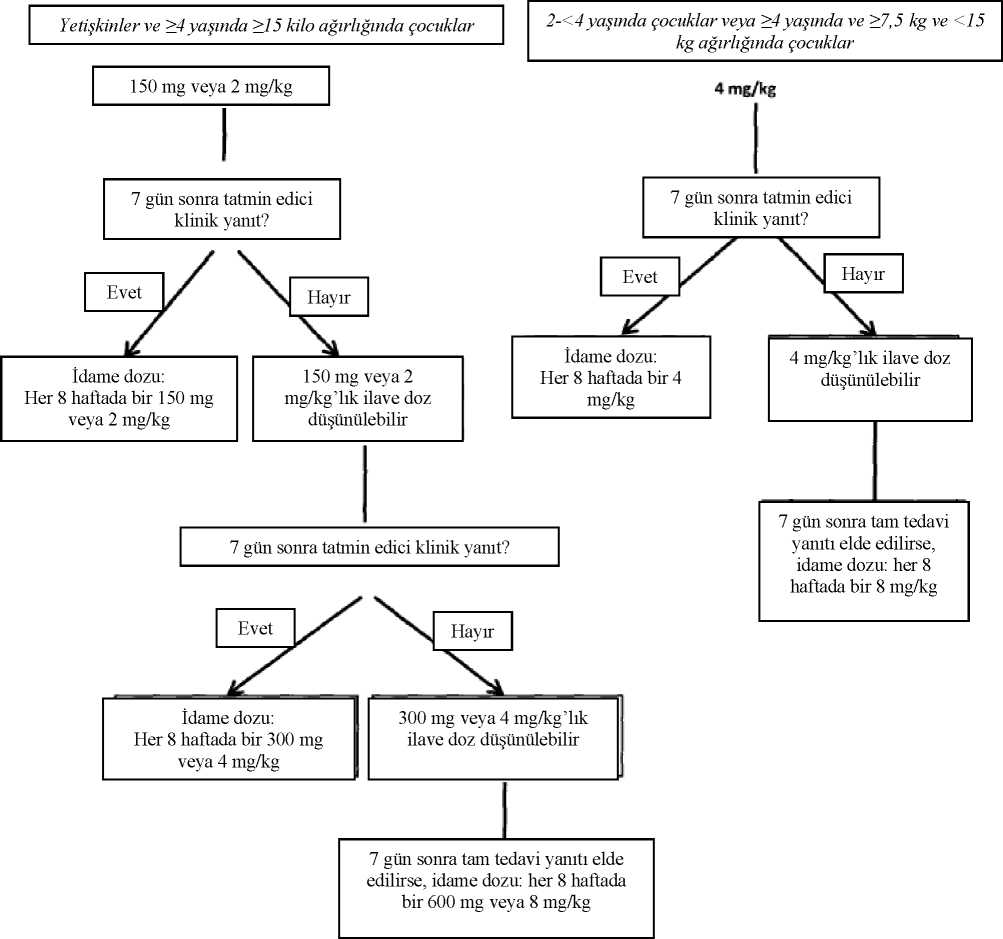
Ilaris 150 Mg/ml Enjeksiyonluk Çözelti İçin Toz İçeren Flakon Kısa Ürün BilgisiKISA ÜRÜN BILGISIBu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERI TIBBİ ÜRÜNÜN ADIILARIS 150 mg/ml enjeksiyonluk çözelti için toz içeren flakon Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her flakon 150 mg canakinumab içermektedir.Canakinumab fare hibridoma Sp2/0 hücrelerinde ekspresyonu yapılan bir rekombinant insan monoklonal antikorudur. Yardımcı maddeler:Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti için toz.Toz, beyaz bir liyofilizattır. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarPeriyodik ateş sendromlarıILARIS, yetişkinlerde, adölesanlarda ve 2 yaş ve üzerindeki çocuklarda aşağıdaki otoenflamatuar periyodik ateş sendromlarının tedavisinde endikedir: Kriyopirin ilişkili Periyodik Sendromlar (CAPS)ILARIS, vücut ağırlığı 7,5 kg ve üzerinde olan 2 yaş ve üstü çocuk ve yetişkinlerde Kriyopirin ilişkili Periyodik Sendromların (CAPS) tedavisinde endikedir: - Ailevi Soğuk Otoinflamatuar Sendrom (FCAS) / Ailevi Soğuk Ürtiker (FCU) - Muckle-Wells Sendromu (MWS) ILARIS, Neonatal Başlangıçlı Multisistem İnflamatuar Hastalık (NOMID) / Kronik İnfantil Nörolojik, Kütanöz, Artiküler Sendrom (CINCA) tedavisinde endikedir. Ailevi Akdeniz ateşi (FMF)ILARIS, kolşisine dirençli FMF olgularında endikedir. ILARIS, uygunsa, kolşisin ile kombinasyon halinde verilmelidir. ILARIS ayrıca Sistemik Jüvenil İdiyopatik Artrit (SJİA) tedavisinde de endikedir: Sistemik Jüvenil İdiyopatik Artrit (SJİA)ILARIS nonsteroidal antiinflamatuvar ilaçlar (NSAİİ) ve sistemik kortikosteroidler ile önceki tedaviye yeterince yanıt vermemiş 2 yaş ve üzerindeki hastalarda aktif Sistemik Jüvenilİdiopatik Artrit (SJİA) tedavisi için endikedir. ILARIS monoterapi olarak veya metotreksat ilekombinasyon halinde verilebilir. 4.2. Pozoloji ve uygulama şekliTedavi, ilgili endikasyonun tanı ve tedavisinde uzman doktorlar tarafından başlatılmalı ve kontrol edilmelidir. Pozoloji/uygulama sıklığı ve süresi:CAPSYetişkinler, ergenler ve 2 yaş veya üzeri çocuklarYetişkinler, ergenler ve > 4 yaşındaki çocuklar- Vücut ağırlığı > 40 kg olan hastalar için 150 mg - Vücut ağırlığı > 15 kg ve <40 kg olan hastalar için 2 mg/kg - Vücut ağırlığı > 7,5 kg ve <<15 kg olan hastalar için 4 mg/kg 2 ila <4 yaşındaki çocuklar:- Vücut ağırlığı > 7,5 kg olan hastalar için 4 mg/kg Bu, her sekiz haftada bir subkutan enjeksiyonla tekli doz olarak uygulanır. Başlangıç dozu 150 mg veya 2 mg/kg olan hastalar için, tedavinin başlatılmasından 7 gün sonra tatmin edici bir klinik yanıt (döküntü ve diğer jeneralize enflamatuar semptomlarda düzelme)elde edilirse her sekiz haftada bir 150mg veya 2mg/kg dozunda idame edilir. Tatmin edici birklinik yanıt elde edilmezse, 150 mg veya 2 mg/kg'da ikinci bir ILARİS dozu düşünülebilir.Ardından tam tedavi yanıtı elde edilirse her 8 haftada bir 300 mg veya 4 mg/kg'lık yoğun dozuygulama rejimi sürdürülmelidir. Dozun bu şekilde artırılmasından 7 gün sonra tatmin edici birklinik yanıt elde edilmezse, 300 mg veya 4 mg/kg'da üçüncü bir ILARIS dozu düşünülebilir.Ardından tam tedavi yanıtı elde edilirse, bireysel klinik değerlendirmeye göre her 8 haftada bir600 mg veya 8 mg/kg'lık yoğun doz uygulama rejimi sürdürülmelidir. Başlangıç dozu 4 mg/kg olan hastalar için, tedavinin başlatılmasından 7 gün sonra tatmin edici bir klinik yanıt (döküntü ve diğer jeneralize enflamatuar semptomlarda düzelme) elde edilirseher sekiz haftada bir 4mg/kg dozunda idame edilir. Tatmin edici bir klinik yanıt elde edilmezse,4 mg/kg'da ikinci bir ILARİS dozu düşünülebilir. Ardından tam tedavi yanıtı elde edilirse,bireysel klinik değerlendirmeye göre her 8 haftada bir 8 mg/kg'lık yoğun doz uygulama rejimisürdürülmelidir. 4 haftadan kısa aralıklarda veya 600 mg ya da 8 mg/kg üzerindeki dozlarda doz uygulama ile klinik deneyim kısıtlıdır. FMFYetişkinler, ergenler ve 2 yaş veya üzeri çocuklarFMF hastalarında önerilen canakinumab başlangıç dozu aşağıdaki gibidir: Vücut ağırlığı > 40 kg olan hastalar için 150 mg Vücut ağırlığı > 7,5 kg ve < 40 kg olan hastalar için 2 mg/kg Bu doz, subkutan enjeksiyon yoluyla tek bir doz olarak dört haftada bir uygulanır. Tedavi başladıktan 4 ay sonra tatmin edici bir klinik ve laboratuvar yanıt elde edilmezse, Canakinumab dozu her 4 haftada bir 300 mg (veya vücut ağırlığı < 40 kg olan hastalar için 4mg/kg) olacak şekilde arttırılmış doza geçilebilir. Klinik iyileşme olmayan hastalarda canakinumab tedavisine devam edilmesi, tedaviyi sürdüren hekim tarafından yeniden değerlendirilmelidir. SJIAVücut ağırlığı > 7,5 kg olan SJİA'lı hastalar için önerilen ILARIS dozu her dört haftada bir subkutan enjeksiyon ile uygulanan 4 mg/kg'dır (maksimum 300 mg'a kadar). Klinik düzelmeolmayan hastalarda ILARIS ile tedaviye devam, tedavi eden doktor tarafından yenidendeğerlendirilmelidir. Uygulama şekli:İlaç deri altına enjeksiyon yoluyla uygulanır. Aşağıdakiler uygun enjeksiyon bölgeleridir: üst uyluk, karın, üst kol veya kalçalar. Ağrıyı önlemek için ürün her enjekte edildiğinde farklı bir enjeksiyon bölgesi seçilmesi önerilir. Çatlakderi ve morarmış veya döküntü ile kaplanmış alanlardan kaçınılmalıdır. ILARIS'e yetersizmaruz kalmaya neden olabileceğinden yara izi dokusuna enjeksiyondan kaçınılmalıdır. Her flakon tek bir hastada tek doz olarak tek kullanımlıktır. Enjeksiyon tekniği ile ilgili uygun eğitim verildikten sonra, hekim uygun görürse ve gerektiği durumlarda tıbbi takiple birlikte hasta veya hastanın bakımı ile ilgilenen kişi ILARIS'i enjekteedebilir (bkz. Bölüm 6.6). Sulandırılmış çözeltinin hazırlanması ve kullanılması ile ilgili talimatlar için bkz. Bölüm 6.6. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek yetmezliği olan hastalarda doz ayarlaması gerekli değildir. Bununla beraber, bu hastalardaki klinik deneyim sınırlıdır. Karaciğer yetmezliği:Karaciğer yetmezliği olan hastalarda özel olarak yapılmış klinik çalışma mevcut değildir. Canakinumab bir insan IgG immünoglobulini olduğundan, karaciğer yetmezliğinincanakinumab farmakokinetiğini etkilemesi beklenmemektedir. Pediyatrik popülasyon:ILARIS'in 2 yaş altındaki CAPS'lı hastalar, FMF'li hastalar ve SJİA'lı hastalarda güvenlilik ve etkililiği belirlenmemiştir. Halihazırda mevcut veriler bölüm 4.8, 5.1 ve 5.2'de açıklanmaklabirlikte pozolojiye ilişkin bir öneride bulunulamaz. Geriyatrik popülasyon:Yaşlı hastalarda doz ayarlaması gerekli değildir. Bununla birlikte, bu hastalardaki klinik deneyim sınırlıdır. 