Imbruvica 420 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA URUN BILGISI^^Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERI TIBBİ ÜRÜNÜN ADIIMBRUVICA 420 mg Film Kaplı Tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her bir film kaplı tablet 420 mg ibrutinib içerir. Yardımcı maddeler:
84 mg
Laktoz monohidrat (sığır sütü kaynaklı) Yardımcı maddeler için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tablet Bir yüzünde ibr diğer yüzünde 420 mg baskısı bulunan sarı-yeşil ile yeşil arası oblong tablet (17,5 mm uzunluk, 7,4 mm genişlik) 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarIMBRUVICA, Mantle Hücreli Lenfoma (MHL): - En az 3 kür rituksimab ve alkilleyici ajan kombinasyonu sonrası nüks eden veya dirençliolan veya otolog kök hücre nakli sonrası nüks eden mantle hücreli lenfomada (MHL)endikedir. Kronik Lenfositik Lösemi (KLL): - Daha önce tedavi edilmemiş kronik lenfositik lösemi (KLL) tanısı olan yetişkinhastaların tedavisinde monoterapi olarak ya da rituksimab veya obinutuzumab veyavenetoklaks ile kombine kullanımda, (bkz. Bölüm 5.1) - Relaps refrakter yetişkin KLL hastalarının tedavisinde monoterapi olarak ya dabendamustin ve rituksimab (BR) ile kombine kullanımda endikedir. Waldenström Makrogloblunemisi (WM): - Waldenström makrogloblunemisi (WM) tanısı olan ve en az bir sıra tedavi sonrasındarelaps yada dirençli erişkin hastaların tedavisinde ya da kemo-immunoterapi için uygunolmayan hastaların ilk sıra tedavisinde monoterapi veya rituksimabla kombinasyon halindeendikedir. 4.2 Pozoloji ve uygulama şekliBu ilaç ile tedavi, anti-kanser ilaçların kullanımında deneyimli bir hekim tarafından başlatılmalı ve gözlem altında tutulmalıdır. Pozoloji uygulama sıklığı ve süresi:Mantle Hücreli Lenfoma (MHL)MHL tedavisi için önerilen doz günde bir kez 560 mg'dır. Kronik Lenfositik Lösemi (KLL) ve Waldenström Makrogloblunemisi (WM)KLL ve WM için önerilen doz, monoterapi veya kombinasyon halinde günde bir kez ağızdan alınan 420 mg'dır (kombinasyon rejimlerine ilişkin detaylı bilgi için bkz. Bölüm 5.1). IMBRUVICA ile tedavi hastalık progresyonuna ya da hasta tarafından artık tolere edilemez hale gelinceye kadar sürdürülmelidir. KLL tedavisi için venetoklaks ile kombinasyon halindeIMBRUVICA, 3 kür (1 kür 28 gündür) için tek bir ajan olarak uygulanmalı, ardından 12 kürIMBRUVICA ve venetoklaks uygulanmalıdır. Venetoklaks dozaj bilgisi için, VenetoklaksKısa Ürün Bilgisi'ne (KÜB) bakınız. IMBRUVICA, anti-CD20 tedavisiyle kombinasyon halinde uygulanacağı zaman, IMBRUVICA'nın aynı gün verilecek anti-CD20 tedavisinden önce uygulanmasıönerilmektedir. Doz ayarlamalarıOrta güçte ya da güçlü CYP3A4 inhibitörleri ibrutinib maruziyetini artırır (bkz. Bölüm 4.4 ve 4.5). Orta güçte CYP3A4 inhibitörleri ile eş zamanlı kullanıldığında ibrutinib dozu günde 280 mg'a düşürülmelidir. Güçlü CYP3A4 inhibitörleri ile eş zamanlı kullanıldığında ibrutinib dozu günde 140 mg'a düşürülmeli ya da tedaviye 7 güne kadar ara verilmelidir. Herhangi bir yeni başlayan ya da kötüleşen Derece 2 kalp yetmezliği, Derece 3 kardiyak aritmiler, Derece 3 ya da üzerindeki non-hematolojik toksisite, ateş veya enfeksiyon ilebirlikte olan Derece 3 ya da üzerindeki nötropeni veya Derece 4 hematolojik toksisitemeydana geldiğinde IMBRUVICA tedavisi durdurulmalıdır. Toksisite semptomları Derece 1veya başlangıç seviyesine döndüğünde (düzelme), IMBRUVICA tedavisine aşağıdakitablolara göre önerilen dozda devam edilmelidir. Kardiyak olmayan olaylar için önerilen doz modifikasyonları aşağıdaki gibidir:
OlaylarDerece 3 veya 4 hematolojik olmayantoksisiteler Enfeksiyon veya ateşle birlikte Derece 3 veya 4nötropeni 4. Derece hematolojik toksisiteler Tedaviye devam ederken, risk-yarar değerlendirmesine göre aynı veya daha düşük dozda yeniden başlanır. Toksisite tekrar oluşursa, günlük dozu 140 mg azaltılır. Kalp yetmezliği veya kardiyak aritmi olayları için önerilen doz modifikasyonları aşağıdaki gibidir:
Unutulan dozlarEğer bir doz planlanan zamanda alınmazsa, aynı gün içerisinde mümkün olan en kısa zamanda alınabilir ve sonraki gün normal uygulama planına devam edilir. Kaçırılan dozutelafi etmek için fazladan tablet alınmamalıdır. Uygulama şekli:IMBRUVICA her gün yaklaşık aynı saatte, günde bir kere ağızdan bir bardak su ile alınmalıdır. Tabletler bütün olarak yutulmalı, kırılmamalı ya da çiğnenmemelidir.IMBRUVICA greyfurt suyu veya turunçgiller ile alınmamalıdır (bkz. Bölüm 4.5). Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek yetmezliği olan hastalarda özel klinik çalışmalar yapılmamıştır. Hafif ya da orta dereceli böbrek yetmezliği olan hastalar IMBRUVICA klinik çalışmalarında tedavi edilmiştir.Hafif ya da orta dereceli böbrek yetmezliği olan (kreatinin klerensi 30 mL/dakikadan büyük)hastalarda doz ayarlaması gerekli değildir. Hidrasyon sağlanmalı ve serum kreatinin düzeyleriperiyodik olarak takip edilmelidir. Şiddetli böbrek yetmezliği (kreatinin klerensi < 30mL/dak) hastalarında, IMBRUVICA ancak fayda riskten ağır bastığı takdirde kullanılmalı vehastalar toksisite belirtileri açısından yakından izlenmelidir. Şiddetli böbrek yetmezliğihastalarında ya da diyaliz hastalarında veri mevcut değildir (bkz. Bölüm 5.2). Karaciğer yetmezliği:İbrutinib karaciğerde metabolize edilir. Bir karaciğer yetmezliği çalışmasında, veriler ibrutinib maruziyetinde artış ortaya koymuştur (bkz. Bölüm 5.2). Hafif dereceli karaciğeryetmezliği olan hastalarda (Child-Pugh sınıfı A), önerilen doz günde 280 mg'dır. Ortadereceli karaciğer yetmezliği olan hastalarda (Child-Pugh sınıfı B), önerilen doz günde 140mg'dır. Hastalar IMBRUVICA toksisitesi belirtileri açısından izlenir ve gerektiği gibi dozmodifikasyon kılavuzları takip edilir. Şiddetli karaciğer yetmezliği olan hastalardaIMBRUVICA kullanılması önerilmez (Child-Pugh sınıfı C). Şiddetli kalp hastalığı:Şiddetli kalp hastalığı olan hastalar IMBRUVICA'nın klinik çalışmalarından dışlanmıştır. Pediyatrik popülasyon:IMBRUVICA'nın 0-18 yaş arası çocuklarda ve adolesanlarda etkililiği ortaya konmadığından kullanımı tavsiye edilmemektedir. Matür B hücreli non-Hodgkin lenfomlı hastalara ilişkinmevcut veriler Bölüm 4.8, 5.1 ve 5.2'de açıklanmaktadır. Geriyatrik popülasyon:Yaşlı hastalarda (65 yaş ve üstü) doz ayarlaması gerekli değildir. 4.3 KontrendikasyonlarEtkin maddeye ya da Bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılık durumunda ve St. John's Wort (Sarı kantaron) içeren ürünler ile birlikte IMBRUVICA kullanımı kontrendikedir. 