Immunine 600 Iu Iv İnfüzyon İçin Liyofilize Toz İçeren Flakon Kısa Ürün BilgisiKISA URUN BILGISIV Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERI TIBBİ ÜRÜNÜN ADIIMMUNINE 600 IU IV infüzyon için liyofilize toz içeren flakon Saflaştırılmış faktör IX konsantresiVirüs inaktivasyonu uygulanmıştır. Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Antihemofilik Faktör IX1 flakon enjeksiyonluk çözelti için toz nominal olarak 600 IU Antihemofilik Faktör IX içerir. 5 mL'lik flakon steril enjeksiyonluk su ile çözüldüğünde 1 mL çözelti yaklaşık 120 IU insan Antihemofilik Faktör IX içerir. F IX potensi (IU), Avrupa Farmakopesi'nde yer alan tek basamaklı pıhtılaşma testiyle belirlenmiştir. İnsan donörlerin plazmasından üretilmiştir. IMMUNINE'ın spesifik etkinliği her 1 mg protein başına > 50 IU'dir. Yardımcı maddeler:IMMUNINE 600 IU bir flakonunda toplam 20 mg sodyum içerir (hesaplanmış değer) Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk veya infüzyonluk çözelti hazırlamak için beyaz ya da soluk sarı liyofilize toz içeren flakon ve 5 mL çözücüsü. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarıHemofili B (konjenital faktör IX eksikliği) hastalarında kanamanın profilaksisinde ve tedavisinde endikedir. IMMUNINE, 6 yaşından büyük çocuklardan yetişkinlere kadar tüm yaş gruplarında endikedir. IMMUNINE'ın 6 yaşından küçük çocuklarda kullamlmasınm faydalı olabileceğini gösteren veriler yetersizdir. 4.2. Pozoloji ve uygulama şekliTedavi, hemofili tedavisinde deneyimli bir hekimin denetiminde başlatılmalıdır. Pozoloji / Uygulama sıklığı ve süresi:Yerine koyma tedavisinin dozu ve süresi, faktör IX eksikliğinin ciddiyetine, kanamanın yeri ve miktarına ve hastanın klinik durumuna bağlıdır. Uygulanan faktör IX ünitelerinin sayısı, faktör IX ürünleri için güncel Dünya Sağlık Örgütü standardı ile ilişkili uluslararası ünite (IU) terimiyle ifade edilir. Plazmadaki faktör IX aktivitesi,ya yüzdesel olarak (normal insan plazmasına göre) ya da uluslarası ünite olarak (plazmadakifaktör IX konsantreleri için uluslararası standartlara göre) ifade edilir. 1 Uluslararası Ünite (IU) faktör IX aktivitesi, 1 mL normal insan plazmasındaki faktör IX miktarına eşdeğerdir. İhtiyaç olduğunda tedaviGereken faktör IX dozunun hesaplanması, 12 yaş ve üzerindeki hastalarda 1 IU/kg faktör IX'un plazma faktör IX düzeyini %1,1 yükselttiği şeklindeki ampirik bulguya dayandırılmıştır.Gereken doz aşağıdaki formülle hesaplanabilir: Gerekli IU miktarı = vücut ağırlığı (kg) x istenen F IX artışı (%)(IU/dL) x 0,9Uygulanacak miktar ve uygulama sıklığı, her hasta için bireysel olarak klinik yararlılık esasına göre düzenlenmelidir. Faktör IX preparatlarının nadiren günde 1 defadan fazla uygulanmagerekliliği vardır. Aşağıda belirtilen kanama olaylarında, faktör IX aktivitesi bu süreçte karşılık gelen plazma faktör IX aktivite düzeyinin (normal düzeyin yüzdesi veya IU/dL olarak) altınadüşürülmemelidir. Aşağıdaki tablo kanama dönemleri ve cerrahide kullanılacak dozu belirlemeye yardımcı olmak için verilmektedir:
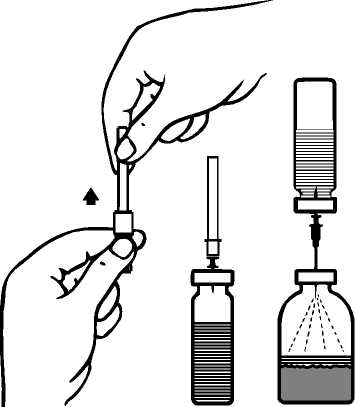
ProfilaksiCiddi hemofili B hastalığı olanlarda kanamaya karşı uzun dönem profilakside, 20-40 IU/kg'lık dozlar 3-4 günlük aralarla verilmelidir. Özellikle genç hastalar olmak üzere bazı olgularda, daha kısa dozaj aralıkları ya da daha yüksek dozlar gerekebilir. Tedavi süresince, tekrarlanan infüzyon sıklığının ve uygulanacak dozun ayarlanmasına rehber olması için faktör IX düzeyi ölçülmesi önerilmektedir. Özellikle majör cerrahi girişimlerde,koagülasyon analizleriyle (plazma faktör IX aktivitesi) yerine koyma tedavisinin sıkı takibigereklidir. Farklı yarılanma zamanı ve in vivoUygulama şekli:İntravenöz yoldan uygulanır. 2 mL/dk'dan daha hızlı bir şekilde uygulanmaması önerilmektedir. Uygulama öncesinde ürünün seyreltilmesine ilişkin talimatlar için bkz. Bölüm 6.6. Özel popülasyonlara ilişkin ek bilgiler:Böbrek/Karaciğer yetmezliği:Böbrek yetmezliği olan hastalara ilişkin ek bir bilgi bulunmamaktadır. Karaciğer yetmezliği olanlarda, trombotik gelişimin ve tüketim koagülopatisinin erken belirtilerinin klinik takibinin uygun biyolojik testlerle yapılması gereklidir (bkz. Bölüm 4.4 Özelkullanım uyarıları ve önlemleri: Tromboembolizm, DIC, Fibrinoliz)Pediyatrik popülasyon:Bu popülasyonla ilgili mevcut veri Bölüm 5.2'de yer almaktadır. Mevcut klinik verilere göre, 12 ila 18 yaş arasındakihastalar için pediyatrik hasta pozolojisi önerisi yapılabilir. 6 ila 12 yaş grubunda, mevcut klinik veriler bir doz önerisi yapmak için yeterlideğildir.IMMUNINE kullanımı pediyatrik hastalarda 6 ila 12 yaşındaki hasta grubunda ve 12 yaşından büyük Hemofili B hastalarında incelenmiştir. Güvenlilik IMMUNINE kullanan yetişkinlerdekigüvenliliğe benzerdir. Ayrıca, IMMUNINE kullanımı sırasıyla 6 yaşına kadar çocuklarda ve 0-64 yaşındaki Hemofili B hastalarında yapılan iki gözlemsel çalışmada incelenmiştir. 6 yaşına kadar çocuklardakigüvenlilik, IMMUNINE kullanan 6 yaşından büyük çocuklarda ve yetişkinlerdeki güvenliliğebenzerdir. Geriyatrik popülasyon:IMMUNINE'ın yaşlı hastalarda kullanımına ilişkin yeterli veri mevcut olmadığından bu popülasyonda dikkatle uygulanmalıdır. 4.3. Kontrendikasyonlar Aktif maddeye veya bileşimindeki diğer maddelere karşı aşırı duyarlılık Heparin alerjisi veya heparine bağlı trombositopeni hikayesi olanlar Bu durumlar yeterli tedavi ile kontrol altına alındığında, IMMUNINE yalnızca yaşamı tehdit eden kanamaları tedavi etmek için uygulanmalıdır. 4.4. Özel kullanım uyarıları ve önlemleriVirüs güvenliliğiIMMUNINE, insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibi, çeşitlihastalıklara yol açabilen enfeksiyon yapıcı ajanlar içerebilirler. IMMUNINE'de VaryantCreutzfeldt-Jacob hastalığının bulaşma riski teorik olarak minimumken, klasikCreutzfeldt-Jacob hastalığının bulaşma riski hiçbir kanıtla desteklenmez. Alınanönlemlere rağmen, bu tür ürünler halen potansiyel olarak hastalık bulaştırabilir.Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıp kalmadığının izlenmesi, belirli virüs enfeksiyonlarınınhalihazırda varlığının test edilmesi ve belirli virüslerin yok edilmesi ve/veyainaktivasyonu ile azaltılmıştır. Bütün bu önlemlere rağmen, bu ürünler hala potansiyelolarak hastalık bulaştırabilirler. Ayrıca, henüz bilinmeyen enfeksiyon yapıcı ajanların buürünlerin içerisinde bulunma ihtimali mevcuttur.HIV, HBV, HCV gibi zarflı virüsler ve HAV gibi zarflı olmayan virüsler için etkili önlemlerin alınmasına dikkat edilmelidir. Parvovirüs B19 gibi zarflı olmayan virüslerekarşı alınan tedbirler sınırlı sayıda olabilir. Parvovirüs B19 enfeksiyonu, gebelikte (fetalinfeksiyon) ve immün yetmezlik ya da kırmızı kan hücre üretiminde artış olan hastalardatehlikeli olabilir (hemolitik anemi gibi).Doktor, bu ilacı hastaya reçete etmeden veya uygulamadan önce hastası ile risk ve yararlarını tartışmalıdır.IMMUNINE kullanılması gerekiyorsa hekim tarafından, hastalık yapıcı etkenlerin hastaya bulaşmasını önlemek için uygun aşıların (Hepatit A, Hepatit B vb.) yaptırılması önerilebilir. Hastalar açısından IMMUNINE her uygulandığında, hastayla ürünün seri numarası arasındaki bağlantının korunabilmesi için, ürünün adı ve seri numarası kaydedilmelidir. Aşırı duyarlılık reaksiyonlarıIMMUNINE ile alerjik tipte aşırı duyarlılık reaksiyonları görülebilir. Ürün, faktör IX dışında eser miktarlarda insan kaynaklı proteinleri içerir. Hastalara aşırı duyarlılık semptomlarının ortaya çıkması durumunda, ürünün kullanımına hemen son vererek hekimlerine danışmaları öğütlenmelidir. Hastalar ve/veya sağlık bakımını üstlenen kişiler kurdeşen, yaygın ürtiker, göğüste sıkışma hissi, hırıltılı solunum, hipotansiyon ve anafilaksi gibi aşırı duyarlılık reaksiyonlarının erken belirtilerikonusunda bilgilendirilmelidir. Şok gelişmesi durumunda, güncel şok tedavisine yönelik tıbbi standartlara uyulmalıdır. İnhibitörlerİnsan koagülasyon faktörü IX ürünleriyle yapılan tekrarlayan tedavilerden sonra, hastalar nötralizan antikorların (inhibitör) gelişme riski açısından izlenmelidir. Oluşan antikorlarınmiktarı, uygun biyolojik test yöntemleri kullanılarak BU (Bethesda Ünitesi) cinsindentanımlanmalıdır. Beklenen faktör IX aktivitesi plazma düzeylerine ulaşılamadıysa ya da uygun bir dozla kanama kontrol altına alınamadıysa, faktör IX inhibitörünün varlığını araştırmak için ölçümyapılmalıdır. Yüksek inhibitör seviyelerine sahip hastalarda, faktör IX tedavisi etkiliolmayabilir ve diğer tedavi seçenekleri değerlendirilmelidir. Bu durumdaki hastaların tedavisi,hemofili olan hastalar konusunda deneyimli hekimler tarafından yürütülmelidir ve bu nedenlebu konuda özelleşmiş bir hemofili merkezi ile iletişim kurulmalıdır. Literatürde, faktör IX inhibitörlerinin oluşumu ve alerjik reaksiyonların gelişmesi arasında ilişki olduğunu gösteren bildirimler vardır. Dolayısıyla, alerjik reaksiyon gelişen hastalar,inhibitör varlığı açısından da