65 yaş üstü hastalarda klinik deneyim sınırlı olduğundan dikkatli olunması tavsiye edilir. 4.3. KontrendikasyonlarILARIS, etkin madde ya da bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı bilinen aşırı duyarlılığı olan hastalarda kontrendikedir. Aktif, ciddi enfeksiyon (bkz Bölüm 4.4) 4.4. Özel kullanım uyarıları ve önlemleriİzlenebilirlik:Biyoteknolojik ürünlerin takip edilebilirliğinin sağlanması için uygulanan ürünün ticari ismi ve seri numarası mutlaka hasta dosyasına kaydedilmelidir. ILARIS ciddi enfeksiyon insidansının artması ile bağlantılıdır. Bu nedenle, ILARIS tedavisi sırasında ve sonrasında enfeksiyon bulguları ve belirtileri için hasta dikkatlice kontroledilmelidir. Enfeksiyonlu, nüksetmiş enfeksiyon öyküsü bulunan ya da hastayı enfeksiyonayatkın hale getiren durumların gözlendiği hastalara ILARIS uygulanırken dikkatli olunmalıdır.Tıbbi girişim gerektiren aktif bir enfeksiyonun gözlendiği hastalarda ILARIS ile tedaviyebaşlanmamalı ya da devam edilmemelidir. ILARIS tedavisi sırasında izole olağandışı veya fırsatçı enfeksiyon vakaları (aspergilloz, atipik mikobakteriyel enfeksiyonlar, herpes zoster dahil) bildirilmiştir. ILARIS'in bu olaylarlanedensel ilişkisi olasılık dışı bırakılamamaktadır. ILARIS'in tümör nekroz faktörü (TNF) inhibitörleri ile birlikte kullanılması önerilmemektedir; çünkü bu ciddi enfeksiyon riskini arttırabilir (bkz. Bölüm 4.5). Tüberküloz taraması:Klinik çalışmalarda bir PPD deri testi ile test edilmiş CAPS hastalarının yaklaşık %12'sinde, ILARIS tedavisi sırasında latent ya da aktif bir tüberküloz enfeksiyonu ile ilgili klinik kanıtarastlanmasa da, takip testinde pozitif bir sonuç elde edilmiştir. ILARIS gibi interlökin-1 (IL-1) inhibitörlerinin kullanılmasının tüberküloz için reaktivasyon riskini artırıp artırmadığı bilinmemektedir. Tedavi başlamadan önce, tüm hastalar hem aktifhem de latent tüberküloz enfeksiyonu açısından değerlendirilmelidir. Özellikle yetişkinlerde budeğerlendirme detaylı bir tıbbi öyküyü kapsamalıdır. Uygun tarama testleri (örn., tüberkülinderi testi, interferon gama salınım analizi [IGRA] veya göğüs röntgeni) tüm hastalardayürütülmelidir (yerel öneriler uygulanabilir). Hastalar ILARIS tedavisi sırasında ve sonrasındatüberküloz işaret ve semptomları açısından yakından takip edilmelidir. Tüm hastalar ILARIStedavisi sırasında tüberkülozu düşündüren belirti veya semptomların (örn., persistan öksürük,kilo kaybı, subfebril vücut sıcaklığı) ortaya çıkması durumunda tıbbi öneriye başvurmaları içinyönlendirilmelidir. PPD testinin negatiften pozitife dönmesi durumunda, özellikle yüksek risklihastalarda tüberküloz enfeksiyonu için alternatif tarama yöntemleri düşünülmelidir. Nötropeni ve lökopeni:ILARIS'in de dahil olduğu IL-1'i inhibe eden ilaçlar ile nötropeni (mutlak nötrofil sayımı [ANC] < 1,5 x 109/L) ve lökopeni gözlenmiştir. Nötropenisi ya da lökopenisi olan hastalardaILARIS tedavisi başlatılmamalıdır. Nötrofil sayımları dahil beyaz kan hücrelerinin tedavibaşlatılmadan önce ve tedaviden 1 ila 2 ay sonra değerlendirilmesi önerilmektedir (bkz. Bölüm4.8). Kronik ya da tekrarlayan tedaviler için, beyaz kan hücrelerinin periyodik olarak tedavisüresince değerlendirilmesi önerilmektedir. Eğer hasta nötropenik ya da lökopenik olursa,beyaz kan hücreleri sayımı yakından izlenmeli ve tedavinin bırakılması düşünülmelidir. Maligniteler:ILARIS ile tedavi edilen hastalarda malignite olayları bildirilmiştir. Anti-interlökin (IL-1) tedavisi ile malignitelerin gelişme riski bilinmemektedir. Aşırı duyarlılık reaksiyonları:ILARIS tedavisi ile aşırı duyarlılık reaksiyonları kaydedilmiştir. Bu vakaların çoğunluğu hafif şiddettedir. ILARIS için 2600'ün üzerinde hasta ile yapılan klinik geliştirme sırasında, hiç biranafilaktik ya da anafilaktoid reaksiyon kaydedilmemiştir. Diğer yandan, enjekte edilenproteinler için nadir olmayan aşırı duyarlılık reaksiyonları riski gözardı edilemez. (bkz. Bölüm4.3). Klinik çalışmalarda geçici ve asemptomatik olarak serum transaminaz ya da bilirubin düzeylerinde yükselme vakaları kaydedilmiştir (bkz. Bölüm 4.8). Aşılar:ILARIS alan hastalarda canlı aşılar aracılığıyla ikincil enfeksiyon bulaşması ile ilgili veri mevcut değildir. Bu nedenle, yararların açık bir şekilde risklerden daha ağır basmadığıdurumlarda, ILARIS ile eş zamanlı olarak canlı aşılar uygulanmamalıdır (bkz. Bölüm 4.5). ILARIS tedavisi başlatılmadan önce, yetişkin ve pediyatrik hastalara pnömokokkal aşı ve inaktive grip aşısı da dahil olmak üzere önerilmiş olan tüm aşılar uygulanmalıdır. CAPS hastalarında NLRP3 genindeki mutasyon:NLRP3 geninde doğrulanmış bir mutasyonun olmadığı CAPS hastalarından elde edilen deneyim sınırlıdır. SJİA'lı hastalarda makrofaj aktivasyon sendromu:Makrofaj aktivasyon sendromu (MAS) özellikle SJİA olmak üzere romatoid durumların görüldüğü hastalarda gelişebilecek yaşamı tehdit eden, bilinen bir hastalıktır. Eğer MASoluşursa veya bundan şüphe edilirse, değerlendirme ve tedaviye mümkün olduğunca erkenbaşlanmalıdır. Hekimler MAS'ı tetiklediği bilindiğinden, enfeksiyon ve SJİA ağırlaşmasınailişkin semptomlara dikkat etmelidir. Klinik çalışma deneyimine dayalı olarak ILARIS, SJİA'lıhastalarda MAS insidansını artırır görünmemekle birlikte, kesin bir sonuç çıkarılamamaktadır. Eozinofili ve sistemik semptomlarla birlikte olan ilaç reaksiyonu (DRESS):Eozinofili ve sistemik semptomlarla birlikte olan ilaç reaksiyonu, özellikle sistemik juvenil idiyopatik artritli (SJIA) hastalarda olmak üzere, ILARIS ile tedavi edilen hastalarda seyreksıklıkla bildirilmiştir. Bu durum ölümcül olabileceğinden DRESS'li hastaların hastaneyeyatırılması gerekebilir. DRESS belirti ve semptomları mevcutsa ve alternatif bir etiyolojibelirlenemiyorsa, ILARIS yeniden uygulanmamalı ve farklı bir tedavi düşünülmelidir. 