipti: yaş 39, hastalık süresi 7.0 yıl, EDSS skoru 2.0. Ek olarak, hastaların %16'smm EDSS puanı >3.5'dir ve %28'i öncekiyıl en az 2 nüks yaşamış ve %42'si halihazırda onaylanmış başka MS tedavileri almıştır. MRGkohortunda, çalışmaya giren hastaların %36'sında başlangıçta Gd+ lezyonları mevcuttur(ortalama Gd+ lezyon sayısı 1,4). CONFIRM'de, hastalar şu özellikleri sergilemiştir: yaş 37, hastalık süresi 6,0 yıl, EDSS skoru 2,5. Buna ek olarak, hastaların %17'si EDSS skoru >3,5'dir ve %32'si önceki yıl en az 2 nüksgeçirmiş ve %30'u önceden başka onaylı MS tedavileri almıştır. MRG kohortunda, çalışmayagiren hastaların %45'inde başlangıçta Gd+ lezyonları mevcuttur (ortalama Gd+ lezyon sayısı2,4). Plaseboyla karşılaştırıldığında, dimetil fumarat ile tedavi edilen hastalarda şu değerlerde klinik açıdan anlamlı ve istatistiksel açıdan önemli bir azalma gözlenmiştir: DEFINE'daprimer sonlanım noktası, 2 yılda relaps nüks yapan hasta oranı; ve Çalışma 2'de primersonlanım noktası, 2 yılda yıllık nüks oranı (ARR). CONFIRM'de; glatiramer asetat ve plasebo için ARR, onaylanmış kısa ürün bilgisi ve kullanma talimatına uygun olarak %29'luk bir azalmaya karşılık gelecek şekilde, sırasıyla0,286 ve 0,401 olarak belirlenmiştir.
a Klinik sonlanım noktalarının bütün analizleri tedaviye gönüllü hastalarda yapılmıştır b MRG analizlerinde MRG kohortu kullanılmıştır * P-değeri <0.05 ** P-değeri <0.01 *** P-değeri <0.0001 ^istatistiksel açıdan anlamlı değil Kontrolsüz 8 yıllık bir uzatma çalışmasına (ENDORSE), pivot çalışmalardan (DEFINE ve CONFIRM) 1.736 uygun RRMS hastasını kaydetmiştir. Çalışmanın birincil amacı, RRMS'lihastalarda Dimetil fumarat'ın uzun vadeli güvenliğini değerlendirmekti. 1.736 hastanınyaklaşık yarısı (909, %52) 6 yıl veya daha uzun süre tedavi görmüştür. 3 çalışmanıntamamında 501 hasta sürekli olarak günde iki kez 240 mg Dimetil fumarat ile tedavi edildi vedaha önce DEFINE ve CONFIRM çalışmalarında plasebo ile tedavi edilen 249 hasta,ENDORSE çalışmasında günde iki kez 240 mg tedavi aldı.Sürekli olarak günde iki kez tedavialan hastalar 12 yıla kadar tedavi edildi. ENDORSE çalışması sırasında, günde iki kez 240 mg Dimetil fumarat ile tedavi edilen tüm hastaların yarısından fazlasında nüks olmadı. 3 çalışmanın tamamında sürekli olarak gündeiki kez tedavi edilen hastalar için, ayarlanmış ARR, DEFINE ve CONFIRM çalışmalarında0.187 (95% GA: 0.156, 0.224) ve ENDORSE çalışmasında 0.141 (95% GA: 0.119, 0.167)'dir. Daha önce plasebo ile tedavi edilen hastalar için, DEFINE ve CONFIRM çalışmalarında0,330 olan düzeltilmiş ARR (95% GA: 0.266, 0.408), ENDORSE çalışmasında 0,149 (95%GA: 0.116, 0.190)'a düşmüştür. ENDORSE çalışmasında, hastaların çoğu (> %75) sakatlığın ilerlemesini doğrulamamıştır (6 aylık kalıcı sakatlık ilerlemesi olarak ölçülmüştür). Üç çalışmadan elde edilen havuzlanmışsonuçlar, Dimetil fumarat ile tedavi edilen hastaların, ENDORSE genelinde ortalama EDSSpuanlarında hafif bir artış ile tutarlı ve düşük doğrulanmış engellilik ilerleme oranlarına sahipolduğunu göstermiştir. MRI değerlendirmeleri (6 yıla kadar, daha önce DEFINE veCONFIRM çalışmalarının MRI kohortuna dahil edilmiş 752 hasta dahil olmak üzere)hastaların çoğunluğunda (yaklaşık %90) Gd'yi artıran lezyonları olmadığını göstermiştir. 