4.4 Özel kullanım uyarıları ve önlemleriKanama ilişkili olaylarIMBRUVICA ile tedavi edilen hastalarda, trombositopeninin eşlik ettiği ve etmediği kanama olay bildirimleri olmuştur. Bunlar arasında kontüzyon, burun kanaması ve peteşi gibi minorkanama olayları ve gastrointestinal kanama, intrakraniyal kanama ve hematüri gibi bazılarıölümcül olan, majör kanama olayları yer alır. Varfarin ya da diğer K vitamini antagonistleri IMBRUVICA ile eş zamanlı olarak kullanılmamalıdır. IMBRUVICA ile birlikte antikoagülan veya trombosit fonksiyonunu inhibe eden (antiplatelet ajanlar) tıbbi ürünlerin eş zamanlı kullanılması majör kanama riskini arttırır.Antikoagülanlarda, antiplatelet ajanlardan daha fazla majör kanama riski gözlendi.IMBRUVICA ile birlikte uygulandığında, antikoagülan veya antiplatelet tedavisinin risklerinive yararlarını göz önünde bulundurun. Kanama bulgu ve semptomları için hasta izlenmelidir. Balık yağı ve E vitamini gibi gıda takviyelerinin kullanımından kaçınılmalıdır. IMBRUVICA tedavisi cerrahinin tipine ve kanama riskine bağlı olarak, cerrahi öncesinde ve sonrasında en az 3 ila 7 gün kesilmelidir. Kanama ile ilişkili olayların mekanizması tam olarak anlaşılamamıştır. Konjenital kanama diyatezi olan hastalar incelenmemiştir. LökostazIMBRUVICA ile tedavi edilen hastalarda lökostaz vakaları bildirilmiştir. Dolaşımdaki lenfosit sayısının yüksek olması (> 400.000/mcL) riskte artışa neden olabilir. IMBRUVICAtedavisinin geçici olarak ara verilmesi düşünülmelidir. Hastalar yakından izlenmelidir. Belirtildiği şekilde hidrasyon ve/veya sitoredüksiyon dahil, destekleyici bakım uygulanmalıdır. Dalak rüptürüIMBRUVICA tedavisinin kesilmesinin ardından dalak rüptürü vakaları bildirilmiştir. IMBRUVICA tedavisine ara veya son verildiğinde, hastalık durumu ve dalak boyutu dikkatleizlenmelidir (örn. klinik muayene, ultrason). Sol üst karın veya omuz ucu ağrısı gelişenhastalar değerlendirilmeli ve dalak rüptürü tanısı düşünülmelidir. EnfeksiyonlarIMBRUVICA ile tedavi edilen hastalarda enfeksiyonlar gözlenmiştir (sepsis, nötropenik sepsis, bakteri, virüs ya da mantar enfeksiyonları dahil). Bu enfeksiyonların bazıları hastaneyeyatma ve ölümle sonuçlanmıştır. Ölümcül enfeksiyonları olan hastaların çoğunda nötropeni devardır. Hastalar ateş, anormal karaciğer fonksiyon testleri, nötropeni ve enfeksiyonlaraçısından izlenmeli ve endike olduğu gibi uygun anti-enfektif tedavi başlatılmalıdır. Fırsatçıenfeksiyon açısından artmış risk altında olan hastalar için standart tedaviye göre profilaksidüşünülmelidir. İbrutinib kullanımını takiben Aspergillozis, Kriptokokkozis ve Pnömosistis jiroveci enfeksiyonları vakaları dahil olmak üzere invazif fungal enfeksiyon vakaları bildirilmiştir.Rapor edilen invaziv mantar enfeksiyonu vakaları ölümcül sonuçlarla ilişkilendirilmiştir. Geçmişte ya da eşzamanlı alınan immünosupresif tedavi koşullarında ölümcül olanları dahil olmak üzere Progresif Multifokal Lökoensefalopati (PML) vakaları rapor edilmiştir. Hekimleryeni ya da kötüleşen nörolojik, bilişsel ya da davranışsal belirti veya semptomları olanhastalarda PML'yi ayırıcı tanıda değerlendirmelidir. PML'den şüphelenildiği takdirde, uyguntanısal değerlendirmeler gerçekleştirilmeli ve PML ekarte edilene kadar tedavidurdurulmalıdır. Herhangi bir şüphe varsa, hastanın bir nöroloji uzmanına sevk edilmesi vePML için tercihen kontrastlı MRG, JC Viral DNA için beyin-omurilik sıvısı (BOS) testi vetekrarlayan nörolojik değerlendirmeler gibi uygun tanısal ölçümler düşünülmelidir. Hepatik olaylarIMBRUVICA ile tedavi edilen hastalarda hepatotoksisite, hepatit B reaktivasyonu ve kronik olabilen hepatit E vakaları meydana gelmiştir. IMBRUVICA ile tedavi edilen hastalardaölümcül olaylar dahil karaciğer yetmezliği meydana gelmiştir. IMBRUVICA ile tedaviyebaşlamadan önce karaciğer fonksiyonu ve viral hepatit durumu değerlendirilmelidir. Hastalar,tedavi sırasında karaciğer fonksiyon parametrelerindeki değişiklikler için periyodik olarakizlenmelidir. Klinik olarak belirtildiği gibi, enfeksiyöz hepatit için viral yük ve serolojiktestler, yerel tıbbi kılavuzlara göre yapılmalıdır. Hepatik olay teşhisi konan hastalar için,yönetim için bir karaciğer hastalığı uzmanına danışmayı düşünün. SitopenilerIMBRUVICA ile tedavi sırasında ortaya çıkan Derece 3 ya da 4 sitopeniler (nötropeni, trombositopeni ve anemi) bildirilmiştir. Ayda bir tam kan sayımları izlenmelidir. İnterstisyel Akciğer Hastalığı (İAH)IMBRUVICA ile tedavi edilen hastalarda İAH vakaları bildirilmiştir. Hastalar, İAH'na işaret eden akciğer semptomları bakımından izlenmelidir. Semptomların ortaya çıkması durumunda,IMBRUVICA'ya ara verilmeli ve İAH uygun şekilde tedavi edilmelidir. Semptomlarındevam etmesi halinde, IMBRUVICA tedavisinin risk ve faydaları hesaplanmalı ve dozmodifikasyonu kılavuzuna uyulmalıdır. Kardiyak aritmiler ve kardiyak yetmezlikIMBRUVICA ile tedavi edilen hastalarda ölümcül ve ciddi kardiyak aritmiler ve kalp yetmezliği meydana gelmiştir. İleri yaşlı, Eastern Cooperative Oncology Group (ECOG)performans durumu >2 olan veya kardiyak komorbiditeleri olan hastalar ani ölümcül kardiyakolaylar açısından daha yüksek riske sahip olabilirler.Özellikle akut enfeksiyonlar ya dakardiyak risk faktörleri, hipertansiyon dahil, diyabet hastalığı ve geçmiş bir kardiyak aritmiöyküsü olan hastalarda atriyal fibrilasyon, atriyal flutter ve ventriküler taşiaritmi ve kardiyakyetmezlik vakaları bildirilmiştir. IMBRUVICA'ya başlamadan önce klinik olarak uygun kardiyak öykü ve fonksiyonun değerlendirmesi yapılmalıdır. Hastalar, tedavi sırasında kardiyak fonksiyonda klinik kötüyegitme belirtileri açısından dikkatle izlenmeli ve klinik olarak değerlendirilmelidir.Kardiyovasküler açıdan endişeler olan hastalar için ileri değerlendirme (örn., EKG,ekokardiyogram) düşünülmelidir. Kardiyak olaylar için ilgili risk faktörleri olan hastalarda, alternatif tedavi IMBRUVICA ile tedaviye başlamadan önce dikkatlice yarar/risk değerlendirilerek düşünülebilir. Ventriküler taşiaritmi belirti ve/veya semptomları gelişen hastalarda, IMBRUVICA geçici olarak kesilmeli ve tedaviye yeniden başlamadan önce kapsamlı bir klinik yarar/riskdeğerlendirmesi gerçekleştirilmelidir. Antikoagülan tedavisi gerektiren, halihazırda atriyal fibrilasyonu olan hastalarda, IMBRUVICA'ya alternatif tedaviler düşünülmelidir. IMBRUVICA ile tedavi sırasında atriyalfibrilasyon gelişirse, hastalar tromboembolik hastalık riski açısından incelenmelidir. Yüksekriski olan ve IMBRUVICA alternatiflerinin uygun olmadığı hastalarda, antikoagülanlar ileçok yakından takip ederek tedavi düşünülmelidir. IMBRUVICA tedavisi sırasında hastalar kardiyak yetmezlik bulgu ve belirtileri açısından izlenmelidir. Bu vakaların bazılarında, IMBRUVICA'nın kesilmesinden veya dozunazaltılmasından sonra kalp yetmezliği düzelmiş veya