4.5 Dier tıbbi ürünler ile etkileimler ve dier etkileimekilleriILARIS ile diğer tıbbi ürünler arasındaki etkileşimler resmi çalışmalarda incelenmemiştir. Başka bir IL-1 blokeri ile TNF inhibitörlerinin birlikte uygulandığı durumda ciddi enfeksiyon insidansının yükseldiği bildirilmiştir. ILARIS'in TNF inhibitörleri ile birlikte kullanılmasıönerilmez; çünkü bu durum ciddi enfeksiyon riskini arttırabilir. Karaciğer CYP450 enzimlerinin ekspresyonu, IL-1 beta gibi kronik enflamasyonu uyaran sitokinler tarafından baskılanabilir. Böylece, canakinumab gibi güçlü sitokin inhibitör tedavisiuygulandığında CYP450 ekpresyonu geri döndürülebilir. Bu, dozun kişiye göre ayarlandığı darbir terapötik indekse sahip CYP450 substratları için klinik açıdan önemlidir. Bu tip bir tıbbiürünle tedavi edilen hastalarda canakinumabın başlatılmasını takiben, etki ya da ilaç etkenmaddesinin konsantrasyonu terapötik olarak izlenmeli ve tıbbi ürünün dozu kişiye göre gereklioldukça ayarlanmalıdır. ILARIS alan hastalarda canlı aşıların etkileri ya da canlı aşılar aracılığıyla ikincil enfeksiyon bulaşması ile ilgili veri mevcut değildir. Bu nedenle yararların açık bir şekilde risklerden dahaağır basmadığı durumlarda, ILARIS ile eş zamanlı olarak canlı aşılar uygulanmamalıdır.ILARIS tedavisi başlatıldıktan sonra canlı aşılarla aşılama yapılacaksa, son ILARISenjeksiyonundan sonra ve bir sonrakinden önce en az 3 ay beklenmesi önerilmektedir (bkzBölüm 4.4). Eğer mümkünse, ILARIS ile tedavi başlatılmadan önce pediyatrik ve yetişkin hastaların tüm immünizasyonlarınm tamamlanması önerilmektedir (bkz bölüm 4.4). Sağlıklı yetişkin insanlarda yapılan bir çalışmanın sonuçları, ILARIS'in 300 mg'lık tek dozunun, grip ve meningococcus bazlı glikolize protein aşıları ile bağışıklama sonrasındaantikor cevaplarının başlamasını ve devamlılığını etkilemediğini göstermiştir. Özel popülasyonlara ilişkin ek bilgiler:Pediyatrik popülasyon:4 yaş ve daha küçük CAPS hastalarında yapılan 56 haftalık, açık etiketli bir çalışmadan elde edilen sonuçlar, standart bakım kapsamındaki canlı olmayan çocukluk dönemi aşıları yapılmıştüm hastalarda koruyucu antikor düzeylerinin geliştiğini göstermiştir. 4.6. Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi:C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü(Kontrasepsiyon).ILARIS tedavisi süresince ve son dozu takiben 3 ay etkili kontraseptikf kullanılmalıdır. Gebelik dönemiCanakinumab'ın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir .Hayvanlar üzerinde yapılan çalışmalar, gebelik/ve-veya/embriyonal/fetal gelişim/ve-veya/doğum/ve-veya doğum sonrası gelişim üzerindeki etkileri bakımından yetersizdir (bkz. Bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. Hayvan üreme çalışmalarında her zaman insandaki yanıt öngörülemediğinden, ILARIS gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Gebe olan ya da gebe kalmayı planlayan kadınlar sadece yarar-risk değerlendirmesinden sonra tedavi edilmelidir. Hayvan çalışmaları, canakinumabın plasentayı geçtiğini ve fetüste saptanabildiğini göstermektedir. İnsanda herhangi bir veri mevcut değildir, ancak canakinumab G sınıfından(IgG1) bir immünoglobulin olduğundan, insanda transplasental aktarma beklenir. Bunun kliniketkisi bilinmemektedir. Bununla birlikte, rahimde canakinumaba maruz kalan yeni doğanbebeklere, annenin doğumdan önceki son canakinumab dozunu takip eden 16 hafta boyuncacanlı aşı uygulanması önerilmez. Gebelik sırasında canakinumab alan kadınlara, yeni doğanbebeklerine herhangi bir aşı yapılmadan önce bebeğin sağlık uzmanına bilgi vermeleri talimatıverilmelidir. LaktasyonCanakinumab'ın insan sütüyle atılıp atılmadığı bilinmemektedir. Hayvanlar üzerinde yapılan çalışmalar canakinumab'ın süt ile atıldığını göstermektedir.Hayvan çalışmaları, bir mürin anti-mürin IL-1 beta antikorunun emzirilen fare yavrularının gelişimi üzerinde herhangi biristenmeyen etki yaratmadığını ve antikorun yavrulara geçtiğini göstermiştir (bkz. Bölüm 5.3).Emzirmenin durdurulup durudurulmayacağına ya da ILARIS tedavisinin durdurulupdurdurulmayacağına/tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verirken emzirmeninçocuk açısından faydası ve ILARIS tedavisinin emziren anne açısından faydası dikkatealınmalıdır. FertiliteILARIS'in insan fertilitesi üzerindeki potansiyel etkisi ile ilgili resmi çalışmalar yapılmamıştır. Canakinumabın marmosetlerdeki (C. jacchus)erkek fertilite parametreleri üzerinde bir etkisibulunmamıştır. Bir mürin anti-mürin IL-1 beta antikorunun erkek ve dişi farelerin fertilitesiüzerinde istenmeyen bir etkisi bulunmamıştır (bkz. Bölüm 5.3).4.7. Araç ve makine kullanımı üzerindeki etkilerILARIS ile tedavi baş dönmesi/vertigo ya da asteniye sebep olarak araç ve makine kullanma yeteneğini azaltabilir. ILARIS tedavisi sırasında bu tarz semptomlar yaşayan hastalar araç vemakine kullanmadan önce bu durumun tamamen ortadan kalkmasını beklemelidir. 4.8. İstenmeyen etkilerGüvenlilik profilinin özeti En sık bildirilen advers ilaç reaksiyonları başlıca üst solunum yolu olmak üzere enfeksiyonlardır. Ciddi enfeksiyonlar gözlenmekle birlikte, olayların çoğu hafif ila ortaşiddettedir. Doz ve tedavi süresinin advers ilaç reaksiyonlarının tip ya da sıklığı üzerinde biretkisi olmamıştır. ILARIS ile tedavi edilen hastalarda aşırı duyarlılık reaksiyonları bildirilmiştir (bkz. Bölüm 4.3 ve Bölüm 4.4). ILARIS ile tedavi edilen hastalarda fırsatçı enfeksiyonlar bildirilmiştir (bkz. Bölüm 4.4). İstenmeyen etkiler MedDRA sistemi organ sınıfına göre listelenmiştir. Her bir sistem organ sınıfı içinde, advers ilaç reaksiyonları ilk sırada en yaygın olan yer alacak şekilde sıklıkkategorisine göre sınıflandırılır. Sıklık kategorileri şu düzen kullanılarak belirlenmiştir: Çokyaygın (> 1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek(>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketletahmin edilemiyor). İstenmeyen etkiler her bir sıklık grubunda azalan ciddiyete göre verilmiştir. CAPS, FMF ve SJIA'da advers olayların tablo halinde özeti
SJIA havuzlanmış analiziKlinik çalışmalarda 2 ila <12 yaş arası 321 hasta, 12 ila <16 yaş arası 88 hasta ve 16 ila <20 yaş arası 36 hasta dahil olmak üzere 2 ila <20 yaş arası toplam 445 SJIA hastası ILARISalmıştır. Tüm SJIA hastalarının havuzlanmış güvenlilik analizi, 16 ila <20 yaşları arasındakigenç erişkin SJIA hastalarından oluşan alt kümede, ILARIS'in güvenlik profilinin, 16 yaşındanküçük SJIA hastalarında gözlenenlerle tutarlı olduğunu göstermiştir. Seçilmiş advers reaksiyonlarla ilgili açıklamalarCAPS hastalarında uzun vadeli veriler ve laboratuar anormallikleriCAPS hastaları üzerinde ILARIS ile gerçekleştirilen klinik çalışmalar sırasında, hemoglobin için ortalama değerler artmış ve lökosit, nötrofil ve trombosit değerleri azalmıştır. CAPS hastalarında nadiren transaminaz yükselmeleri gözlenmiştir. ILARIS ile tedavi edilmiş CAPS hastalarında, transaminazlarda eş zamanlı yükselmeler olmaksızın serum bilirubinde asemptomatik ve hafif yükselmeler gözlenmiştir. Doz yükseltme ile birlikte yapılan uzun vadeli, açık etiketli çalışmalarda, enfeksiyon (gastroenterit, solunum yolu enfeksiyonu, üst solunum yolu enfeksiyonu), kusma ve sersemlik,diğer doz grupları ile karşılaştırıldığında 600 mg ya da 8 mg/kg doz grubunda daha sıkbildirilmiştir. FMF hastalarında