6 yılboyunca, yeni veya yeni genişleyen T2 ve yeni T1 lezyonlarının yıllık düzeltilmiş ortalamasayısı düşük kalmıştır. Yüksek hastalık aktivitesi gösteren hastalarda etkililik- 3 aylık uzamış engellilik progresyonuna kadar geçen zamana etkisi net bir şekildebelirlenmemişken, yüksek hastalık aktivitesine sahip bir hasta alt grubunda nüksler üzerineuygun bir tedavi etkisi gözlenmiştir. Çalışmaların dizaynı nedeni ile, yüksek hastalıkaktivitesi aşağıdaki şekilde tanımlanmıştır: - Bir yılda 2 veya daha fazla nüks yaşayan ve beyin MRI'sında bir veya daha fazla Gd tutanlezyon gösteren hastalar (DEFINE'da n=42; CONFIRM'de n=51) veya - Tedavi sırasında önceki yıl en az 1 nüks yaşayan ve kraniyal MRI'da en az 9 T2-hiperintenslezyonu veya en az 1 Gd-tutan lezyonu olan, tam ve yeterli bir beta-interferon tedavisine(en az bir yıllık tedavi) yanıt vermemiş olan hastalar veya önceki 2 yıla kıyasla son biryılda nüks oranı aynı kalan veya artan hastalar (DEFINE'da n=177; CONFIRM'de n=141). Pediyatrik popülasyonDimetil fumarat'ın pediyatrik RRMS'deki güvenliliği ve etkililiği, 10 ila 18 yaş arası RRMS'li hastalarda randomize, açık etiketli, aktif kontrollü (interferon beta-1a) paralel grupçalışmasında değerlendirilmiştir. Yüz elli hasta, 96 hafta boyunca dimetil fumarata (240 mgBID oral) veya interferon beta-1a'ya (haftada bir 30 gg IM) randomize edilmiştir. Birincil sonnokta, 96. haftada beyin MRI taramalarında yeni veya yeni büyüyen T2 hiperintens lezyonlarıolmayan hastaların oranıydı. Ana ikincil son nokta, 96. haftada beyin MRI taramalarında yeniveya yeni genişleyen T2 hiperintens lezyonlarının sayısıydı. Tanımlayıcı istatistikler, birincilson nokta için önceden planlanmış doğrulayıcı bir hipotez olmadığı için sunulmuştur. Başlangıca göre 96. haftada yeni veya yeni genişleyen T2 MRI lezyonu olmayan ITT popülasyonunda hastaların oranı dimetil fumarat için %12.8 iken interferon beta-1a grubunda%2.8 idi. 96. Haftada başlangıca göre yeni veya yeni genişleyen T2 lezyonlarının ortalamasayısı, başlangıç T2 lezyon sayısı ve yaşa göre ayarlanmış (MRI ölçümleri olmayan hastalarhariç ITT popülasyonu), dimetil fumarat için 12.4 ve interferon beta-1a için 32.6'dır.96haftalık açık etiketli çalışma döneminin sonunda klinik nüks olasılığı dimetil fumaratgrubunda %34 ve interferon beta-1a grubunda %48 idi. Dimetil fumarat alan pediyatrikhastalarda (13 yaşından 18 yaşına kadar) güvenlilik profili, daha önce yetişkin hastalardagözlemlenenle niteliksel olarak tutarlıydı (bkz. bölüm 4.8). 