değerlendirilmelidir. Faktör IX inhibitörü olan hastaların, dahasonraki faktör IX uygulamalarında anafilaksi açısından daha büyük bir risk taşıdığı akıldatutulmalıdır. Faktör IX konsantreleriyle görülebilecek alerjik reaksiyon riski nedeniyle, faktör IX preparatının ilk uygulamaları, tedavi eden hekimin kararı doğrultusunda, alerjik reaksiyonlarıntıbbi tedavisinin uygun bir şekilde sağlanabileceği bir merkezde yapılmalıdır. Tromboembolizm, DIC, FibrinolizFaktör IX kompleks konsantrelerinin kullanımı tarihsel olarak tromboembolik komplikasyonlarla ilişkilidir. Düşük saflıktaki preparatlarla komplikasyon gelişme riski dahayüksektir. Bu nedenle, faktör IX içeren ürünlerin kullanımı fibrinoliz ve yaygın damar içipıhtılaşma (DIC) belirtileri gösteren hastalarda kullanımı tehlikeli olabilir. Trombotik komplikasyon potansiyel riski nedeniyle karaciğer hastalığı olanlarda, trombofilisi olanlarda, hiperkoagülopati durumlarında, anjina pektoriste, koroner hastalığı ya da akutmiyokard enfarktüsü durumunda, post-operatif dönemdeki hastalarda, prematüre veyenidoğanlarda ya da trombotik olay veya DIC gelişme riski bulunan hastalarda bu ürünkullanılırken trombotik ve tüketim koagülopatisinin erken belirtilerinin klinik takibinin, uygunbiyolojik testlerle başlatılması gereklidir. Bu koşulların her birinin varlığında, IMMUNINE iletedavinin sağlayacağı yarar, komplikasyon gelişme riskiyle karşılaştırılmalıdır. Sodyum içeriğiBu tıbbi ürün her flakonunda 20 mg sodyum içermektedir. Bu miktar, bir yetişkin için Dünya Sağlık Örgütü (WHO) tarafından önerilen maksimum günlük 2 g sodyum alımının %1'ineeşdeğerdir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriIMMUNINE ile gerçekleştirilen herhangi bir etkileşim çalışması bulunmamaktadır. 4.6. Gebelik ve laktasyonGenel tavsiye:Gebelik Kategorisi: C. Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon)Faktör IX ile hayvan üreme çalışmaları yapılmamıştır. Hemofili B hastalığı kadınlarda nadiren görülen bir hastalık olduğundan çocuk doğurma potansiyeli bulunan kadınlarda faktör IX kullanımıyla ilgili bir deneyim de bulunmamaktadır. Gebelik dönemiHayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. Bölüm5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. Hemofili B hastalığı kadınlarda nadiren görülen bir hastalık olduğundan gebelik döneminde IMMUNINE kullanımıyla ilgili bir deneyim bulunmamaktadır. Bu nedenle gebelik dönemindekesin gerekli olmadıkça kullanılmamalıdır. Parvovirüs B19 enfeksiyonu riski açısından 4.4 Özel kullanım uyarıları ve önlemleri: " Virüs güvenliliğiLaktasyon dönemiEmziren kadınlarda IMMÜNINE'ın anne sütüne geçip geçmediği bilinmemektedir. Hemofili B hastalığı kadınlarda nadiren görülen bir hastalık olduğundan emzirme döneminde IMMUNINE kullanımıyla ilgili bir deneyim bulunmamaktadır. Bu nedenle emzirmedöneminde kesin gerekli olmadıkça kullanılmamalıdır. Üreme yeteneği / fertiliteFaktör IX ile hayvan üreme çalışmaları yürütülmemiştir. İnsanlardaki üreme yeteneği / fertiliteyi etkileyip etkilemediği bilinmemektedir. 