laboratuar anormallikleriNötrofillerHastaların % 6,5'inde (yaygın) nötrofil sayısında > Derece 2 seviyesine düşme ve hastaların % 9,5'inde Derece 1 seviyesine düşme meydana gelmiş olmakla birlikte, düşüşler genelliklegeçicidir ve nötropeni ile ilişkili enfeksiyon bir advers reaksiyon olarak tanımlanmamıştır. TrombositlerHastaların % 0,6'sında trombosit sayısında düşüş (> Derece 2) meydana gelmiş olmakla birlikte, kanama bir advers reaksiyon olarak tanımlanmamıştır. Trombositlerde hafif ve geçiciDerece 1 seviyesine düşüş hastaların % 15,9'unda meydana gelmiş ancak ilişkili herhangi birkanama advers olayı olmamıştır. SJIA hastalarında laboratuar anormallikleri HematolojiSJIA programının genelinde, 33 hastada (%16,5) lökosit (WBC) sayımlarında geçici azalma (< 0,8 x LLN) bildirilmiştir. SJIA programının genelinde, 12 hastada (%6) mutlak nötrofil sayımlarında (ANC) 1 x 109/L'den daha düşük düzeylere geçici azalmalar bildirilmiştir. SJIA programının genelinde, 19 hastada (%9,5) trombosit sayımlarında geçici azalmalar (<LLN) gözlenmiştir. ALT/ASTSJIA programının genelinde, 19 hastada (%9,5) >3 x normalin üst sınırı (ULN) düzeyinde yüksek ALT ve/veya ALT bildirilmiştir. Gözlemsel çalışmadan uzun süreli verilerToplam 243 CAPS hastası (> 2 ila < 17 yaş arası 85 pediyatrik hasta ve 18 yaş ve üzeri 158 erişkin hasta), uzun süreli bir kayıt çalışmasında (ortalama 3.8 yıl canakinumab maruziyeti)rutin klinik uygulamada canakinumab ile tedavi edilmiştir. Canakinumabın bu ortamda uzunsüreli tedaviyi takiben gözlemlenen güvenlilik profili, CAPS hastalarında yapılan girişimselçalışmalarda gözlemlenen profillerle tutarlı olmuştur. Pediyatrik popülasyonÇalışmalara dahil edilmiş 80 pediyatrik CAPS hastası (2-17 yaşları arasında) bulunmaktadır. Genel olarak, genel CAPS popülasyonu ile karşılaştırıldığında (yetişkin ve pediyatrikhastalardan oluşmaktadır, N=211), pediyatrik hastalarda enfeksiyon epizotlarının sıklığı veşiddeti de dahil olmak üzere ILARIS'in güvenlilik ve tolerabilite profilinde klinik açıdananlamlı farklılıklar yoktur. Üst solunum yolu enfeksiyonları en sık bildirilen enfeksiyonolaylarıdır. 16 haftalık bir çalışmada canakinumab alan 102 TRAPS, H IDS/MKD ve FMF hastası (2-17 yaş) yer almıştır. Genel olarak, genel popülasyona kıyasla pediyatrik hastalarda canakinumabıngüvenlilik ve tolere edilebilirlik profilinde klinik olarak anlamlı bir fark olmamıştır.Yaşlı popülasyon65 yaş ve üzeri hastalarda gözlenen güvenlilik profilinde anlamlı farklılıklar yoktur. Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr[email protected]. Doz aımıAşırı doz ile deneyim sınırlıdır. Erken klinik çalışmalarda, hastalar ve sağlıklı gönüllüler akut toksisite kanıtına rastlanmaksızın intravenöz veya subkutan yolla uygulanan 10 mg/kg'a varanyüksek dozlar almıştır. Doz aşımı durumunda hastanın advers reaksiyonlar veya etkilerinsemptomları veya belirtileri açısından izlenmeleri ve gerekli olduğunda uygun semptomatiktedavinin başlatılması önerilmektedir. 5.FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grup: İmmünosupresanlar, interlökin inhibitörleri ATC Kodu: L04AC08 Etki mekanizmasıCanakinumab, IgG1/K izotipinin tamamen insan monoklonal bir anti-human interlökin-1 beta (IL-1 beta) antikorudur. Canakinumab yüksek bir afinite ile spesifik olarak insan IL-1 betayabağlanır ve insan IL-1 betanın biyolojik aktivitesini onun IL-1 reseptörleri ile etkileşiminiengelleyerek nötralize eder; bu şekilde IL-1 beta ile indüklenen gen aktivasyonu ve interlökin-6 ya da siklooksigenaz-2 gibi enflamatuar aracıların üretimi engellenir. Farmakodinamik etkilerCAPS ve FMF:Klinik çalışmalarda, kontrolsüz aşırı IL-1 beta üretimine sahip CAPS ve FMF hastaları canakinumab ile tedaviye hızlı ve sürekli bir yanıt göstermiştir. (Örneğin; yüksek C-reaktifprotein (CRP) ve serum amiloid A (SAA) yüksek nötrofil ve trombosit sayıları ve lökositozhızla normale dönmüştür.) SJİA:Sistemik Jüvenil İdiopatik Artrit başlıca IL-1-beta olmak üzere pro-inflamatuvar sitokinler aracılığıyla doğal immünitenin neden olduğu şiddetli bir otoinflamatuar hastalıktır. SJİA'nın yaygın özellikleri ateş, döküntü, hepatosplenomegali, lenfadenopati, poliserozit ve artrittir. Canakinumab ile tedavi, SJİA'nın hem artiküler hem de sistemik özelliklerinde hızlıve kalıcı bir iyileşme sağlamış ve buna çoğu hastada inflamasyonlu eklem sayısında anlamlı birazalma, ateşte hızlı düzelme ve akut faz reaktanlarında azalma eşlik etmiştir (bkz. Kliniketkililik ve güvenlilik). Klinik etkililik ve güvenlilikCAPS:Canakinumab etkililiği ve güvenliliği, çeşitli şiddetlerde hastalığa ya da farklı CAPS fenotiplerine (FCAS/FCU, MWS ve NOMID/CINCA gibi) sahip 211 yetişkin ve pediyatrikhastalarda gösterilmiştir. Sadece NLRP3 mutasyonu doğrulanan hastalar pivotal çalışmalaradahil edilmiştir. Faz I/II çalışmada, canakinumab tedavisinin etkileri hemen ortaya çıkmış ve doz uygulamasından bir gün sonra semptomlar ortadan kalkmış ya da semptomlarda klinik olarakanlamlı iyileşme sağlanmıştır. Yüksek CRP ve SAA gibi laboratuar parametreleri, yükseknötrofil ve trombosit sayıları, canakinumab enjeksiyonundan sonraki birkaç gün içinde hızlıcanormale dönmüştür. Pivotal çalışma, 48 haftalık, üç bölümlü çok merkezli bir çalışmadır; yani, 8 haftalık açık etiketli dönem (Kısım I), 24 haftalık randomize, çift kör, plasebo kontrollü bırakma dönemi(Kısım II) ve bunu takip eden 16 haftalık açık etiketli dönem (Kısım III). Çalışmanın amacı,CAPS'li hastalarda canakinumabın (her 8 haftada 150 mg ya da 2 mg/kg) etkililik, güvenlilikve tolere edilebilirliğinin değerlendirilmesidir. Kısım I: Tedavi başlatıldıktan sonra 7 gün içinde, hastaların %97'sinde canakinumaba tam bir klinik yanıt ve biyomarker yanıtı (hekimin otoenflamatuar ve cilt hastalığı üzerindeki bileşikdeğerlendirmesi < minimal ve CRP ya da SAA değerleri < 10 mg/litre olarak tanımlanmıştır)gözlenmiştir. Hekimin otoenflamatuar hastalık aktivitesi ile ilgili klinik değerlendirmesindeanlamlı iyileşmeler olmuştur: genel otoenflamatuar hastalık aktivitesi değerlendirmesi, cilthastalığı (ürtiker tipi cilt döküntüsü), artralji, miyalji, baş ağrısı/migren, konjonktivit,yorgunluk/huzursuzluk değerlendirmesi, diğer bağlantılı semptomlarla ilgili değerlendirme,hastanın semptom değerlendirmesi. Kısım II: Pivotal çalışmanın bırakma döneminde, birincil sonlanım noktası hastaların hastalık nüksü/alevlenme oranı olarak tanımlanmıştır; canakinumaba randomize hastaların hiçbirinde(%0) alevlenme görülmezken, plaseboya randomize edilenlerin %81'inde alevlenme meydanagelmiştir. Kısım III: Kısım II'de plaseboyla tedavi edilmiş ve canakinumab tedavisinin uygulandığı açık etiketli uzatma dönemine kaydolmuş hastalarda da, sürekli canakinumab ile tedavi edilenhastalarla karşılaştırıldığında, hastalık aktivitesinde anlamlı klinik ve serolojik iyileşmelermeydana gelmiştir. Faz III çalıma, pivotal plasebo kontrollü geri çekme döneminde (II. Kısım) gözlenen etkililiin tablo haline getirilmiözeti