iyileşmiştir. Serebrovasküler olaylarEşlik eden atriyal fibrilasyon ve/veya hipertansiyonu olan ve olmayan IMBRUVICA ile tedavi edilen hastalarda, serebrovasküler olay, geçici iskemik atak ve ölümleri içeren iskemikinme vakaları bildirilmiştir. Bildirilen gecikmeli vakalar arasında, IMBRUVICA ile tedaviyebaşlanmasından merkezi sinir iskemik vasküler durumlarının başlangıcına kadar çoğu vakadabirkaç ay bulunması (%78'inde 1 aydan fazla ve vakaların %44'ünde 6 aydan fazla) hastalarındüzenli olarak izlenmesine ihtiyaç olduğunu vurguladı (bkz. Bölüm 4.4 Kardiyak aritmi veHipertansiyon ve Bölüm 4.8). Tümör lizis sendromuTümör lizis sendromu (TLS) IMBRUVICA tedavisinde bildirilmiştir. Tümör lizis sendromu riski taşıyan hastalar, tedavi öncesi tümör yükü yüksek olan hastalardır. Hastalar yakındantakip edilmeli ve uygun önlemler alınmalıdır. Non-melanom cilt kanseriHavuzlanmış randomize Faz 3 karşılaştırmalı çalışmalarda, IMBRUVICA ile tedavi edilen hastalarda karşılaştırma kollarına oranla daha sık non-melanom cilt kanseri bildirilmiştir.Hastalar non-melanom cilt kanseri açısından yakından takip edilmelidir. HipertansiyonIMBRUVICA ile tedavi edilen hastalarda hipertansiyon meydana gelmiştir (bkz. Bölüm 4.8). IMBRUVICA ile tedavi edilen hastalarda kan basıncı düzenli olarak izlenmelidir.IMBRUVICA ile tedavi boyunca uygun görüldüğü takdirde antihipertansif ilaçlar başlatılmalıveya dozu ayarlanmalıdır. Hemofagositik lenfohistiyositoz (HLH)IMBRUVICA ile tedavi edilen hastalarda HLH vakaları (ölümcül vakalar dahil) bildirilmiştir. HLH, aşırı sistemik inflamasyonun klinik belirti ve semptomları ile karakterize, yaşamı tehditeden bir patolojik immün aktivasyon sendromudur. HLH ateş, hepatosplenomegali,hipertrigliseridemi, yüksek serum ferritin ve sitopeniler ile karakterizedir. Hastalar HLHsemptomları hakkında bilgilendirilmelidir. Patolojik immün aktivasyonun erken belirtilerinigeliştiren hastalar derhal değerlendirilmeli ve HLH tanısı düşünülmelidir. İlaç-ilaç etkileşimleriIMBRUVICA'nın güçlü ya da orta dereceli CYP3A4 inhibitörleri ile eş zamanlı kullanımı ibrutinib maruziyetini artırabilir ve dolayısıyla toksisite için yüksek risk oluşturabilir. Öteyandan, CYP3A4 enzimini indükleyen ilaçların eş zamanlı kullanımı IMBRUVICAmaruziyetini azaltabilir ve dolayısıyla etkililikte azalma riski ortaya çıkabilir. Bu sebeple,IMBRUVICA'nın güçlü CYP3A4 inhibitörleri ve güçlü ya da orta dereceli CYP3A4indükleyicileri ile eş zamanlı kullanımından mümkün olduğunca kaçınılmalı ve eş zamanlıkullanım sadece potansiyel yarar potansiyel zarardan üstün olduğunda düşünülmelidir.CYP3A4 inhibitörü kullanılması zorunlu olan hastalar, IMBRUVICA toksisite belirtileriaçısından yakından izlenmelidir (bkz. Bölüm 4.2 ve 4.5). Eğer bir CYP3A4 indükleyicisininkullanılması gerekiyorsa, hastalar IMBRUVICA etkisizlik riski belirtileri açısındanizlenmelidir. Çocuk doğurma potansiyeline sahip kadınlarÇocuk doğurma potansiyeline sahip kadınlar IMBRUVICA kullanırken yüksek düzeyde etkili bir doğum kontrol yöntemi kullanmalıdır (bkz. Bölüm 4.6). Yardımcı maddelere intoleransNadir kalıtımsal galaktoz intolerans problemi, lapp laktaz yetmezliği ya da glukoz -galaktoz malabsorsiyonu olan hastaların bu ilacı kullanmamaları gerekir. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriİbrutinib primer olarak sitokrom P450 enzim 3A4 (CYP3A4) ile metabolize edilir. İbrutinibin plazma konsantrasyonlarını yükseltebilen ilaçlar:Orta güçte ya da güçlü CYP3A4 inhibitörlerinin IMBRUVICA ile eş zamanlı kullanımı ibrutinib maruziyetini arttırabilir ve güçlü CYP3A4 inhibitörlerinin kullanımdankaçınılmalıdır. Güçlü CYP3A4 inhibitörleri18 sağlıklı gönüllüde, çok güçlü bir CYP3A4 inhibitörü olan ketokonazolün aç karnına eş zamanlı uygulaması ibrutinib maruziyetini (Cmaks ve EAA) sırasıyla 29 ve 24 kat arttırmıştır.Aç karnına koşullarının kullanıldığı simülasyonlar güçlü bir CYP3A4 inhibitörü olanklaritromisinin ibrutinib EAA'sını 14 kat etkilediğini göstermektedir. IMBRUVICA'yı yemekile birlikte alan B-hücreli malignitesi olan hastalarda, güçlü CYP3A4 inhibitörü olanvorikonazolün eş zamanlı uygulanması Cmaks'ı 6,7 kat ve EAA'yı 5,7 kat artırmıştır. GüçlüCYP3A4 inhibitörlerin (örneğin ketokonazol, indinavir, nelfinavir, ritonavir, sakuinavir,klaritromisin, telitromisin, itrakonazol, nefazadon, kobisistat, vorikonazol ve posakonazol)kullanımından kaçınılmalıdır. Eğer fayda riskten fazlaysa ve güçlü bir CYP3A4 inhibitörükullanımı zorunluysa IMBRUVICA dozu inhibitör kullanımı süresince 140 mg'a indirilir yada IMBRUVICA geçici bir süre kesilir (7 gün ya da daha kısa bir süre). Hastalar toksisiteaçısından yakından izlenmeli ve gerektiği gibi doz modifikasyon kılavuzları takip edilmelidir(bkz. Bölüm 4.2 ve 4.4). Orta güçte CYP3A4 inhibitörleriIMBRUVICA'yı yemek ile birlikte alan B-hücreli malignitesi olan hastalarda CYP3A4 inhibitörü olan eritromisinin eş zamanlı uygulanması Cmaks'ı 3,4 kat ve EAA'yı 3 katartırmıştır. Eğer orta güçte bir CYP3A4 inhibitörü (örneğin flukonazol, eritromisin,amprenavir, aprepitant, atazanavir, siprofloksasin, krizotinib, diltiazem, fosamprenavir,imatinib, verapamil, amiodaron ve dronedaron) endike ise, IMBRUVICA dozu inhibitörkullanımı süresince 280 mg'a indirilmelidir. Hastalar toksisite açısından yakından izlenmelive gerektiği gibi doz modifikasyon kılavuzları takip edilmelidir (bkz. Bölüm 4.2 ve 4.4). Zayıf CYP3A4 inhibitörleriAç karnına koşullarının kullanıldığı simülasyonlar zayıf CYP3A4 inhibitörleri olan azitromisin ve fluvoksaminin ibrutinib EAA'sını 2 kattan az yükseltebileceğini göstermiştir.Zayıf güçte inhibitörler ile kombinasyonunda herhangi bir doz ayarlaması gerekli değildir.Hastalar toksisite açısından yakından izlenmeli ve gerektiği gibi doz modifikasyon kılavuzlarıtakip edilmelidir. CYP3A4 inhibitörü olan greyfurt suyunun eş zamanlı kullanımının değerlendirildiği 8 sağlıklı hastada ibrutinib maruziyeti (Cmaks ve EAA) sırasıyla 4 ve 2 kat artmıştır. Orta güçte CYP3A4inhibitörleri içerdikleri için, IMBRUVICA tedavisi sırasında greyfurt suyu ve turunçgillertüketilmemelidir (bkz. Bölüm 4.2). İbrutinibin plazma konsantrasyonlarını azaltabilen ilaçlar:IMBRUVICA'nın CYP3A4 indükleyicileri ile birlikte kullanılması ibrutinibin plazma konsantrasyonlarını azaltabilir. 