4.7. Araç ve makine kullanımı üzerindeki etkilerAraç ve makine kullanımı üzerinde herhangi bir etki bildirilmemiştir. 4.8. İstenmeyen etkilerGüvenilirlik profili özetiFaktör IX içeren ürünlerle tedavi edilen hastalarda seyrek olarak aralarında anjiyoödem, infüzyon uygulanan bölgede yanma ve batma, üşüme/ürperme, yüz ve boyunda kızarma(flushing), yaygın ürtiker, baş ağrısı, kurdeşen, hipotansiyon, letarji, bulantı, huzursuzluk,taşikardi, göğüste baskı hissi, karıncalanma, kusma, hırıltılı solunum gibi belirtileri olabilenaşırı duyarlılık veya alerjik reaksiyonlar gözlenmiştir. Bazı vakalarda bu reaksiyonların şiddetli anafilaksiye kadar ilerlediği ve anafilaksi oluşumunun faktör IX'a karşı inhibitör gelişimiyle yakından ilişkili olduğu bildirilmiştir (aynızamanda bkz. Bölüm 4.4). Faktör IX inhibitörleri ve alerjik reaksiyon hikayesi olan hemofili B hastalarında immün tolerans indüksiyonu girişimini takiben nefrotik sendrom gelişebildiği bildirilmiştir. Seyrek olarak ateş gözlenmiştir. Hemofili B hastalarında faktör IX'a karşı nötralizan antikorlar (inhibitör) gelişebilir (bkz. Bölüm 4.4). Bu tür inhibitörler oluşursa, durum yetersiz klinik yanıtla ortaya çıkar. Bu türdurumlarda bu konuda özelleşmiş bir hemofili merkezi ile iletişim kurulmalıdır. Düşük saflıktaki preparatlar kullanıldığında risk daha yüksek olmak üzere faktör IX ürünlerinin kullanımında tromboembolik ataklara ilişkin potansiyel risk bulunmaktadır. Düşük saflıktakifaktör IX ürünlerinin kullanımı miyokard enfarktüsü, yaygın damar içi pıhtılaşma, venöztromboz ve pulmoner emboli olaylarıyla ilişkili olmuştur. Yüksek saflıktaki faktör IX ile butür yan etkiler seyrektir. Virüs güvenilirliği ile ilgili olarak bkz. Bölüm 4.4. Advers reaksiyonların listesiIMMUNINE kullanımı sırasında görülen advers reaksiyonlar MedDRA sistem-organ sınıflamasına (SOC ve tercih edilen terminoloji) göre aşağıda listelenmiştir. Listede raporlanan istenmeyen etkiler, 197 denekte IMMUNINE ile yürütülen 6 klinik çalışmadan elde edilen rapora ve pazarlama sonrası deneyimlere dayanmaktadır. İstenmeyen etkilerin görülme sıklığı, izleyen kriterler kullanılarak değerlendirilmiştir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek(>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketletahmin edilemiyor). Kan ve lenf sistemi hastalıklarıBilinmiyor: Faktör IX inhibisyonu, yaygın damar içi pıhtılaşma Bağışıklık sistemi hastalıklarıBilinmiyor: Alerjik reaksiyon, anafilaktik / anafilaktoid reaksiyon, anjiyoödem, ürtiker, inhibitörlerin varlığı durumunda serum hastalığı, aşırı duyarlık reaksiyonu Sinir sistemi hastalıklarıBilinmiyor: Baş ağrısı, huzursuzluk, karıncalanma hissi Kardiyak hastalıklarBilinmiyor: Miyokard enfarktüsü, taşikardi Vasküler hastalıklarBilinmiyor: Hipotansiyon, tromboembolik ataklar, (örn., pulmoner emboli, venöz tromboz, arteriyel tromboz, serebral arter trombozu), yüz ve