İki açık etiketli, kontrollü olmayan, uzun vadeli faz III çalışma yürütülmüştür. Biri, CAPS hastalarında canakinumaba ilişkin bir güvenlilik, tolerabilite ve etkililik çalışmasıdır. Toplamtedavi süresi 6 ay ila 2 yıl arasında değişmiştir. Diğeri Japon CAPS hastalarında 48 haftayavaran uzatma fazı ile birlikte, 24 hafta boyunca etkililik ve güvenliliği değerlendirmek üzerecanakinumab ile yürütülen açık etiketli bir çalışmadır. Birincil hedef dozun artırıldığı hastalarda dahil olmak üzere 24. haftada relapssız hasta oranını değerlendirmektir. Bu iki çalışma için birleştirilmiş etkililik analizinde, daha önce canakinumab ile tedavi edilmemiş hastaların %65,6'sı 150 mg veya 2 mg/kg'da tam yanıt elde ederken, hastaların%85,2'si hiçbir dozda tam yanıt elde etmemiştir. 600 mg veya 8 mg/kg (veya hatta daha yüksek)ile tedavi edilen hastalardan %43,8'i tam yanıt elde etmiştir. Daha büyük pediatrik ve yetişkinhastalara kıyasla 2 ila <4 yaşındaki daha az hasta tam yanıt (%57,1) elde etmiştir. Tam yanıtelde eden hastalardan %89,3'ünde yanıt relapssız korunmuştur. Dozun her 8 haftada bir 600 mg'a (8 mg/kg) artırılmasını takiben tam yanıt elde eden bireysel hastalardan edinilen deneyim, önerilen dozlar ile (>15 kg ve <40 kg ağırlığındaki hastalar için150 mg veya 2 mg/kg) tam yanıt elde etmeyen ya da tam yanıtı koruyamayan hastalarda dahayüksek bir dozun faydalı olabileceğini düşündürmektedir. Artırılmış bir doz FCAS veyaMWS'ye kıyasla NOMID/CINCA semptomları olan hastalara ve 2 ila <4 yaşındaki hastalaradaha sık uygulanmıştır. Rutin klinik uygulamada pediyatrik ve yetişkin CAPS hastalarında ILARIS tedavisinin uzun süreli güvenliliği ve etkililiği hakkında veri sağlamak için 6 yıllık bir gözlemsel veritabanıçalışması yapılmıştır. Çalışmaya 243 CAPS hastası dahil edilmiştir (18 yaşından küçük 85 hastadahil). Hastalık aktivitesi, çalışmadaki tüm başlangıç sonrası zaman noktalarında hastaların %90'ından fazlasında mevcut değil veya hafif/orta dereceli olarak değerlendirilmiştir ve ortalamaserolojik enflamasyon belirteçleri (CRP ve SAA) başlangıç sonrası tüm zaman noktalarındanormaldir (<10 mg / litre). ILARIS alan hastaların yaklaşık % 22'sinde doz ayarlamasıgerekmesine rağmen, terapötik etkinin olmaması nedeniyle hastaların sadece küçük bir bölümü(% 1,2) ILARIS'i kesmiştir. Pediyatrik popülasyonILARİS ile yürütülen CAPS çalışmaları 2 ila 17 yaş aralığında toplamda 80 pediyatrik hasta içermiştir (yaklaşık yarısı mg/kg bazında tedavi edilmiştir). Genelde, genel CAPSpopülasyonuna kıyasla pediyatrik hastalarda ILARİS'in etkililik, güvenlilik ve tolerabiliteprofilinde klinik olarak anlamlı farklar söz konusu değildir. Pediyatrik hastaların çoğu kliniksemptomlarda ve objektif enflamasyon belirteçlerinde (örn., SAA ve CRP) iyileşme eldeetmiştir. 4 yaş ve altındaki pediyatrik CAPS hastalarında canakinumabın etkililiğini, güvenliliğini ve tolere edilebilirliğini değerlendirmek için 56 haftalık, açık etiketli bir çalışma yapılmıştır. 2-8mg/kg'lık ağırlığa dayalı başlangıç dozları kullanılarak 17 hasta (2 yaşın altındaki 6 hasta dahil)değerlendirilmiştir. Çalışmada ayrıca canakinumabın standart çocukluk aşılarına karşıantikorların gelişimi üzerindeki etkisi de incelenmiştir. 2 yaşın altındaki hastalarda, 2 yaş veüzerindeki hastalara kıyasla güvenlilik veya etkililik açısından hiçbir fark gözlemlenmemiştir.Canlı olmayan, standart bakım çocukluk aşıları alan tüm hastalarda (N=7) koruyucu antikorseviyeleri gelişmiştir. FMF:Canakinumabın TRAPS, HIDS/MKD ve FMF tedavisi için etkililiği ve güvenliliği, üç ayrı hastalık kohortundan oluşan tek, pivotal, faz III, 4 kısımlı bir çalışmada (N2301) gösterilmiştir.- Kısım I: Her hastalık kohortunda 2 yaş ve üzerindeki hastalar, hastalık alevlenmesininbaşlangıcı açısından değerlendirildikleri 12 haftalık bir tarama periyoduna girmişlerdir. - Kısım II: Alevlenme başlangıcındaki hastalar, 16 haftalık çift kör, plasebo kontrollü bir tedavi periyoduna randomize edilmiş, bu süre boyunca 4 haftada bir 150 mg canakinumab(vücut ağırlığı < 40 kg olan hastalar için 2 mg/kg) subkutan (s.c.) veya plasebo almışlardır.28 günden büyük ancak 2 yaşından küçük hastaların, randomize olmayan hastalar olarakdoğrudan Kısım II'nin açık koluna çalışmaya girmelerine izin verilmiştir (ve birinciletkinlik analizinden çıkarılmışlardır). - Kısım III: 16 haftalık tedaviyi tamamlayan ve yanıt verenler olarak sınıflandırılan hastalar,24 haftalık, çift-kör geri çekme periyodu için her 8 haftada bir canakinumab 150 mg (< 40kg hastalar için 2 mg/kg) S.C. veya plasebo almak üzere yeniden randomize edilmiştir. - Kısım IV: Canakinumab ile tedavi edilen tüm Kısım III hastaları, 72 haftalık açık etiketli bir tedavi uzatma periyoduna girmeye uygun olmuşlardır. 28 günlük ve üzeri yaşta toplam 185 hasta çalışmaya alınmış ve çalışmanın II. kısmına 2 yaş ve üzeri toplam 181 hasta randomize edilmiştir. Randomize tedavi periyodunun (Kısım II) birincil etkililik sonlanım noktası, her bir kohort içinde 15. günde başlangıçtaki hastalık alevlenmesi düzelen ve 16 haftalık tedavi döneminingeri kalanında yeni bir alevlenme yaşamayan (tam bir yanıt olarak tanımlanır) yanıt veren hastaoranıdır. Başlangıçtaki hastalık alevlenmesinin düzelmesi, Hekimin Hastalık Aktivitesi KüreselDeğerlendirmesi (PGA) < 2 (minimum veya hastalık yok) ve CRP'nin normal aralıkta (<10mg/L) olması veya başlangıca göre azalmanın > %70 olması olarak tanımlanmıştır. Yenialevlenme, PGA skoru > 2 (hafif, orta veya şiddetli hastalık) ve CRP > 30 mg/L olaraktanımlanmıştır. Tamamı 16. hafta sonuçlarına (Kısım II'nin sonu) dayanan ikincil sonlanımnoktaları, PGA skoru < 2 olan hastaların oranını, serolojik remisyona giren hastaların oranını(CRP < 10 mg/L olarak tanımlanır) ve normalleştirilmiş SAA düzeyine sahip hastaların oranını(SAA < 10 mg/L olarak tanımlanır) içermiştir. Birincil etkililik sonlanım noktası açısından canakinumab, üç hastalık kohortunun tümü için plasebodan üstün olmuştur. Canakinumab ayrıca her üç kohortta da PGA < 2 ve CRP < 10 mg/Likincil sonlanım noktalarında plaseboya kıyasla üstün etkililik göstermiştir. Her üç kohorttaplaseboya kıyasla canakinumab tedavisi ile 16. haftada daha yüksek hasta oranlarında SAAnormalize olmuş (<10 mg/L), TRAPS hastalarında istatistiksel olarak anlamlı bir farkgözlemlenmiştir (aşağıda, çalışma sonuçlarını içeren Tabloya bakınız). Faz III çalışmanın pivotal, randomize, plasebo kontrollü tedavi periyodunda (Kısım II) etkililiğinin tablo halinde özeti