5.2 Farmakokinetik özelliklerOral olarak uygulanan dimetil fumarat esterazlar tarafından presistemik olarak hızlıca hidrolize uğrar ve aktif olan monometil fumarat adlı primer metabolitine dönüştürülür.Dimetil fumarat, TENİPRA'nın oral olarak alınmasından sonra plazmada ölçülemez. Bunedenle dimetil fumarat ile ilgili tüm farmakokinetik analizler plazma monometil fumaratkonsantrasyonlarıyla gerçekleştirilmiştir. Farmakokinetik veriler, multipl sklerozlu ve sağlıklıgönüllülerden oluşan deneklerden elde edilmiştir. Emilim:Monometil fumaratın Tmaks değeri 2 ila 2,5 saattir. Dimetil fumarat, enterik sert kapsül, enterik bir kaplama tarafından korunan mikrotabletler içerdiğinden, emilim kapsüller mideyi terkedene kadar başlamaz (genellikle 1 saatten az). Günde iki kez yiyeceklerle birlikte alınan 240mg'ın ardından, multipl sklerozlu deneklerde medyan pik (Cmaks) 1,72 mg/L ve genel eğrialtındaki alan (EAA) maruziyet 8,02 saat.mg/L olmuştur. Genel olarak, çalışılan dozaralığında (120 mg ila 360 mg) Cmaks ve EAA, yaklaşık olarak dozla orantılı şekilde artmıştır.Multipl sklerozlu hastalarda, iki 240 mg'lık doz bir günlük doz rejimi olarak 4 saat araylagünde üç kez uygulanmıştır. Bu durum, hiçbir güvenlik önlemi olmaksızın günde iki kezuygulanan dozlamaya kıyasla %12'lik medyan Cmaks'ında bir artış sağlayarak maruziyetinminimum birikimi ile sonuçlanmıştır (günde üç kez yapılan uygulama için 1,93 mg/L'ye karşı,günde iki kez yapılan uygulama için 1,72 mg/L). Yiyecek, dimetil fumaratın maruziyeti üzerine klinik açıdan bir etki göstermemektedir. Bununla birlikte, dimetil fumarat kızarma veya gastrointestinal advers olaylar ile ilgiligelişmiş tolere edilebilirlik nedeni ile yiyeceklerle birlikte alınmalıdır (bkz. Bölüm 4.2). Dağılım:240 mg dimetil fumaratın oral yolla alınmasının ardından görülen dağılım hacmi 60 L ve 90 L arasında değişmektedir. Monometil fumaratın insan plazma proteinine bağlanma oranıgenelde %27 ile %40 arasındadır. Biyotransformasyon:İnsanlarda, dimetil fumarat büyük ölçüde metabolize olup %0,1'den azı idrarda değişmemiş olarak atılmaktadır. Dimetil fumarat, sistemik dolaşıma ulaşmadan önce ilk olarakgastrointestinal kanal, kan ve dokularda bulunan esterazlarla metabolize olur. Başka birmetabolizma, sitokrom P450 (CYP) sisteminin dahil olmadığı, trikarboksilik asit döngüsüaracılığı ile meydana gelmektedir. Tek bir 240 mg 14C-dimetil fumarat dozu ile yapılançalışmada, glukoz insan plazmasındaki baskın metabolit olarak tanımlanmıştır. Dolaşımdakidiğer metabolitleri fumarik asit, sitrik asit ve monometil fumarattır. Fumarik asitin daha sonrametabolizması trikarboksilik asitten CO2'in uzaklaştırılması ile olur ve bu eliminasyonunprimer yoludur. Eliminasyon:CO2 uzaklaştırılması, dozun %60'ının eliminasyonundan sorumlu olan primer dimetil fumarat eliminasyon yoludur. Renal ve fekal eliminasyon, sırasıyla dozun %15,5'i ve %0,9'unuelimine eden sekonder eliminasyon yollarıdır. Monometil fumaratın terminal yarı ömrü kısadır (yaklaşık 1 saat) ve monometil fumarat bireylerin çoğunda 24 saatte dolaşımda kalmaz. Terapötik rejimde çoklu dimetil fumaratdozları ile ana ilaç veya monometil fumarat birikimi meydana gelmez. Doğrusallık / Doğrusal olmayan durumTek ve çoklu 120 mg ve 360 mg'lık doz aralığında çalışılmış olan dimetil fumaratın maruziyeti, dozla orantılı bir biçimde artmaktadır. Özel hasta gruplarındaki farmakokinetik özelliklerVaryans Analizi (ANOVA) bulgularına dayalı olarak, Relapsing Remitting Multipl Skleroz (RRMS) hastalarında maruziyetin ana orta değişkeni (Cmaks ve EAA'ya göre) vücut ağırlığıdır.Ancak klinik çalışmalarda değerlendirilen güvenlilik ve etkililik ölçümlerini etkilememiştir. Cinsiyet ve yaş, dimetil fumaratın farmakokinetik özellikleri üzerine klinik açıdan önemli bir etki göstermemiştir. 