18 sağlıklı gönüllüde, güçlü bir CYP3A4 indükleyicisi olan rifampisinin aç karnına eş zamanlı uygulaması ibrutinib maruziyetini (Cmaks ve EAA) sırasıyla %92 ve %90 azaltmıştır.Güçlü veya orta güçte CYP3A4 indükleyicilerinin (örneğin karbamazepin, rifampisin,fenitoin) eş zamanlı kullanımından kaçınılmalıdır. St. John's Wort (Sarı kantaron) içerenpreparatlar ile IMBRUVICA'nın eş zamanlı kullanımı etkililik azalabileceğindenkontrendikedir. Daha düşük bir CYP3A4 indüksiyonu sağlayan alternatif ilaçlardüşünülmelidir. Eğer fayda riskten fazlaysa ve güçlü veya orta güçte bir CYP3A4indükleyicisinin kullanımı zorunluysa hastalar etkisizlik açısından yakından takip edilmelidir(bkz. Bölüm 4.3 ve 4.4). Zayıf indükleyiciler IMBRUVICA ile eş zamanlı kullanılabilir amayine de hastalar etkisizlik açısından yakından takip edilmelidir. İbrutinib pH'a bağlı çözünürlük sergiler ve daha yüksek pH değerinde daha düşük çözünürlüğe sahiptir. 5 gün süreyle günde bir kere 40 mg omeprazol aldıktan sonra aç karnına560 mg'lık tek bir ibrutinib dozu uygulanan sağlıklı gönüllülerde daha düşük bir Cmaks değerigözlenmiştir (bkz. Bölüm 5.2). Daha düşük bir Cmaks değerinin klinik anlamı olduğuna dairhiçbir bulgu mevcut değildir ve midenin pH değerini artıran tıbbi ürünler (örneğin protonpompası inhibitörleri) pivot klinik çalışmalarda hiçbir kısıtlama olmaksızın kullanılmıştır. Plazma konsantrasyonları ibrutinib ile değişen ilaçlar:İbrutinib in vitrobir P-gp ve meme kanseri rezistan proteini (BCRP) inhibitörüdür. Bu etkileşimi kanıtlayan bir klinik çalışma olmadığından, ibrutinibin terapötik bir dozdaintestinal P-gp'yi ve BCRP'yi inhibe edebileceği de olasılık dışı bırakılamaz. Gastrointestinalkanalda bir etkileşim potansiyelini en aza indirmek üzere, digoksin veya metotreksat gibi oraldar bir terapötik aralığına sahip P-gp veya BCRP substratları IMBRUVICA'dan en az 6 saatönce ya da sonra kullanılmalıdır. İbrutinib aynı zamanda karaciğerde BCRP'yi inhibe edebilirve BCRP-aracılı hepatik atılıma uğrayan tıbbi ürünlerin, rosuvastatin gibi, maruziyetini artırır.Venetoklaks (400 mg) ile kombinasyon halinde ibrutinib (420 mg) çalışmalarındaki KLL hastalarında, venetoklaks için monoterapi verilerine kıyasla venetoklaks maruziyetinde birartış (EAA bazında yaklaşık 1,8 kat) gözlenmiştir. B-hücreli maligniteleri olan hastalarda yapılan bir ilaç etkileşimi çalışmasında, tek seferlik 560 mg ibrutinib dozunun, CYP3A4 substratı midazolamın maruziyeti üzerinde klinik olarakanlamlı bir etkisi olmamıştır. Aynı çalışmada, günde 560 mg ibrutinib ile 2 haftalık tedavininoral kontraseptiflerin (etinilestradiol ve levonorgestrel), CYP3A4 substratı midazolamın veyaCYP2B6 substratı bupropionun farmakokinetiği üzerinde klinik olarak anlamlı bir etkisiolmamıştır. Özel popülasyonlara ilişkin ek bilgilerHerhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:Herhangi bir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve LaktasyonGenel tavsiyeGebelik kategorisi:Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Hayvanlardan elde edilen bulgulara dayalı olarak, IMBRUVICA gebe kadınlara uygulandığında fetusa zarar verebilir. IMBRUVICA kullanırken ve tedavinin tamamlanmasınıtakiben 3 ay süresince kadınlar gebe kalmaktan kaçınmalıdır. Bu sebeple çocuk doğurmapotansiyeline sahip kadınlar IMBRUVICA kullanırken ve tedavinin tamamlanmasını takiben3 ay süresince yüksek düzeyde etkili bir doğum kontrol yöntemi kullanmalıdır. İbrutinibinhormonal doğum kontrol yöntemlerini etkileyip etkilemediği henüz bilinmemektedir. Busebeple, hormonal doğum kontrol yöntemi kullanan hastalar ikinci bir bariyer yöntemi deilave etmelidir. Gebelik dönemiIMBRUVICA gebelik sırasında kullanılmamalıdır. IMBRUVICA'nın gebe kadınlarda kullanımına ilişkin veri mevcut değildir. Hayvan çalışmaları üreme toksisitesi ortayakoymuştur (bkz. Bölüm 5.3). Laktasyon dönemiİbrutinibin veya metabolitlerinin anne sütüyle atılıp atılmadığı bilinmemektedir. Emzirilen bebeğe olan riskler göz ardı edilemez. IMBRUVICA tedavisi sırasında emzirme kesilmelidir. FertiliteErkek ya da dişi sıçanlarda test edilen maksimum doza kadar (100 mg/kg/gün - insana eşdeğer doz 16 mg/kg/gün) fertilite ya da üreme kapasitesine bir etki görülmemiştir (bkz.Bölüm 5.3). İbrutinibin insan fertilitesine etkileri üzerine veri bulunmamaktadır. 4.7 Araç ve makine kullanımı üzerindeki etkilerIMBRUVICA'nın araç ve makine kullanma yeteneği üzerinde minör bir etkisi bulunmaktadır. IMBRUVICA kullanan bazı hastalarda yorgunluk, baş dönmesi ve asteni bildirilmiş olup, bir hastanın taşıt ya da makine kullanma kapasitesini değerlendirirken bu dikkate alınmalıdır. 4.8 İstenmeyen etkilerGüvenlilik profili özetiGüvenlilik profili, dört Faz 2 klinik çalışmada ve sekiz randomize Faz 3 çalışmada IMBRUVICA ile tedavi edilen 1981 hastadan ve pazarlama sonrası deneyimlerdenbirleştirilen verilere dayanmaktadır. Klinik çalışmalarda MHL ile tedavi edilen hastalar gündebir kez 560 mg IMBRUVICA almış ve klinik çalışmalarda KLL ya da Waldenströmmakroglobulinemisi (WM) için tedavi edilen hastalar da günde bir kez 420 mg IMBRUVICAalmıştır. Hastalara IMBRUVICA ile venetoklaksın kombinasyon halinde sabit süreli verildiğiçalışmalar (CLL3011 ve PCYC-1142-CA çalışmaları) haricinde klinik çalışmalardaki tümhastalar IMBRUVICA'yı hastalık progresyonuna ya da artık tolere edilemez hale gelenekadar almıştır. Birleştirilmiş veri seti genelinde IMBRUVICA tedavisinin medyan süresi 14,7aydı. KLL/SLL için medyan tedavi süresi 14,7 aydı (52 aya kadar); MHL için medyan tedavisüresi 11,7 aydı (28 aya kadar); WM için medyan tedavi süresi 21,6 aydı (37 aya kadar). En yaygın görülen advers ilaç reaksiyonları (> %20) diyare, nötropeni, kas-iskelet ağrısı, döküntü, hemoraji (örneğin morarma), döküntü, bulantı, trombositopeni, artralji ve üstsolunum yolu enfeksiyonu olmuştur. En yaygın Derece 3 veya 4 advers ilaç reaksiyonları (>%5) arasında nötropeni, lenfositoz, trombositopeni, hipertansiyon ve pnömoni yer almıştır. Tablolaştırılmış advers ilaç reaksiyonlarıB-hücreli maligniteleri olan hastaların tedavilerinde advers ilaç reaksiyonları ve pazarlama sonrası advers reaksiyonlar sistem organ sınıfına ve sıklık derecelerine göre aşağıdalistelenmiştir. Sıklık dereceleri şöyledir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10);yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek(<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). İstenmeyen etkilerher sıklık gruplamasında ciddiyette azalma sırasına göre sunulmaktadır. Tablo 1: B-hücreli Maligniteleri Olan Hastalarda Klinik Çalışmalarda ve Pazarlama Sonrası Raporlanan Advers Reaksiyonlar^