Doz yükseltmeÇalışmanın II. kısmında, hastalığı aktif devam eden canakinumab ile tedavi edilen hastalar, ilk ay içinde 150 mg'lık (veya < 40 kg hastalar için 2 mg/kg) ek bir doz almıştır. Bu ek doz, ilktedavi dozundan en erken 7 gün sonra uygulanabilmiştir. Dozu yükseltilen tüm hastalar, her 4haftada bir 300 mg'lık (veya < 40 kg hastalar için 4 mg/kg) artan dozda kalmıştır. Birincil sonlanım noktasının keşifsel analizinde, ilk dozdan sonra yetersiz yanıt alan hastalarda, ilk ay içinde 4 haftada bir 300 mg (veya 4 mg/kg) doza yükseltmenin alevlenme kontrolünüdaha da artırdığı, hastalık aktivitesinin azaldığı ve CRP ve SAA seviyelerini normalleştirdiğigözlemlenmiştir. Pediyatrik hastalar:Yaşları > 28 gün ve < 2 yıl olan randomize olmayan iki HIDS/MKD hastası çalışmaya dahil edilmiş ve canakinumab almıştır. Bir hastada tek doz 2 mg/kg canakinumab aldıktan sonra 15.günde başlangıçtaki alevlenme düzelmiş fakat hasta, bu ilk dozdan sonra ciddi advers olaylar(pansitopeni ve karaciğer yetmezliği) nedeniyle tedaviyi bırakmıştır. Bu hastanın, çalışmabaşlangıcında immün trombositopenik purpura öyküsü ve anormal hepatik fonksiyonları vardı.İkinci hasta canakinumab 2 mg/kg başlangıç dozu ve 3. haftada 2 mg/kg ek dozu almıştır ve 5.haftada, çalışmanın II. kısmının sonuna 4 haftada bir 4 mg/kg dozu alacak şekilde titreedilmiştir. Hastalık alevlenmesi 5. haftada düzelmiştir ve hasta çalışmanın II. kısmının sonunda(16. hafta) herhangi bir yeni alevlenme yaşamamıştır. SJİA:ILARIS'in aktif SJİA tedavisi için etkililiği iki pivotal çalışmada (G2305 ve G2301) değerlendirilmiştir. Kaydedilen hastalar 2 ila 20 yaşın altındadır (başlangıçta ortalama yaş 8,5ve ortalama hastalık süresi 3,5 yıl) ve aktif artritli >2 eklem, ateş ve yükselmiş CRP iletanımlanan aktif hastalığa sahiptir. Çalışma G2305:Çalışma G2305 4 mg/kg'lık (300 mg'a kadar) tek bir subkutan ILARIS dozu veya plasebo kullanımına randomize edilmiş 84 hastada ILARIS'in kısa vadeli etkililiğini değerlendirmeküzere yürütülen randomize, çift kör, plasebo kontrollü, tek dozlu, 4 haftalık bir çalışmadır (43hasta ILARIS ve 41 hasta plasebo kullanmıştır). Bu çalışmanın primer amacı, 15. günde ateşgörülmemesini (ateş <38°C) içerecek şekilde uyarlanmış pediyatrik ACR(Amerikan Romatoloji Derneği) yanıt kriterlerinde en az %30 iyileşme elde eden hasta oranı açısından ILARIS'in plaseboya karşı üstünlüğünü göstermektir. Ayrıca inaktif hastalık (SJİA'ya bağlanabilecek aktif artrit, ateş, döküntü, serozit, hepatomegali veya lenfadenopati yokluğu, normal CRP ve hekimin, hastalık aktivitesine işaretetmeyen genel değerlendirmesi olarak tanımlanır) değerlendirilmiştir. Pediyatrik ACR yanıtları 6 temel sonuç değişkeninin 3'ünde başlangıca göre yüzde iyileşme (%30, %50, %70, %90 ve %100) ve geri kalan değişkenlerden >1'inde >%30 kötüleşme olaraktanımlanır. Temel sonuç değişkenleri hekimin genel hastalık aktivitesi değerlendirmesi,ebeveynin veya hastanın genel sağlık değerlendirmesi, aktif artrit görülen eklem sayısı, kısıtlıhareket açıklığına sahip eklem sayısı, CRP ve fonksiyonel kapasiteyi (Çocukluk Çağı SağlıkDeğerlendirme Anketi - CHAQ) içermiştir. Tüm primer ve sekonder sonlanım noktaları karşılanmıştır. Pediyatrik ACR yanıtı ve inaktif hastalığa göre hasta yüzdesi aşağıda sunulmaktadır. ILARIS tedavisi, 15 ve 29. günlerde plasebo ile karşılaştırıldığında tüm pediatrik ACR yanıt puanlarını iyileştirmiştir. 15. ve 29. günde pediyatrik ACR yanıtı ve hastalık durumu
Sistemik ve artritik bileşenleri içeren uyarlanmış pediatrik ACR bileşenleri için bulgular genel yanıt bulguları ile tutarlıdır. 15. günde, aktif artrit ve kısıtlı hareket açıklığı görülen eklemsayısında başlangıca göre medyan değişiklik ILARIS için (n=43) sırasıyla %67 ve %73 iken,plasebo için (N=41) medyan değişiklik %0 ve %0'dır. 15. günde hasta ağrı skorundaki ortalamadeğişiklik (0 100 mm görsel analog ölçeği) plasebo (N=25) için +4.5 mm'ye kıyasla ILARIS(N=43) için 50 mm'dir. ILARIS ile tedavi edilen hastalarda ağrı skorunda ortalama değişiklik29. günde tutarlıdır. Çalışma G2301:Çalışma G2301 aktif SJİA'lı hastalarda ILARIS ile alevlenmelerin önlenmesine ilişkin randomize, çift kör, plasebo kontrollü bir geri çekme çalışmasıdır. Çalışma iki bağımsız primersonlanım noktasına sahip iki ana kısımdan oluşmuştur. Kısım I'e (açık etiket) 177 hastakaydedilmiş ve 32 haftaya kadar 4 haftada bir uygulanan 4 mg/kg (300 mg'a kadar) ILARISalmışlardır. Kısım II'deki (çift kör) hastalar, 37 alevlenme olayı meydana gelene kadar her 4haftada bir ILARIS 4 mg/kg ya da plasebo almıştır. Kortikosteroid dozunun azaltılmasıKısım I'de kortikosteroid kullanan 128 hastadan 92'si kortikosteroid azaltma girişiminde bulunmuş, 57'sinde (%62) kortikosteroidler başarılı bir şekilde azaltılmış, 42'si (%46)kortikosteroid kullanmayı bırakmıştır. Alevlenmeye kadar geçen süreKısım Il'de canakinumab alan hastalarda alevlenme riski plasebo grubuna kıyasla %64 azalmıştır (tehlike oranı 0,36; %95 GA: 0,17 ila 0,75; p=0,0032). Plasebo veya canakinumabaatanmış olmaları fark etmeksizin, Kısım lI'ye giren 100 hastanın 63'ü gözlem süresi boyunca(maksimum 80 haftaya kadar) bir alevlenme yaşamamıştır. Çalışma G2305 ve G2301'de sağlıkla bağlantılı yaşam kalitesi sonuçları:ILARIS ile tedavi edilen hastaların yaşam kalitesi ve günlük işlev gösterme kapasitelerinde hızlı, kalıcı ve klinik olarak ilgili iyileşmelerle sonuçlanmıştır. Çocukluk Çağı SağlıkDeğerlendirme Anketi En Küçük Kareler ortalamalarındaki iyileşme plaseboya karşı ILARISiçin 0,69 olup, bu değer 0,19'luk minimum klinik olarak önemli farkın 3,6 katını temsiletmektedir (p=0,0002). Çalışma G2301'in I. Kısmının sonunda başlangıca göre medyaniyileşme 0,88'dir (%79). Çalışma G2305'te plaseboya karşı ILARIS için Çocuk Sağlık Anketi-PF50 skorlarında istatistiksel olarak anlamlı iyileşmeler bildirilmiştir (fiziksel p=0,0012;psikososyal sağlık p =0,0017). Havuzlanmış etkililik analizi:Çalışma G2305, G2301 ve uzatma çalışmalarından ILARIS tedavisinin ilk 12 haftasına ilişkin veriler, etkililiğin korunup korunmadığını değerlendirmek üzere birleştirilmiştir. Bu verilerpediyatrik ACR yanıtları ve her bir pediyatrik ACT temel bileşeninde ayrı ayrı çalışmalardagözlenene benzer iyileşmeleri göstermiştir (Aşağıdaki tablo). 