65 yaş ve üzeri hastalarda farmakokinetik özellikler çalışılmamıştır. Pediyatrik popülasyonGünde iki kez 240 mg kullanılan dimetil fumaratın farmakokinetik profili, RRMS'li 13-17 yaş arası pediyatrik hastada yapılan küçük, açık uçlu, kontrolsüz bir çalışmada değerlendirilmiştir(n=21). Dimetil fumaratın bu adolesan hastalardaki farmakokinetiği daha öncesinde yetişkinhastalarda gözlenenler ile tutarlıdır. (Cmaks: 2,00±1,29 mg/l; EAAo-i2sa: 3,62±1,16 sa.mg/l, tambir günlük EAA: 7,24 sa.mg/l'ye denk gelen). Böbrek yetmezliğiRenal yolak, dimetil fumarat için uygulanan dozun %16'sından azının atıldığı sekonder bir eliminasyon yolu olduğundan, böbrek yetmezliği olan bireylerde farmakokinetik özelliklerideğerlendirilmemiştir. Karaciğer yetmezliğiDimetil fumarat ve monometil fumarat, CYP450 sistemi dahil olmadan esterazlar tarafından metabolize edildiğinden, karaciğer bozukluğu olan bireylerde farmakokinetik özellikdeğerlendirmesi yürütülmemiştir. 5.3 Klinik öncesi güvenlilik verileriAşağıdaki toksikoloji ve üreme toksisitesi bölümlerinde açıklanan advers reaksiyonlar, klinik çalışmalarda gözlenmemiştir. Ancak klinik maruziyet düzeylerine benzer maruziyetdüzeylerinde hayvanlarda görülmüştür. Mutajenezis:Dimetil fumarat ve monometil fumarat bir seri in vitroin vivomikroçekirdektestinde negatif sonuç vermiştir.Karsinojenezis:Dimetil fumaratın karsinojenite çalışmaları, fare ve sıçanlarda 2 yıla varan bir süre boyunca gerçekleştirilmiştir. Dimetil fumarat farelere 25, 75, 200 ve 400 mg/kg/gün ve sıçanlara 25,50, 100, ve 150 mg/kg/gün'lük dozlarda oral yolla uygulanmıştır. Farelerde, önerilen insandozuna eşdeğer maruziyette (EAA) 75 mg/kg/gün dozunda renal tübüler karsinom insidansıartmıştır. Sıçanlarda, renal tübüler karsinom ve testiküler Leydig hücre adenomu insidansı,önerilen insan dozundan yaklaşık 2 kat daha yüksek, 100 mg/kg/gün'de artmıştır. Bubulguların insandaki risk ile ilişkisi bilinmemektedir. Farelerde, önerilen insan dozu ile eşdeğer maruziyette ve sıçanlarda önerilen insan dozu maruziyetinin altında, nonglandüler midede (ön mide) skuamöz hücre papilloma vekarsinomu insidansı artmıştır (EAA baz alınarak). Kemirgenlerdeki ön midenin insanlarda birkarşılığı yoktur. Toksikoloji:Kemirgenlerde, tavşanlarda ve maymunlarda oral gavaj yoluyla uygulanan dimetil fumarat süspansiyonu (%0,8 hidroksipropil metilselülozda dimetil fumarat) ile klinik dışı çalışmalaryürütülmüştür. Kronik köpek çalışması, dimetil fumarat kapsülün oral yolla uygulanmasıylagerçekleştirilmiştir. Farelerde, sıçanlarda, köpeklerde ve maymunlarda dimetil fumaratın tekrarlı şekilde oral alınmasından sonra böbrek üzerinde etkileri gözlenmiştir. Hasarı düşündüren renal tübülepitelyum rejenerasyonu tüm türlerde gözlenmiştir. Renal tübüler hiperplazisi yaşam boyudozlama ile (2 yıllık çalışma) sıçanlarda gözlenmiştir. 