* Çoklu advers reaksiyon şartlarını içerir. * Bazı durumlarda görme kaybı ile ilişkili. * Ölümle sonuçlanan olayları da içerir. @ Seçim için kullanılan düşük düzey terimi (LLT). Seçilmiş advers reaksiyonların tanımıAdvers reaksiyonlara bağlı tedavi sonlandırması ve doz azaltımı B-hücreli maligniteler için IMBRUVICA ile tedavi olan 1981 hastadan %6'sı temelde advers ilaç reaksiyonlarına bağlı olarak tedaviyi kesmiştir. Bu advers ilaç reaksiyonlarının başındapnömoni, atriyal fibrilasyon, nötropeni, döküntü, trombositopeni ve kanama gelir. Hastalarınyaklaşık %8'inde dozun azaltılmasına neden olan advers ilaç reaksiyonları görülmüştür. Geriatrik popülasyon IMBRUVICA ile tedavi gören 1981 hastanın %50'si 65 yaş veya daha üzerinde idi. IMBRUVICA ile tedavi edilen yaşlı hastalarda Derece 3 veya daha yüksek pnömoni (65 yaşve üzeri hastaların %11'ine karşılık 65 yaş altındaki hastaların %4'ünde) ve trombositopeni(65 yaş ve üzeri hastaların %11'ine karşılık 65 yaş altındakilerin %5'i) daha sık meydanagelmiştir. Uzun dönem güvenlilik verileri IMBRUVICA ile tedavi edilen 1284 hastada (tedavi görmemiş KLL/SLL n=162, nüks/dirençli KLL/SLL n=646, nüks/dirençli MHL n=370 ve WM n=106) 5 yıllık bir süredeelde edilen uzun süreli güvenlilik verileri analiz edilmiştir. KLL/SLL için medyan tedavisüresi 51 aydır (aralık; 0,2 ila 98 ay) ve hastaların %70'i 2 yıl ve %52'si ise 4 yıldan uzun birsüreyle tedavi görmüştür. MHL için medyan tedavi süresi 11 aydır (aralık; 0 ila 87 ay) vehastaların %31'i 2 yıl ve %17'si ise 4 yıldan uzun süreyle tedavi görmüştür. WM için medyan tedavi süresi 47 aydır (aralık; 0,3 ila 61 ay) ve hastaların %78'i 2 yıl ve %46'si ise 4 yıldan uzun süreyle tedavi görmüştür. IMBRUVICA'ya maruz kalan hastaların bilinen genelgüvenlik profili tutarlı kalırken hipertansiyon prevalansındaki bir artış dışında hiçbir yenigüvenlilik endişesi belirlenmemiştir. Derece 3 veya üzeri hipertansiyon prevalansı %4 (0-1yıl), %7 (1-2 yıl), %9 (2-3 yıl), %9 (3-4 yıl) ve %9 (4-5 yıl) ve 5 yıllık dönemdeki insidansı%11 olmuştur. Pediatrik popülasyon Güvenlik değerlendirmesi, rituksimab, ifosfamid, karboplatin, etoposid ve deksametazon (RICE) rejimi veya rituksimab, vinkristin, ifosfamid, karboplatin, idarubisin ve deksametazon(RVICI) rejimi ile kombinasyon halinde arka plan tedavisi olarak ya da nüks eden veyarefrakter matür B hücreli non-Hodgkin lenfomalı pediyatrik ve genç erişkin hastalarda (3 ila19 yaş arası) tek başına arka plan tedavisi olarak IMBRUVICA'nın bir Faz 3 çalışmasındanelde edilen verilere dayanmaktadır (bkz. bölüm 5.1). Bu çalışmada hiçbir yeni adversreaksiyon gözlenmemiştir. Şüpheli advers reaksiyonların raporlanması Ruhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99). 4.9 Doz aşımı ve tedavisiIMBRUVICA doz aşımının etkilerine ilişkin veriler sınırlıdır. Hastaların 12,5 mg/kg/gün'e kadar dozlar kullandığı (1.400 mg/gün) Faz 1 çalışmasında maksimum tolere edilen bir dozaulaşılmamıştır. Başka bir çalışmada, 1.680 mg dozunda ilaç kullanan sağlıklı bir birey Derece4 geri dönüşümlü hepatik enzim artışı [aspartat aminotransferaz (AST) ve alaninaminotransferaz (ALT)] deneyimlemiştir. IMBRUVICA'nın spesifik bir antidotu yoktur.Önerilen dozdan fazlasını alan hastalar yakından izlenmeli ve uygun destekleyici tedavialmalıdır. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: Antineoplastik ajanlar, protein kinaz inhibitörleri, ATC kodu: L01EL01. Etki mekanizmasıİbrutinib küçük moleküllü, güçlü bir BTK (Bruton tirozin kinaz) inhibitörüdür. İbrutinib, BTK aktif bölgesindeki bir sistein molekülüyle (Cys-481) kovalent bağ oluşturarak, BTKenzimatik aktivitesinin sürekli inhibisyonuna yol açar. Tec kinaz ailesinin bir üyesi olan BTK,B hücresi antijen reseptörünün (BCR) ve sitokin reseptörü yolaklarının önemli bir sinyalmolekülüdür. BCR yolağı, Mantle hücreli lenfoma (MHL), Diffüz Büyük B Hücreli Lenfoma(DLBCL), foliküler lenfoma ve KLL dahil, çeşitli B-hücreli malignitelerin patogenezinde roloynamaktadır. BTK'nin B hücresi yüzey reseptörleri aracılı gerçekleşen sinyal iletimindekirolü, B-hücre trafiği, kemotaksis ve adhezyon için gerekli yolakların aktivasyonuylasonuçlanır. Preklinik çalışmalar, ibrutinibin in-vivoin-vitrohücre göçü ve substrat adezyonunu engellediğini göstermektedir.Klinik öncesi tümör modellerinde, ibrutinib ve venetoklaks kombinasyonu, her iki ajanın tek başına kullanılmasına kıyasla artmış hücresel apoptoz ve anti-tümör aktivite ilesonuçlanmıştır. İbrutinib tarafından gerçekleştirilen BTK inhibisyonu, KLL hücrelerinin birhücre sağkalım yolu olan BCL-2'ye bağımlılığını arttırırken, venetoklaks da BCL-2'yi inhibeederek apoptoza öncülük eder. LenfositozTedaviye başlanmasıyla birlikte, IMBRUVICA ile tedavi edilen KLL hastalarının yaklaşık dörtte üçünde sıklıkla lenfadenopatide küçülme ile ilişkili, geri dönüşümlü bir lenfosit artışı(yani başlangıca göre %50 ve üzerinde bir artış ve mutlak sayısı 5.000/mcL'nin üzerinde)gözlenmiştir. Bu etki ayrıca IMBRUVICA ile tedavi gören relaps ya da refrakter MHLhastalarının da üçte birinde görülmüştür. Gözlenen bu lenfositoz farmakodinamik bir etkidirve diğer klinik bulguların yokluğunda progresif hastalık olarak değerlendirilmemelidir. Heriki hastalık tipinde de lenfositoz tipik olarak IMBRUVICA tedavisinin ilk ayında meydanagelir ve tipik olarak MHL hastalarında medyan 8,0 hafta ve KLL hastalarında medyan 14haftada düzelir. Bazı hastalarda dolaşımdaki lenfosit sayısında büyük artışlar görülmüştür(örneğin >400.000/mcL). IMBRUVICA ile tedavisi gören WM'li hastalarda lenfositoz gözlenmemiştir. In vitroplatelet agregasyonuBir in vitroçalışmada, ibrutinib kolajen etkisi ile oluşan platelet agregasyon inhibisyonu göstermiştir. İbrutinib, platelet agregasyonunun diğer agonistlerini kullanarak anlamlı birplatelet agregasyon inhibisyonu göstermemiştir.Kardiyak elektrofizyoloji ve QT/QTc aralığı üzerindeki etkiİbrutinibin QTc aralığı üzerindeki etkisi, plasebo ve pozitif kontrollerle randomize, çift kör ayrıntılı bir QT çalışmasında 20 sağlıklı erkek ve kadın gönüllüde değerlendirilmiştir.İbrutinib 1.680 mg'lik supraterapötik bir dozda QTc aralığını klinik olarak ilgili bir derecedeuzatmamıştır. İbrutinib ile plasebo arasındaki başlangıçtaki düzeltilmiş ortalama bakımındanfarklılıklar için 2 yanlı %90 GA'nın en büyük üst sınırı 10 ms'nin altında olmuştur. Aynıçalışmada, QTc aralığında konsantrasyona bağlı bir kısalma gözlenmiştir (1.680 mg'lıksupraterapötik dozu takiben 719 ng/mL Cmaks değerinde -5,3 ms [%90 GA: -9,4, -1,1]). Klinik etkililik ve güvenlilikMHLRelaps ya da refrakter MHL hastalarında IMBRUVICA'nın güvenliliği ve etkililiği, 111 hastayı kapsayan açık-etiketli, çok-merkezli bir Faz 2 çalışmada değerlendirilmiştir (PCYC-1104-CA Çalışması). Medyan yaş 68 yıl idi (aralık: 40 ila 84 yıl), hastaların %77'si erkek ve%92'si beyaz ırktandı. ECOG performans durumu 3 ya da üzerinde olan hastalar çalışmayaalınmamıştır. Tanıdan itibaren geçen medyan süre 42 aydı; önceki tedavilerin medyan sayısı 3olup (aralık: 1 ila 5 tedavi), hastaların %35'ine daha önceden yüksek doz kemoterapi,%43'üne bortezomib, %24'üne lenalidomid ve %11'ine de daha önce otolog veya allojenikkök hücre nakli uygulanmıştır. Başlangıçta, taramada hastaların %39'unda kitlesel (bulky)hastalık (>5 cm), %49'unda Basitleştirilmiş MHL Uluslararası Prognostik İndeks (MIPI) ileölçülen yüksek riskli skor ve %72'sinde de ilerlemiş hastalık mevcuttu (ekstranodal tutulumve/veya kemik iliği tutulumu). IMBRUVICA hastalık progresyonu veya kabul edilemez toksisite meydana gelene kadar oral olarak günde bir kez 560 mg dozda uygulandı. Tümör yanıtı, non-Hodgkin lenfoma (NHL)için revize edilmiş Uluslararası Çalışma Grubu (IWG) kriterlerine göre değerlendirildi. Buçalışmadaki birincil sonlanım noktası, araştırmacı tarafından değerlendirilen genel yanıtoranıydı (ORR). IMBRUVICA'ya verilen yanıtlar Tablo 2'de gösterilmektedir. Tablo 2: Relapslı ya da Refrakter MHL Hastalarında ORR ve DOR (PCYC-1104-CAÇalışması)