12. haftada uyarlanmış pediyatrikACR30, 50, 70, 90 ve 100 cevapları sırasıyla %70, %69, %61, %49 ve %30'du, hastaların%28'inde inaktif hastalık mevcuttu (N=178). Sınırlı olmakla birlikte klinik çalışmalardan elde edilen kanıtlar, tocilizumab veya anakinraya yanıt vermeyen hastaların ILARIS'e yanıt verebileceğini göstermiştir. Çalışma G2301E1:G2305 ve G2301 çalışmalarında gözlemlenen etkililik, açık etiketli uzun süreli uzatma çalışması G2301E1'de korunmuştur. Çalışmadaki 270 SJIA hastasının 147'si, G2305 veyaG2301 (Kohort I) çalışmalarında canakinumab ile tedavi görmüştür ve 123 hasta önceden hiçcanakinumab kullanmamıştır (Kohort II). Kohort I'deki hastalar ortanca 3,2 yıl (5,2 yıla kadar)ve Kohort II'deki hastalar ortanca 1,8 yıl (2,8 yıla kadar) süreyle tedavi edilmiştir. Uzatmaçalışmasında, tüm hastalar 4 haftada bir 4 mg/kg (maksimum 300 mg'a kadar) canakinumabalmıştır. Her iki kohortta da, iyi kontrollü yanıt veren hastaların (geriye dönük olarakuyarlanmış pediatrik ACR > 90 olarak tanımlanır) ve eş zamanlı kortikosteroid gerektirmeyenhastaların canakinumab dozlarını 4 haftada bir 2 mg/kg'a düşürmelerine izin verilmiştir(62/270; %23). Çalışma G2306:Çalışma G2306, 4 haftada bir 4 mg/kg canakinumab alan SJIA hastalarında canakinumab dozunun azaltılması (4 haftada bir 2 mg/kg) veya doz aralığının uzatılması (8 haftada bir 4mg/kg) ile tedavi yanıtının sürdürülmesini değerlendirme amaçlı açık etiketli bir çalışmadır Enaz 4 hafta boyunca eşzamanlı kortikosteroid ve/veya metotreksat kullanımının kesilmesiyleinaktif hastalık durumunu sürdürebilen hastalar dahil olmak üzere canakinumab monoterapisiile arka arkaya en az 6 ay boyunca inaktif hastalık durumunu sürdüren (klinik remisyon) 2 ila22 yaşları arasındaki 75 hasta, 4 haftada bir 2 mg/kg canakinumab (N=38) veya her 8 haftadabir 4 mg/kg canakinumab (N=37) almak üzere randomize edilmiştir. 24 hafta sonra, azaltılmışdoz (4 haftada bir 2 mg/kg) alan hastaların %71'i (27/38) ve uzatılmış doz aralığı uygulananhastaların %84'ü (31/37) (8 haftada bir 4 mg/kg) 6 ay boyunca inaktif hastalık durumunukoruyabilmiştir. Daha fazla doz azaltma (4 haftada bir 1 mg/kg) veya doz aralığı uzatma (12haftada bir 4 mg/kg) ile devam eden ve klinik remisyonda olan hastaların %93'ü (26/28) ve%91'i (30/33) 6 ay boyunca inaktif hastalık durumunu koruyabilmiştir. Bu en düşük dozrejiminde 6 ay daha inaktif hastalık durumunu koruyan hastaların canakinumabı kesmesine izinverilmiştir. Genel olarak, doz azaltma veya doz aralığı uzatma kollarına randomize edilenhastaların %33'ü (25/75) canakinumab tedavisini sonlandırmayı ve 6 ay boyunca inaktifhastalık durumunu korumayı başarmıştır. Her iki tedavi kolundaki advers olayların oranı, 4haftada bir 4 mg/kg canakinumab ile tedavi edilen hastalarda görülen orana benzer olmuştur. İmmünojenisite:CAPS ve SJİA için ILARIS ile tedavi edilen hastaların sırasıyla yaklaşık %1.5 ve %3'ünde ILARIS'e antikor gelişmiştir. SJİA klinik çalışmalarının çoğunda daha yüksek duyarlılığa sahipköprüleme analizi kullanılmıştır. Nötralizan antikor tespit edilmemiştir. Antikor gelişimi veklinik yanıt veya advers olaylar arasında belirgin bir korelasyon gözlenmemiştir. 16 haftalık tedavi boyunca 150 mg ve 300 mg'lık dozlarla tedavi edilen TRAPS, HIDS/MKD ve FMF hastalarında hastalarında canakinumaba karşı antikor gözlemlenmemiştir. 5.2. Farmako kinetik özelliklerGenel ÖzelliklerCAPSEmilimYetişkin CAPS hastalarında 150 mg tek bir cilt altı uygulamayı takiben yaklaşık 7 gün içinde pik serum canakinumab konsantrasyonlarına (Cmaks) ulaşılmıştır. Ortalama terminal yarı ömür26 gündür. Tipik bir yetişkin CAPS hastasında (70 kg) tek bir subkutan 150 mg dozdan sonraCmaks ve EAAinf için ortalama değerler 15,9 mcg / mL ve 708 mcg *d / mL olarak belirlenmiştir.CAPS popülasyonunda yürütülen popülasyon farmakokinetik analizine dayalı olarak, cilt altıyoldan uygulanan canakinumabın mutlak biyoyararlanımı %66 olarak hesaplanmıştır.Maruziyet parametreleri (EAA ve Cmaks) intravenöz infüzyon olarak verilen 0,3 ila 10 mg/kgveya subkutan enjeksiyon olarak verilen 150 ila 600 mg doz aralığında dozla orantılı olarakartmıştır. 8 haftada bir 150 mg (ya da 2 mg/kg) subkutan uygulamadan sonra öngörülen kararlıdurum maruziyet değerleri (Cmin,ss, Cmaks,ss, EAAss,8w) <40 kg (4 mcg / mL, 19,9 mcg / mL, 566mcg *d/ mL) ve >70 kg (4,6 mcg / mL, 17,8 mcg / mL, 545 mcg *d/ mL) kilo kategorileri ilekarşılaştırıldığında 40-70 kg kilo kategorisinde (6,6 mcg / mL, 24,3 mcg / mL, 767 mcg *d/mL) biraz daha yüksek olmuştur. 6 ay boyunca 8 haftada bir 150 mg subkutan canakinumabuygulamasını takiben beklenen birikim oranı 1,3 kattır. DağılımCanakinumab serum IL-1 betaya bağlanır. Canakinumabın dağılım hacmi (Vss) vücut ağırlığına göre değişiklik göstermiştir. 70 kg vücut ağırlığına sahip bir CAPS hastasında 6,2litre olduğu hesaplanmışır. EliminasyonCanakinumabın klirensi (CL) vücut ağırlığı ile artış göstermiştir. Vücut ağırlığı 70 kg olan bir CAPS hastasında 0,17 L/gün ve 33 kg ağırlığında bir SJIA hastasında 0,11 L/gün olduğu hesaplanmıştır. Vücut ağırlığındaki farklılıklar hesaba katıldığında, CAPS ve SJİA hastaları arasında canakinumabın farmakokinetik özellikleri açısından klinik olarak anlamlı farklılıklargözlenmemiştir. Canakinumabın farmakokinetik özelliklerinde, tekrarlanan uygulama sonrasında hızlandırılmış klirens veya zamana bağlı değişiklik belirtisi yoktur. Vücut ağırlığı için düzeltme yapıldıktansonra cinsiyet veya yaşa bağlı farmakokinetik farklılık gözlenmemiştir. Doğrusallık/ doğrusal olmayan durumTekrarlı uygulamayı takiben canakinumabın farmakokinetik parametrelerinde hızlanmış klirens ya da zamana bağlı değişim işaretlerine rastlanmamıştır. Vücut ağırlığına göre düzeltmeyapıldıktan sonra, cinsiyet ya da yaşa bağlı farmakokinetik değişiklikler gözlenmemiştir. FMFFMF hastalarında biyoyararlanım bağımsız olarak belirlenmemiştir. 55 kg (0,14 L/d) vücut ağırlığındaki FMF popülasyonundaki görünür klirens (CL/F), 70 kg (0,7 L/d) vücutağırlığındaki CAPS popülasyonu ile benzerdir. Görünür dağılım hacmi (V/F), 55 kg vücutağırlığında 4,96 L bulunmuştur. 