11 ay boyunca dimetil fumaratın günlükoral dozlarını alan köpeklerde kortikal atrofi için hesaplanan marj, EAA'ya dayalı olarakönerilen dozun 3 katında gözlenmiştir. 12 ay boyunca dimetil fumaratın günlük oral dozlarınıalan maymunlarda tek hücre nekrozu, EAA'ya dayalı olarak önerilen dozun 2 katındagözlenmiştir. İnterstisyel fibrozis ve kortikal atrofi, EAA'ya dayalı olarak önerilen dozun 6katında gözlenmiştir. Bu bulguların insanlarla ilişkisi bilinmemektedir. Testislerdeki seminiferöz epitelin dejenerasyonu sıçanlarda ve köpeklerde görülmüştür. Bulgular sıçanlarda yaklaşık olarak önerilen dozda ve köpeklerde önerilen dozun 3 katında(EAA'ya dayalı olarak) gözlenmiştir. Bu bulguların insanlarla ilişkisi bilinmemektedir. 3 ay ya da daha uzun süreli çalışmalarda, farelerin ve sıçanların ön midesinde skuamöz epitel hiperplazisi ve hiperkeratozu; inflamasyonu ve skuamöz hücre papilloma ve karsinomuolduğu gözlenmiştir. Fare ve sıçanlardaki ön midenin insanlarda bir karşılığı yoktur. Üreme toksisitesi:Çiftleşme öncesinde veya sırasında 75, 250 ve 375 mg/kg/gün'de erkek sıçanlara oral dimetil fumarat uygulaması, test edilen en yüksek doza kadar (EAA'ya dayalı olarak önerilen dozunen az 2 katı) erkek fertilitesi üzerine etki göstermemiştir. Çiftleşme öncesinde ve sırasında vegebeliğin 7. gününe kadar devam eden 25, 100 ve 250 mg/kg/gün'de dişi sıçanlara oral dimetilfumarat uygulaması, 14 günlük östrus evresi sayısını azaltmış ve test edilen en yüksek dozda(EAA'ya dayalı olarak önerilen dozun 11 katı) uzamış diöstrus gözlenen hayvan sayısınıarttırmıştır. Bununla birlikte, bu değişimler fertiliteyi veya oluşan canlı fetüs sayısınıetkilememiştir. Sırasıyla 0,48 ila 0,64 ve 0,1'lik fetal/maternal plazma konsantrasyonu oranları ile dimetil fumaratın sıçanlarda ve tavşanlarda plasenta membranından geçerek fetal kana karıştığıgösterilmiştir. Sıçanlarda veya tavşanlarda uygulanan herhangi bir dimetil fumarat dozundahiçbir malformasyon gözlenmemiştir. Organogenez dönemi boyunca gebe sıçanlara 25, 100ve 250 mg/kg/gün'lük oral dozlarda yapılan dimetil fumarat uygulaması EAA'ya dayalı olarakönerilen dozun 4 katında maternal advers etkilere yol açmış ve EAA'ya dayalı olarak önerilendozun 11 katında düşük fetüs ağırlığına ve gecikmiş osifikasyona (metatarsallar ve arka bacakfalenksleri) neden olmuştur. Daha düşük fetüs ağırlığı ve gecikmiş osifikasyon maternaltoksisiteye (azalmış vücut ağırlığı ve gıda tüketimi) sekonder kabul edilmiştir. Organogenez sırasında gebe tavşanlara 25, 75 ve 150 mg/kg/gün'lük oral dozlarda yapılan dimetil fumarat uygulaması, embriyo-fetal gelişim üzerine etki göstermemiştir. EAA'ya dayalıolarak önerilen dozun 7 katında azalmış maternal vücut ağırlığına neden olmuş ve önerilendozun 16 katında da düşük artışına yol açmıştır. Gebelik ve laktasyon sırasında sıçanlara 25, 100 ve 250 mg/kg/gün'lük oral dozlarda yapılan dimetil fumarat uygulaması EAA'ya dayalı olarak önerilen dozun 11 katında F1 nesliyavrularında daha düşük vücut ağırlıklarına ve F1 nesli erkeklerinde cinsel olgunlaşmadagecikmelere neden olmuştur. F1 nesli yavrularında fertilite üzerine etkiler