Bir Bağımsız İnceleme Komitesi (IRC) etkililik verilerini ilave olarak değerlendirmiş ve %69'luk bir ORR, %21'lik bir CR ve %48'lik bir PR oranı ortaya koymuştur. IRC DOR'yi19,6 ay olarak saptanmıştır. IMBRUVICA'ya verilen genel yanıt bortezomib ve lenalidomidi kapsayan geçmiş tedaviden ya da temelde yatan risk/prognoz faktörlerinden, kitlesel (bulky) hastalık, cinsiyet ya dayaştan bağımsız olarak gözlenmiştir. IMBRUVICA'nın güvenliliği ve etkililiği, daha önce en az bir tedavi görmüş 280 MHL hastasını kapsayan randomize, açık etiketli, çok merkezli Faz 3 çalışmada ortaya konmuştur(Çalışma MCL3001). Hastalar, 21 gün süreyle günde bir kez oral yoldan 560 mgIMBRUVICA ya da ilk kürün 1, 8 ve 15. günlerinde 175 mg ve onu takiben her 21 günlükkürün 1, 8 ve 15. günlerinde intravenöz yoldan 75 mg temsirolimus alacak şekilde 1:1oranında randomize edilmiştir. Her iki kolda da tedaviye, hastalık progresyonuna veya kabuledilemez toksisiteye kadar devam edilmiştir. Medyan hasta yaşı 68 idi (aralık: 34 ila 88);hastaların %74'ü erkek ve %87'si beyaz ırktandı. Tanıdan itibaren geçen medyan süre 43 aydıve önceki tedavilerin medyan sayısı 2 olup (aralık: 1 ila 9 tedavi), hastaların %51'ine dahaönceden yüksek doz kemoterapi, %18'ine bortezomib, %5'ine lenalidomid ve %24'üne dedaha önce kök hücre nakli uygulanmıştır. Başlangıçta, taramada hastaların %53'ünde kitlesel(bulky) hastalık (>5 cm), %21'inde Basitleştirilmiş MIPI ile ölçülen yüksek riskli skor,%60'ında ekstranodal hastalık ve %54'ünde de kemik iliği tutulumu mevcuttu. Progresyonsuz sağkalım (PFS), IRC tarafından non-Hodgkin lenfoma (NHL) için revize edilmiş Uluslararası Çalışma Grubu (IWG) kriterlerine göre değerlendirildi. MCL3001çalışmasına yönelik etkililik sonuçları Tablo 3'te ve PFS için Kaplan-Meier eğrisi Şekil 1'degösterilmektedir. Tablo 3: Relaps veya Refrakter MHL Hastalarında Etkililik Sonuçları (Çalışma MCL3001)
İbrutinib ile tedavi edilen daha az hasta temsirolimusa karşı lenfoma semptomlarında klinik olarak anlamlı kötüleşme yaşamış (%27 karşısında %52) ve semptomların kötüleşmesinekadar geçen süre ibrutinib ile temsirolimus karşısında daha yavaş olmuştur (HR 0,27, p<0,0001). Şekil 1: MCL3001 Çalışmasına Ait Kaplan-Meier PFS (ITT Popülasyonu) Eğrisi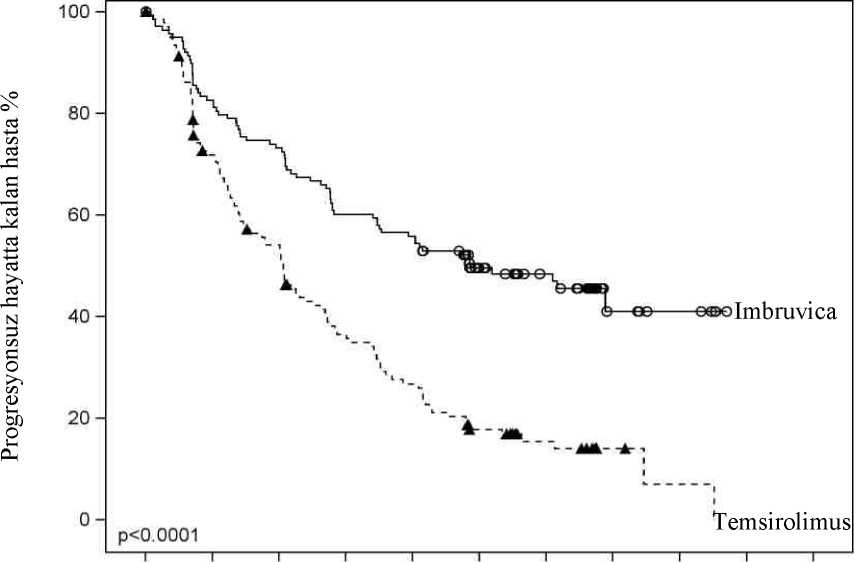
KLL KLL için daha önce tedavi almamış hastalar Tek ajan Daha önce tedavi almamış 65 yaş ve üzeri KLL hastalarında IMBRUVICA'nın klorambusil ile karşılaştırıldığı randomize, çok merkezli, açık etiketli bir Faz 3 çalışması (PCYC-1115-CA) gerçekleştirilmiştir. 65-70 yaş arasındaki hastalarda fludarabin, siklofosfamid verituksimab ile birinci basamak kemoimmünoterapi kullanımına engel olan en az birkomorbidite olması koşulu konmuştur. Hastalar (n=269) hastalık progresyonu veya kabuledilemez toksisiteye kadar günde 420 mg IMBRUVICA veya her bir 28 günlük kürün 1. ve15. günlerinde 0,5 mg/kg başlangıç dozunda klorambusil almak ve 0,8 mg/kg'lık doz artışlarıtolere edildiği sürece maksimum 12 kür için izin verilir şekilde 1:1 oranda kollara randomizeedildiler. Hastalık progresyonunun doğrulanması ardından klorambusil grubundaki hastalaribrutinib grubuna geçiş yapabilmiştir. Medyan hasta yaşı 73 idi (aralık 65 ila 90); hastaların %63'ü erkek ve %91'i beyaz ırktandı. Hastaların %91'inin başlangıçtaki ECOG performans durumu 0 ya da 1 ve %9'unun ECOGperformans durum 2'ydi. Çalışmaya 269 KLL hastası kaydedilmiştir. Başlangıçta, hastaların%45'inde ileri klinik evre (Rai Evre III veya IV), %35'inde > 5 cm'lik en az bir tümör,%39'unda başlangıçta anemi, %23'ünde başlangıçta trombositopeni, %65'inde > 3500 mcg/LP2 mikroglobulin yüksekliği, %47'sinde < 60 mL/dak CrCL, %20'sinde del11q, %6'sında del17p/tümör protein 53 (TP53) mutasyonu ve %44'ünde mutasyonsuz immünoglobulin ağırzincir değişken bölgesi (IGHV) mevcuttu. Uluslararası KLL Çalışma Grubu (IWCLL) kriterlerine göre Bağımsız Değerlendirme Kurulu (IRC) tarafından değerlendirilen progresyonsuz sağkalım (PFS), IMBRUVICA kolunda ölümya da progresyon riskinde %84 oranında istatistiksel olarak anlamlı bir azalma göstermiştir.PCYC-1115-CA çalışmasına ait etkililik sonuçları Tablo 4'te ve PFS ve OS için Kaplan-Meier eğrileri sırasıyla Şekil 2 ve 3'te gösterilmektedir. ITT popülasyonunda klorambusil karşısında ibrutinib lehine istatistiksel olarak anlamlı bir sürekli trombosit veya hemoglobin düzelmesi gözlenmiştir. Başlangıçta sitopenileri olanhastalarda sürekli hematolojik düzelme aşağıdaki gibi olmuştur: ibrutinib ve klorambüsil içinsırasıyla trombosit için %77,1'e %42,9, hemoglobin için %84,3'e %45,5'tir. Tablo 4: PCYC-1115-CA Çalışmasındaki Etkililik Sonuçları

48 aylık takip PCYC-ni5-CA ve ona ait uzatma çalışmasında 48 aylık medyan takip süresi ile IMBRUVICA kolundaki hastalar için, araştırmacı değerlendirmesine göre ölüm veyaprogresyon riskinde %86 oranında bir azalma gözlenmiştir. Araştırmacı tarafındandeğerlendirilen medyan PFS'ye IMBRUVICA kolunda ulaşılmazken, klorambusil kolunda15 ay [%95 GA (10,22, 19,35)] olarak bulunmuştur (HR=0,14 [%95 GA (0,09, 0,21)]).Dört yıllık tahmini PFS, IMBRUVICA kolunda %73,9 ve klorambusil kolunda %15,5olarak bulunmuştur. Güncellenmiş Kaplan-Meier PFS eğrisi Şekil 4'te gösterilmektedir. Araştırmacı tarafından değerlendirilen ORR, IMBRUVICA kolunda %91,2 ve klorambusil kolunda %36,8 olmuştur. IWCLL kriterlerine göre CR oranı, IMBRUVICA kolunda %16,2ve klorambusil kolunda %3 olmuştur. Uzun süreli takip sırasında, ilk başta klorambusilkoluna randomize edilen toplam 73 hasta (%54,9) daha sonra çapraz geçiş tedavisi olarakibrutinib almıştır. Kırk sekizinci aydaki Kaplan-Meier OS dönüm noktası tahminiIMBRUVICA kolunda %85,5 olmuştur. PCYC-1115-CA çalışmasında ibrutinib ile görülen tedavi etkisi, del 17p/TP53 mutasyonu, del 11q ve/veya mutasyonsuz IGHV olan yüksek riskli hastalar genelinde tutarlılıkgöstermiştir. Şekil 4: PCYC-1115-CA Çalışmasında 48 Aylık Takip ile Kaplan-Meier PFS Eğrisi(ITT Popülasyonu)