boyunda kızarma (flushing) Solunum, göğüs bozuklukları ve mediastinal hastalıklarYaygın olmayan: Boğaz tahrişi, orofarengeal ağrı, kuru öksürük Bilinmiyor: Hırıltılı solunum, dispne Gastrointestinal hastalıklarBilinmiyor: Bulantı, kusma Deri ve deri altı doku hastalıklarıYaygın olmayan: Döküntü, kaşıntı Bilinmiyor: Ürtiker Böbrek ve idrar yolu hastalıklarıBilinmiyor: Nefrotik sendrom1 Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarYaygın olmayan: Pireksi Bilinmiyor: Titreme, infüzyon bölgesinde yanma ve batma, letarji, göğüste sıkışma hissi. 'bağışıklık tolerans indüksiyonunu takibenFaktör IX'a karşı inhibitörlerIMMUNINE ile klinik çalışmalarda faktör IX inhibitörü gelişimi bildirilmemiştir. IMMUNINE klinik çalışmalarına, daha önceden hiç tedavi almamış hastalar dahiledilmemiştir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99) 4.9. Doz aşımı ve tedavisiİnsan koagülasyon faktör IX ile doz aşımında ortaya çıkan herhangi bir belirti bildirilmemiştir. 5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik Grubu:ATCkodu:B02BD04Faktör IX, molekül kütlesi yaklaşık 68.000 Dalton olan tek zincirli bir glikoproteindir. K vitaminine bağlı bir pıhtılaşma faktörüdür ve karaciğerde sentezlenmektedir. Faktör IX,intrensek pıhtılaşma yolunda, faktör XIa tarafından ve ekstrensek pıhtılaşma yolunda faktörVII/doku faktörü kompleksi tarafından aktif hale getirilmektedir. Aktif hale gelmiş faktör IX,aktif faktör VIII'le birlikte, faktör X'u aktif hale getirir. Aktif hale gelmiş faktör X, protrombinitrombine çevirir. Trombin daha sonra, fibrinojeni fibrine çevirir ve pıhtı oluşumu sağlanır.Hemofili B, kan pıhtılaşmasının cinsiyete bağlı kalıtımsal bir hastalığıdır; faktör IXdüzeylerinin azalması sonucunda, eklem içi, kaslar ya da iç organlarda kendiliğinden ya dayaralanmaya veya cerrahi girişime bağlı travma sonucunda ağır kanama ile sonuçlanır.Replasman tedavisi ile faktör IX'un plazma düzeyi yükselir ve dolayısıyla, faktör IX eksikliğive kanama eğiliminde geçici bir düzelme sağlanır. Pediyatrik popülasyonIMMUNINE'ın 6 yaşından küçük çocuklarda kullanımım önerebilecek yeterli veri bulunmamaktadır. 5.2. Farmakokinetik özelliklerGenel özelliklerEmilimUygulama yeri açısından intravenöz yolla uygulanan ilaç doğrudan kana karışır. Dağılım26 hastada yapılan bir farmakokinetik çalışmada ortalama rezidüel süresi 23,86 saat olarak bulunmuştur (SD: 5,09; %95GA: 1,85-25,88). Daha önceden tedavi görmüş hastalarda gerçekleştirilen bir faz-4 çalışmasında, 12 yaş ve üzeri olanlarda (hasta sayısı=27) ortalama aşamalı-geri kazanım (IR) her IU/kg başına 1,1 (±0,27)olmuştur (0,6 ile 1,7 IU/dL arası değişen değerler). Aynı çalışmada ortalama IR, daha önceden tedavi görmüş 11 yaş ve altı hastalarda (hasta sayısı=4) 0,9 (± 0,12) olarak bulunmuştur (0,8 ile 1,1 IU/dL arası değişen değerler). BiyotransformasyonMetabolizmasına ait bilgi bulunmamaktadır. EliminasyonBiyolojik yarılanma ömrü yaklaşık 17 saattir. 