4 haftada bir 150 mg'lık tekrarlanan subkutan uygulamadan sonra 16. haftadaki canakinumab minimum konsantrasyonu (Cmin) 15,4 ± 6,6 pg/mL olarak hesaplanmıştır. Tahmini kararlıdurum EAAtau, 636,7 ± 260,2 pg*d/mL bulunmuştur. SJIASJIA hastalarında biyoyararlanım bağımsız olarak belirlenmemiştir. Kg vücut ağırlığı başına belirgin klirens (kg başına CL/F) SJIA ve CAPS popülasyonunda benzerdir (kg başına 0,004L/d). Kg başına belirgin dağılım hacmi (kg başına V/F) 0,14 L/kg'dır. 4 haftada bir 4 mg/kg tekrarlı uygulamadan sonra, SJIA hastalarında canakinumab birikim oranı 1,6 kat olmuştur. Kararlı duruma 110 gün sonra ulaşılmıştır. Cmin,ss, Cmaks,ss ve EAA,ss4w içingenel öngörülen ortalama (± SD) sırasıyla 14,7 ± 8,8 mcg/ mL, 36,5 ± 14,9 mcg/ mL ve 696,1± 326,5 mcg*d/ mL olarak belirlenmiştir. Her yaş grubundaki EAAss4w, 2-3, 4-5, 6-11 ve 12-19 yaşları için 692, 615, 707 ve 742 mcg *d/ mL 'dir. Ağırlığa göre sınıflandırıldığında, daha yüksek vücut ağırlığı (>40 kg) kategorisi ilekarşılaştırıldığında, daha düşük vücut ağırlığı kategorisinde (<40 kg), Cmin,ss (11,4 karşısında19 mcg/ mL) ve EAAss (594 karşısında 880 mcg*d/ mL) için daha düşük bir (%30-40) medyanmaruziyet gözlenmiştir. Popülasyon farmakokinetik modelleme analizine dayanarak, 16 ila 20 yaş arasındaki genç yetişkin SJIA hastalarındaki canakinumab farmakokinetiği, 16 yaşından küçük hastalardakinebenzerdir. 20 yaşın üzerindeki hastalarda 4 mg/kg (maksimum 300 mg) doz düzeyindeöngörülen canakinumab kararlı durum maruziyetleri, 20 yaşın altındaki SJIA hastalarındagözlenenle benzer olmuştur. Pediyatrik popülasyon4 yaş ve üstü pediyatrik hastalarda tek bir subkutan canakinumab 150 mg veya 2 mg/kg uygulamasının ardından, 2 ila 7. günler (Tmaks) arasında pik canakinumab konsantrasyonlarınaulaşılmıştır. Terminal yarı ömür aralığı 22,9-25,7 gün olarak belirlenmiştir ve bu değerleryetişkinlerde gözlenen farmakokinetik özelliklere benzerdir. Popülasyon farmakokinetikmodelleme analizine dayanarak, 2-4 yaş arası çocuklardaki canakinumabın farmakokinetiği, 4yaş ve üstü hastalardakine benzerdir. Subkutan emilim oranının yaşla birlikte azaldığı ve engenç hastalarda en hızlı olduğu görülmüştür. Buna göre, daha genç SJIA hastalarında (2-3 yıl) 19 / 22Tmaks, daha yaşlı SJIA hastalarına (12-19 yıl; Tmaks 6 gün) göre daha kısa olmuştur (3.6 gün). Biyoyararlanım (EAAss) etkilenmemiştir. İlave bir farmakokinetik analiz, 2 yaşın altındaki 6 pediyatrik CAPS hastasındaki canakinumabın farmakokinetiğinin 2-4 yaş arası pediyatrik hastalarda gözlenenfarmakokinetiğe benzer olduğunu göstermiştir. Popülasyon farmakokinetik modellemeanalizine dayanarak, 2 mg/kg'lık bir dozdan sonra beklenen maruziyetler CAPS pediyatrik yaşgrupları boyunca benzerdir; ancak çok düşük vücut ağırlığına sahip pediyatrik hastalarda(örneğin, 10 kg) yetişkin hastalardakine (150 mg doz) göre yaklaşık %40 daha düşüktür. Bu,CAPS hastalarında daha yüksek vücut ağırlığı gruplarında daha yüksek maruziyetlerinolduğuna dair gözlemlerle tutarlıdır. FMF'de, 4 haftada bir 2 mg/kg canakinumab subkutan uygulamasının ardından 2 ila < 20 yaş arasındaki yaş gruplarında maruziyet parametreleri (çukur konsantrasyonlar) benzerbulunmuştur. CAPS, FMF ve SJİA pediyatrik popülasyonlarında farmakokinetik özellikler benzerdir. Yaşlı popülasyonYaşlı hastalar ve 65 yaşın altındaki yetişkin hastalar arasında klirense dayalı farmakokinetik parametreler açısından değişiklik gözlenmemiştir. 5.3. Klinik öncesi güvenlilik verileriKlinik dışı veriler, çapraz reaktivite, tekrarlanan doz toksisitesi, immünotoksisite, üreme ve gelişme toksisitesi ile ilgili geleneksel çalışmalara dayalı olarak insanlar için özel bir tehlikeortaya koymamaktadır. Canakinumab ile resmi karsinojenisite çalışmaları yapılmamıştır. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiSükroz L-histidin L-histidin HCl monohidrat Polisorbat 80 Hidroklorik asit %25 (pH ayarlayıcısı olarak, y.m.) Enjeksiyonluk su (Çözücü, dondurarak kurutma sırasında uzaklaştırılmaktadır.) 6.2. GeçimsizliklerGeçimlilik çalışmaları mevcut olmadığından, bu tıbbi ürün uygulanırken diğer tıbbi ürünlerle karıştırılmamalıdır. 6.3. Raf ömrüAçılmamış flakon: 2°C - 8°C'de 36 ay Sulandırılmış çözelti: 2°C - 8°C'de 24 saat Kullanıma hazırlandıktan sonra mikrobiyolojik açıdan ürün hemen kullanılmalıdır. Hemen kullanılmazsa, kullanım sırasındaki saklama süreleri ve kullanımdan önceki koşullarkullanıcınm sorumluluğundadır ve normalde 2°C-8°C'de 24 saatten uzun olmaması gerekir. 6.4. Saklamaya yönelik özel tedbirlerSulandırılmış tıbbi ürünün saklama koşulları için bkz. Bölüm 6.3. Açılmamış flakonlar 2°C - 8°C'de saklanmalıdır. Dondurmayınız. Işıktan korumak için orijinal ambalajında saklayınız. Ürününü hazırlanmasından sonraki saklama koşulları için Bölüm 6.3'e bakınız. 6.5. Ambalajın niteliği ve içeriğiKaplanmış klorobütil kauçuktan tıpa ve alüminyum geçme kapak bulunan renksiz 6 mL cam flakon Ambalaj büyüklüğü: 1 flakon içeren paket ya da 4 (4x1) flakon içeren çoklu paket. 6.6. Beşeri tıbbi ürünlerden arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış ürün ya da artık materyaller ''Tıbbi Ürünlerin Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. ILARIS 150 mg/ ml enjeksiyonluk çözelti için toz içeren flakon bireysel kullanıma yönelik tek kullanımlık bir flakonda tedarik edilir. Hazırlama ve kullanma talimatlarıSteril teknik kullanarak, her bir ILARIS flakonunu, ,1 mL enjektör ve 18 G x 2'' iğne aracılığıyla 1 mL enjeksiyonluk suyu enjekte ederek sulandırın. Flakonu 45 derecelik bir açıdayaklaşık 1 dakika boyunca çevirin ve daha sonra 5 dakika bekleyin. Daha sonra yavaşça on kereters çevirin ve eski haline getirin. Eğer mümkünse kauçuk tıpaya parmaklarınızla dokunmayın.Berrak ila opalesan bir çözelti elde etmek için flakonu oda sıcaklığında 15 dakika bekletin.Çalkalamayın. Çözeltide parçacıklar varsa, çözeltiyi kullanmayın. Tıpada sıvı kalmışsa, flakonun kenarlarına vurarak sıvıyı uzaklaştırın. Çözeltide görünür parçacık olmamalı ve berrak ile bulanık arasında bir görünüşe sahip olmalıdır. Çözelti berrakya da hafif kahverengimsi sarı olmalıdır. Eğer sulandırıldıktan sonra 60 dakika içindekullanılmazsa, çözelti 2°C - 8°C'de saklanmalı ve 24 saat içinde kullanılmalıdır. Uygulanacak doza bağlı olarak (0,1 mL'den 1 mL 'ye kadar) gerekli hacmi dikkatli bir şekilde çekin ve 27 G x 0,5 (13 mm) iğne kullanarak cilt altına enjekte edin. Yaralı dokuya enjeksiyondan kaçınılmalıdır; çünkü bu ILARIS'e yetersiz maruziyete yol açabilir. 7. RUHSAT SAHİBİNovartis Sağlık, Gıda ve Tarım Ürünleri San. ve Tic. A.Ş. Kavacık/Beykoz/İstanbul 8. RUHSAT NUMARASI131/25 9. İLK RUHSAT TARİHİ/ RUHSAT YENİLEME TARİHİİlk ruhsat tarihi:Ruhsat yenileme tarihi:-10. KUB'UN YENILENME TARİHİ |
İlaç BilgileriIlaris 150 Mg/ml Enjeksiyonluk Çözelti İçin Toz İçeren FlakonEtken Maddesi: Canakinumab Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.