Kombinasyon tedavisi IMBRUVICA'nın daha önce tedavi görmemiş KLL/SLL hastalarındaki güvenliliği ve etkililiği, obinutuzumabla kombinasyon halinde IMBRUVICA'nın obinutuzumablakombinasyon halinde klorambusille karşılaştırıldığı randomize, çok merkezli, açık etiketli,Faz 3 çalışmasında (PCYC-1130-CA) daha ayrıntılı biçimde incelenmiştir. Çalışmaya 65yaşında veya daha büyük ya da <65 yaşında olan ve mevcut hastalıklar olan, kreatininklirensine göre böbrek işlevinde düşüş olan (<70 mL/dk.) veya del 17p/TP53 mutasyonu olanhastalar kaydedilmiştir. Hastalar (n=229) 6 döngü boyunca 28 günlük her bir döngünün 1. ve15. günlerinde hastalık ilerlemesine veya kabul edilemez toksisiteye kadar günlükIMBRUVICA 420 mg'a ya da 0,5 mg/kg klorambusil dozuna 1:1 oranında randomizeedilmiştir. Her iki kolda da hastalara birinci döngünün 1., 8. ve 15. gününde 1000 mgobinutuzumab verilmiş, daha sonra izleyen 5 döngünün birinci gününde tedavi uygulanmıştır(toplam 6 döngü, her biri 28 günlük). İlk obinutuzumab dozu 1. gün (100 mg) ve 2. gün (900mg) arasında bölünmüştür. Medyan yaş 71'dir (aralık, 40 ila 87) ve hastaların %64'ü erkek, %96'sı beyazdır. Tüm hastaların bazal ECOG performans statüsü 0 (%48) ve 1-2'dir (%52). Başlangıçta hastaların%52'si ileri klinik dönemdedir (Rai Evre III veya IV), %32'sinde kitlesel hastalık (>5 cm), %44'ünde bazal anemi, %22'sinde bazal trombositopeni, %28'sinde CrCL <60 mL/dk vardır ve medyan Yaşlılarda Kümülatif Hastalık Değerlendirme Ölçeği (CIRS-G) 4'tür (aralık, 0 ila12). Başlangıçta, hastaların %65'inde yüksek risk faktörleriyle KLL/SLL görülmüştür(del17p/TP53 mutasyonu [%18], del11q [%15] veya mutasyona uğramamış IGHV [%54]). İlerlemesiz sağkalım (PFS), IWCLL kriterlerine göre IRC tarafından incelenmiştir ve IMBRUVICA kolunda ölüm ve ilerleme riskinde istatistik açıdan anlamlı %77 oranında birdüşüş olduğuna işaret etmiştir. Çalışmadaki 31 aylık medyan izlem süresinde, IMBRUVICA+ obinutuzumab kolunda medyan PFS'ye ulaşılmamıştır ve klorambusil + obinutuzumabkolunda 19 aydır. PCYC-1130-CA için etkililik sonuçları Tablo 5'te gösterilmektedir ve PFSiçin Kaplan-Meier eğrisi Şekil 5'te gösterilmektedir. Tablo 5: PCYC-1130-CA Çalışmasındaki Etkililik Sonuçları
a IRC tarafından değerlendirilmiştir. b IMBRUVICA + obinutuzumab kolunda tam yanıt veren ve tamamlanamamış kemik iliği iyileşmesi ile tam yanıt (CRi) veren 1 hasta dahil.c PR = PR + nPR Şekil 5: PCYC-1130-CA Çalışmasında PFS İçin Kaplan-Meier Eğrisi (ITTPopülasyonu)
Risk altındaki gönüllüler IBR+Ob Klb+Ob İbrutinibin tedavi etkisi yüksek riski KLL/SLL popülasyonda tutarlıdır (del 17p/TP53 mutasyonu, del 11q ve/veya mutasyona uğramamış IHGV) ve Tablo 6'da gösterildiği gibi, PFS HR 0,15'tir [%95 GA (0,09; 0,27)]. Yüksek riskli KLL/SLL popülasyonu için 2 yıllık PFS oranı tahminleri IMBRUVICA + obinutuzumab ve klorambusil + obinutuzumabkollarında sırasıyla %78,8 [%95 GA (67,3; 86,7)] ve %15,5'tir [%95 GA (8,1; 25,2)]. Tablo 6: PFS Alt Grup Analizi (PCYC-1130-CA Çalışması)
IMBRUVICA + obinutuzumabla tedavi edilen hastaların %25'inde, klorambusil ve obinutuzumabla tedavi edilen hastaların %58'inde tüm derecelerde infüzyon ilişkili reaksiyongözlemlenmiştir. IMBRUVICA + obinutuzumabla tedavi edilen hastaların %3'ünde,klorambusil ve obinutuzumabla tedavi edilen hastaların %9'unda Derece 3 veya daha ciddiinfüzyon ilişkili reaksiyon gözlemlenmiştir. IMBRUVICA'nın daha önce tedavi edilmemiş KLL veya SLL hastalarındaki güvenliliği ve etkililiği randomize, çok merkezli, açık etiketli bir Faz 3 çalışmasında (E1912), rituksimablakombinasyon halinde IMBRUVICA (IR) ile standart fludarabin, siklofosfamid ve rituksimab(FCR) kemoimmünoterapisi karşılaştırılarak incelenmiştir. Çalışmaya daha önce tedaviedilmemiş ve 70 yaşında veya daha küçük KLL veya SLL hastaları kaydedilmiştir. Hastalar(n=529) 2:1 oranında IR veya FCR'ye atanmıştır. IMBRUVICA, hastalık ilerlemesi veyakabul edilemez toksisite olana kadar 420 mg/gün dozunda uygulanmıştır. Fludarabin 25mg/m2 dozunda ve siklofosfamid 250 mg/m2 dozunda, her ikisi de 1.-6. Döngülerin 1., 2. ve3. günlerinde uygulanmıştır. Rituksimab IR kolunda toplam 6 döngü boyunca, 2. döngüde, FCR kolunda 1. döngüde başlatılmıştır ve birinci döngünün 1. gününde 50 mg/m2 dozunda, birinci döngünün 2. gününde 325 mg/m2 dozunda, izleyen 5 döngünün 1. gününde 500 mg/m2dozunda uygulanmıştır. Her bir döngü 28 gündür. Medyan yaş 58'dir (aralık 28 ila 70) ve hastaların %67'si erkek, %90'ı beyazdır. Tüm hastaların bazal ECOG performans statüsü 0 veya 1 (%98) veya 2'dir (%2). Bazalda,hastaların %43'ünde Rai evresinin 3. veya 4. evrede olduğu görülmüştür ve hastaların%59'unda yüksek risk faktörlü KLL/SLL görülmüştür (TP53 mutasyonu [%6], del11q [%22]veya mutasyonsuz IGHV [%53]). 37 aylık medyan takip süresi olan E1912 çalışmasının etkililik sonuçları Tablo 7'de gösterilmektedir. PFS (IWCLL kriterlerine göre değerlendirilmiştir) ve OS için Kaplan-Meiereğrileri sırasıyla Şekil 6 ve Şekil 7'de gösterilmektedir. Tablo 7: E1912 Çalışmasındaki Etkililik Sonuçları
a P değeri katmanlaştırılmamış log-sıra testine göredir. b Araştırmacı değerlendirmeli. HR = tehlike oranı; NE = değerlendirilemez Şekil 6: E1912 Çalışmasında PFS'nin Kaplan-Meier Eğrisi (ITT Popülasyonu)10090607060
fludarabin,^I I siklofosfamid +rituximabe 50uı40302010o
İbrutinibin tedavi etkisi yüksek riskli KLL/SLL popülasyonunda (TP53 mutasyonu, del11q veya mutasyonsuz IGHV) tutarlıdır; PFS HR değeri 0,23 [%95 GA (0,13; 0,4)], p <0,0001,Tablo 8'de gösterilmektedir. Yüksek riskli KLL/SLL popülasyonu için 3 yıllık PFS oranıtahminleri IR ve FCR kollarında sırasıyla %90,4 [%95 GA (85,4; 93,7)] ve %60,3'tür [%95GA (46,2; 71,8)]. Tablo 8: PFS Alt Grup Analizi (E1912 Çalışması)
p=0.0007