26 hastada yapılan bir farmakokinetik çalışmada ortalama klerens 8,89 ml/saat/kg olarak bulunmuştur (SD: 2,91; %95GA: 7,72-10,06). Doğrusallık/ Doğrusal olmayan durum5.3. Klinik öncesi güvenlilik verileriIMMUNINE, faktör II, VII ve X'u ancak eser miktarlarda içeren ileri derecede saflaştırılmış bir faktör IX konsantresidir. Laboratuvar hayvanlarına tek doz uygulanan IMMUNINE'ıntoksikolojik ya da trombojenik potansiyeli görülmemiştir. Laboratuvar hayvanlarındaki insan proteinlerinin heterolog karakterinden dolayı, tekrarlayan doz uygulamayla klinik olmayan çalışma yapılması anlamlı değildir. Faktör IX, fizyolojik koşullarda plazmada dolaşan insan kaynaklı bir protein olduğundan üreme üzerinde toksik, mutajenik ya da karsinojenik etki göstermesi beklenmemektedir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiKuru toz: - Sodyum klorür - Trisodyum sitrat-2H2OÇözücü: - Steril Enjeksiyonluk Su 6.2. GeçimsizliklerGeçimlilik çalışmalarının olmaması durumunda, bu tıbbi ürün bölüm 6.6'da belirtilenler dışında başka tıbbi ürünlerle karıştırılmamalıdır. Yalnızca ambalaj içeriğinde bulunan enjeksiyon/infüzyon setleriyle birlikte kullanılmalıdır çünkü insan koagülasyon faktörü IX'un, bazı enjeksiyon/infüzyon ekipmanının içyüzeylerinden adsorbe olması, tedavide başarısızlığa neden olabilir. 6.3. Raf ömrü24 ay. IMMUNINE'ın rekonstitüsyondan sonra fiziksel ve kimyasal stabilitesinin, 25°C'a kadar olan sıcaklıklarda 3 saat olduğu gösterilmiştir. Mikrobiyolojik açıdan, kontaminasyonuengelleyecek valide bir rekonstitüsyon yöntemi kullanılmadıysa, preparat hemenkullanılmalıdır. Hemen kullanılmadığı koşullarda, kullanılana kadar ve kullanım sırasındakisaklama koşulları kullanıcının sorumluluğundadır. Rekonstitüsyondan sonra tekrarbuzdolabına konulmamalıdır. 6.4. Saklamaya yönelik özel tedbirlerBuzdolabında (2°C- 8°C arasında) saklanmalıdır. Dondurulmamalıdır. Işıktan korumak için orijinal ambalajında saklanmalıdır. Belirtilen raf ömrü süresince, 3 ay boyunca oda sıcaklığında (25 °C) saklanabilir. Oda sıcaklığında saklandığı süre, ambalajın üzerindeki son kullanma tarihinin altına not edilmelidir.Oda sıcaklığında saklanmışsa bu dönemin sonunda yeniden buzdolabında saklanmayabaşlanamaz; ya hemen kullanılmalı ya da imha edilmelidir. Sulandırılmış ilacın saklama koşulları için bkz. Bölüm 6.3. 6.5. Ambalajın niteliği ve içeriğiIMMUNINE toz, tek kullanımlık hidrolitik tip II nötral cam flakonlarda sunulmuştur. Çözücü tek kullanımlık hidrolitik tip I nötral cam flakonlarda sunulmuştur. Ürünün flakonu klorobütillastik tıpa ile kapalıdır. Çözücünün flakonu klorobütil veya bromobütil lastik tıpa ile kapalıdır. Ambalaj içeriği:1flakon IMMUNINE 600 IU1 flakon 5 ml steril enjeksiyonluk su 1 transfer iğnesi 1 havalandırma iğnesi 1 filtre iğnesi 1 tek kullanımlık iğne 1 tek kullanımlık enjektör (5 ml) 1 infüzyon seti Ambalaj miktarı: 1 x 600 IU 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller "Tıbbi Atıkların Kontrolü Yönetmeliği" ve "Ambalaj Atıklarının Kontrolü Yönetmelik"lerine uygun olarak imha edilmelidir. Yalnızca verilen enjeksiyon/infüzyon setleri kullanılmalıdır. IMMUNINE uygulamadan hemen önce sulandırılmalıdır. Çözelti hemen sonra kullanılmalıdır (preparat koruyucu bir madde içermez). İnfüzyon sulandırmadan sonraki 3 saat içindetamamlanmalıdır, bkz. Bölüm 6.4. Sulandırılan ürünler, uygulama öncesi herhangi birpartiküler madde içerip içermediği ya da renk değiştirip değiştirmediği açısından kontrol edilmelidir. Çözelti berrak ya da hafif opak görünümde olmalıdır. Bulanık görünümlü ya da partikül içeren çözeltiler kullanılmamalıdır. Uygulama için kullanılan venöz hattın, IMMUNINE infüzyonu öncesi ve sonrasında izotonik sodyum klorür çözeltisi ile yıkanması önerilir. Enjeksiyonluk çözelti hazırlamak üzere tozun sulandırılması:Aseptik teknik kullanınız! 1. Çözücü (steril enjeksiyonluk su) içeren kapalı flakonu oda sıcaklığına getiriniz (maksimum+ 37°C). 2. Toz flakonunun ve çözücü flakonunun koruyucu kapaklarını çıkarınız (Şekil A) ve herikisinin de lastik tıpalarını dezenfekte ediniz. 3. Ambalaj içeriğindeki 'transfer iğnesinin' koruyucu kapağını, bir ucundan döndürerek veçekerek çıkarınız. Görünür hale gelen iğneyi çözücü flakonunun lastik tıpasına batırınız(Şekil B ve C). 4. Transfer iğnesinin diğer ucundaki koruyucu kapağını, açıkta kalan kısımlarına temasetmemeye dikkat ederek çıkarınız. 5. Çözücü flakonunu toz flakonunun üzerinde ters çevirin ve transfer iğnesinin serbest ucunutoz flakonunun kauçuk tıpasından geçirin (Şekil D). Çözücü vakum ile toz flakonunaçekilecektir. 6. İğneyi toz flakonundan çıkararak iki flakonun ayrılmasını sağlayınız (Şekil E). Çözünmeyihızlandırmak için, toz flakonunu hafifçe çalkalayınız veya döndürünüz. 7. Tozun sulandırılması tamamlanınca, ambalaj içeriğindeki 'havalandırma iğnesini' takınız(Şekil F). Oluşmuş bulunan köpüklenme kaybolacaktır. Havalandırma iğnesini çıkarınız. Enjeksiyon / infüzyonAseptik teknik kullanınız ! 1. Ambalaj içeriğindeki 'filtre iğnesinin' koruyucu kapağını, döndürerek ve çekerek çıkarınız ve steril tek kullanımlık bir enjektöre takınız. Çözeltiyi enjektöre çekiniz (Şekil G). 2. Filtre iğnesini enjektörden çıkarınız ve ambalajdaki kelebek infüzyon setini (ya daambalajdaki tek kullanımlık iğneyi) kullanarak çözeltiyi yavaş olarak (enjeksiyon hızıdakikada 2 mL'yi aşmamalıdır) intravenöz enjeksiyon şeklinde uygulayınız. İnfüzyon şeklinde uygulanacaksa, uygun bir filtreye sahip olan tek kullanımlık infüzyon seti kullanılmalıdır.
   Şek. C Şek. DŞek. EŞek.FŞek.G 7. RUHSAT SAHIBITakeda İlaç Sağlık Sanayi Ticaret Limited Şirketi Levent-Şişli/İSTANBUL 8. RUHSAT NUMARASI(LARI)2016/127 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 01.02.2016 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ |
İlaç BilgileriImmunine 600 Iu Iv İnfüzyon İçin Liyofilize Toz İçeren FlakonEtken Maddesi: Insan Koagülasyon Faktörü Ix Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.