gözlenmemiştir.Daha düşük yavru vücut ağırlığı maternal toksisiteye sekonder kabul edilmiştir. Jüvenil sıçanlarda, doğum sonrası (PND) 28. günden PND 90 ila 93'e kadar (insanlarda yaklaşık 3 yaş ve üzerindekilere eşdeğer) günlük oral dimetil fumarat uygulaması ile yapılaniki toksisite çalışması, yetişkin hayvanlarda gözlenene benzer şekilde böbrek ve ön midedebenzer hedef organ toksisitelerini ortaya çıkarmıştır. İlk çalışmada, dimetil fumarat, 140mg/kg/günlük en yüksek doza kadar gelişimi, nörodavranışı veya erkek ve kadındoğurganlığını etkilememiştir (pediyatrik hastalarda sınırlı EAA verilerine göre önerilen insandozunun yaklaşık 4.6 katı). Benzer şekilde, erkek jüvenil sıçanlarda yapılan ikinci çalışmada(önerilen pediyatrik dozda varsayılan EAA'nın yaklaşık 15 katı) en yüksek 375 mg/kg/gündimetil fumarat dozuna kadar erkek üreme ve aksesuar organları üzerinde hiçbir etkigözlenmemiştir. Bununla birlikte, erkek jüvenil sıçanlarda femur ve lomber vertebralardaazalmış kemik mineral içeriği ve yoğunluğu belirgindi. İn vivo olarak aynı aktif metabolitmonometil fumarata metabolize olan başka bir fumarik ester olan oral diroksimel fumaratuygulamasını takiben jüvenil sıçanlarda kemik dansitometrisi değişiklikleri de gözlenmiştir.Jüvenil sıçanlarda dansitometri değişiklikleri için NOAEL, önerilen pediatrik dozdavarsayılan EAA'nın yaklaşık 1.5 katıdır. Kemik etkilerinin daha düşük vücut ağırlığı ile ilişkisimümkündür, ancak doğrudan bir etkinin dahil edilmesi göz ardı edilemez. Kemik bulgularıyetişkin hastalar için sınırlı bir öneme sahiptir. Pediyatrik hastalar için alaka düzeyibilinmemektedir. 6. FARMASÖTİK ÖZELLİKLER6.1 Yardımcı maddelerin listesiMikrokristalin selüloz Kroskarmelloz sodyumSilikanlandırılmış mikrokristalin selülozKolloidal silikon dioksitMagnezyum stearat Metakrilik asit - Metil metakrilat kopolimer (1:1) Trietil sitrat Talk Metakrilik asit - Etil akrilat kopolimer (1:1) dispersiyon %30 Kapsül içeriği:Jelatin (sığır jelatini) Titanyum dioksit Parlak mavi FCF-FD&C Blue 1 Siyah demir oksit Sarı demir oksit 6.2 GeçimsizliklerUygulanabilir değildir. 6.3 Raf ömrü24 ay 6.4 Saklamaya yönelik özel uyarılar25°C altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriğiTENİPRA 240 mg Gastrorezistan Sert Kapsül, 56 veya 168 kapsül PVC/PE/PVDC -Alüminyum blister ambalajlarda kullanma talimatı ile karton kutuda piyasaya sunulmaktadır. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerÖzel bir gereklilik bulunmamaktadır. Kullanılmamış olan ürünler ya da atık materyaller "Tıbbi Atıkların Kontrolü Yönetmeliği" ve "Ambalaj Atıklarının Kontrolü Yönetmelikleri"ne uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİAbdi İbrahim İlaç San. ve Tic. A.Ş. Reşitpaşa Mahallesi, Eski Büyükdere Caddesi No:4 34467 Maslak /Sarıyer/ İstanbulTel: 0212 366 84 00Faks: 0212 276 20 20 8. RUHSAT NUMARASI2019/602 9. İLK RUHSAT TARİHİİlk ruhsat tarihi: 21.11.2019 Ruhsat yenilenme tarihi: - 10. KÜB'ÜN YENİLENME TARİHİ |
İlaç BilgileriTenipra 240 Mg Gastrorezistan Sert KapsülEtken Maddesi: Dimetil Fumarat Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.