o 0 3691 21 51821242 7303336394245485154(Month)Risk altındaki gönüHüler IMBRUVICA+rituximabfludarabin,I siklofosfamid +rituximab354 351349348347346344343343308263225195152100733980175 16315815715515114814414312310286725337211010Sabit süreli kombinasyon tedavisi Önceden tedavi edilmemiş KLL hastalarında IMBRUVICA'mn venetoklaks ile kombinasyon halinde sabit süreli tedavisinin güvenliliği ve etkililiği, randomize, açık etiketli bir faz 3çalışmasında (CLL3011) değerlendirilmiştir. Çalışmaya, daha önce tedavi edilmemiş, CIRSskoru >6 veya CrCL >30 ila <70 mL/dak olan <65 yaş KLL hastaları ve 65 yaş ve üstüyetişkin hastalar dahil edilmiştir. Del 17p veya bilinen TP53 mutasyonları olan hastalarçalışma dışı bırakılmıştır. Hastalar (n=211), venetoklaks ile kombinasyon halindeIMBRUVICA veya obinutuzumab ile kombinasyon halinde klorambusil almak üzere 1:1oranında randomize edilmiştir. IMBRUVICA + venetoklaks kolundaki hastalar, 3 kürboyunca tek ajan IMBRUVICA ve ardından 12 kür (5 haftalık doz titrasyon programı dahil)venetoklaks ile kombinasyon halinde IMBRUVICA almıştır. Her kür 28 gündür.IMBRUVICA günde 420 mg dozda uygulanmıştır. Venetoklaks, 1 hafta boyunca 20 mg ilebaşlanarak, ardından 1 hafta boyunca her bir 50 mg, 100 mg ve 200 mg doz seviyesinde,ardından önerilen günlük doz olan 400 mg olarak uygulanmıştır. Klorambusil +obinutuzumab koluna randomize edilen hastalar, 6 kür boyunca tedavi görmüştür.Obinutuzumab, 1. kür 1., 8. ve 15. günlerinde 1.000 mg dozunda uygulanmıştır. 2 ila 6.kürlerde, 1. günde 1.000 mg obinutuzumab verilmiştir. Klorambusil, 1 ila 6. kürlerin 1. ve 15.günlerinde 0.5 mg/kg vücut ağırlığı dozunda uygulanmıştır. Sabit süreli rejimlerden herhangibirinin tamamlanmasının ardından IWCLL kriterlerine göre progrese olduğu tespit edilenhastalar, tek ajan IMBRUVICA ile tedavi edilebilir. Medyan yaş 71 (47 ila 93 yaş arası), %58'i erkek ve %96'sı beyaz ırktandır. Tüm hastaların başlangıç ECOG performans durumu 0 (%35), 1 (%53) veya 2 (%12)'dir. Başlangıçta,hastaların %18'i del 11q'lu KLL ve %52'si mutasyona uğramamış IGHV ile başvurmuştur.Tümör lizis sendromu riski için temel değerlendirmede, hastaların %25'inde yüksek tümöryükü vardır. 3 kür tek ajan IMBRUVICA öncü tedavisinden sonra, hastaların %2'sindeyüksek tümör yükü görülmüştür. Yüksek tümör yükü, herhangi bir lenf nodu >10 cm veyaherhangi bir lenf nodu >5 cm ve mutlak lenfosit sayısı >25^109/L olarak tanımlanmıştır. 28aylık medyan takip süresinde, IWCLL kriterlerine göre CLL3011 çalışması için IRCtarafından değerlendirilen etkililik sonuçları Tablo 9'da, PFS için Kaplan-Meier eğrisi Şekil8'de ve minimal kalıntı hastalık oranları (MRD) negatifliği oranları Tablo 10'dasunulmaktadır. Tablo 9: CLL3011 Çalışmasındaki Etkililik Sonuçları
a IRC tarafından değerlendirilen b Katmanlı log-sıra testinden alınan P değeri c IMBRUVICA + venetoklaks kolunda, tamamlanamamış kemik iliği iyileşmesi ile tam yanıt (CRi) veren 3 hastayı içerir.d Cochran-Mantel-Haenszel ki-kare testinden alınan P değerie Genel yanıt = CR+CRi+nPR+PR CR = tam yanıt; CRi = tamamlanamamış kemik iliği iyileşmesi ile tam yanıt; HR = tehlike oranı; NE = ulaşılamamıştır; nPR = nodüler kısmi yanıt; PR = kısmi yanıt Şekil 8: CLL3011 Çalışması KLL Hastalarına ait Kaplan-Meier PFS (ITTpopülasyonu) Eğrisi Sc3O
IMBRUVICA + venetoklaksın tedavi etkisi, 0,23 PFS HR [%95 GA (0,13, 0,41)] ile yüksek riskli KLL popülasyonunda (TP53 mutasyonu, del 11q veya mutasyona uğramamış IGHV) iletutarlıdır. Genel sağkalım verileri henüz olgun değildir. 28 aylık medyan takipte, toplam 23 ölümle tedavi kolları arasında anlamlı fark yoktur: IMBRUVICA + venetoklaks kolunda 11 (%10,4)ve klorambusil + obinutuzumab kolunda 12 (%11,4) OS HR 1,048 [%95 GA (0,454, 2,419)].6 aylık ek takibin ardından, IMBRUVICA + venetoklaks kolunda ve klorambusil +obinutuzumab kolunda sırasıyla 11 (%10,4) ve 16 (%15,2) ölüm rapor edilmiştir ve tahminiOS HR 0,76'dır [%95 GA (0,352, 1,642]. Tablo 10: CLL3011 Çalışması MRD Negatiflik Oranları
P değerleri Cochran-Mantel-Haenszel ki-kare testinden alınmıştır. NGS ile kemik iliğinde MRD negatiflik oranı için P değeri birincil MRD analizidir. a Yeni nesil sıralama testi (clonoSEQ) kullanılarak 10-4 eşiğine dayalıdır b MRD, merkezi laboratuvar başına periferik kan veya kemik iliğinin akış sitometrisi ile değerlendirilmiştir. Negatif durumun tanımı, 10.000 lökosit başına <1 KLL hücresidir (<1x104). GA = güven aralığı; NGS = yeni nesil sıralama Tedavinin tamamlanmasından on iki ay sonra, periferik kandaki MRD negatiflik oranları, IMBRUVICA + venetoklaks ile tedavi edilen hastalarda NGS testi ile %49,1 (52/106) ve akışsitometrisi ile %54,7 (58/106) olmuştur ve ilgili zaman noktasında klorambusil +obinutuzumab ile tedavi edilen hastalarda NGS testi ile %12,4 (13/105) ve akış sitometrisi ile%16,2'dir (17/105). Klorambusil + obinutuzumab ile tedavi edilen 6 hastada TLS bildirilmiş ve venetoklaks ile kombinasyon halinde IMBRUVICA'da TLS bildirilmemiştir. Önceden tedavi edilmemiş KLL hastalarında IMBRUVICA'nın venetoklaks ile kombinasyon halinde sabit süreli tedavisinin güvenliliği ve etkililiği, faz 2, çok merkezli, 2 kohortlu(PCYC-1142-CA) çalışmasının bir kohortunda ayrıca değerlendirilmiştir. Çalışmaya dahaönce tedavi edilmemiş 70 yaş veya daha genç KLL hastaları dahil edilmiştir. Çalışmaya 323hasta alınmış, bunlardan 159 hastaya, 3 kür tek ajan IMBRUVICA ve ardından 12 kür (5haftalıkdoz titrasyon programı dahil) venetoklaks ile kombinasyon halinde IMBRUVICA'dan oluşan sabit süreli tedavi verilmiştir. Her kür 28 gündür. IMBRUVICA günde 420 mg dozda uygulanmıştır. Venetoklaks, 1 hafta boyunca 20 mg ile başlanarak,ardından 1 hafta boyunca her bir 50 mg, 100 mg ve 200 mg doz seviyesinde, ardındanönerilen günlük doz olan 400 mg olarak uygulanmıştır. Sabit süreli rejimin tamamlanmasınınardından IWCLL kriterlerine göre progrese olduğu tespit edilen hastalar, tek ajanIMBRUVICA ile yeniden tedavi edilebilirler. Medyan yaş 60 (aralık, 33 ila 71), %67'si erkek ve %92'si beyaz ırktandır. Tüm hastaların temel ECOG performans durumu 0 (%69) veya 1'dir (%31). Başlangıçta, hastaların%13'ünde del 17p, %18'inde del 11q, %17'sinde del 17p/TP53 mutasyonu, %56'sındamutasyona uğramamış IGHV ve %19'unda kompleks karyotip vardır. Tümör lizis sendromuriski için temel değerlendirmede, hastaların %21'inde yüksek tümör yükü vardır. 3 kür tek ajan IMBRUVICA öncü tedavisinden sonra, hastaların %1'inde yüksek tümör yükü görülmüştür. Yüksek tümör yükü, herhangi bir lenf nodu >10 cm veya herhangi bir lenf nodu>5 cm ve mutlak lenfosit sayısı >25^109/L olarak tanımlanmıştır. PCYC-1142-CA çalışmasının 28 aylık medyan takip süresinde IWCLL kriterlerine göre IRC tarafından değerlendirilen etkililik sonuçları Tablo 11'de ve minimal rezidüel hastalık (MRD)negatiflik oranları Tablo 12'de sunulmaktadır. Tablo 11: PCYC 1142-CA Çalışmasındaki Etkililik Sonuçları (Sabit Süreli Kohort)
CR = tam yanıt; Cri = tamamlanamamış kemik iliği iyileşmesi ile tam yanıt; Npr = nodüler kısmi yanıt; PR = kısmi yanıt; NE = ulaşılamamıştır
PCYC-1142-CA'da del 17p/TP53 mutasyonu (n=27) olan hastalarda, IRC değerlendirmesine dayalı genel yanıt oranı %96,3'tür; tam yanıt oranı %55,6'dır ve medyan tam yanıt süresineulaşılmamıştır (aralık, 4,3 ila 22,6 ay). Tedavi bitiminden 3 ay sonra del 17p/TP53 mutasyonuolan hastalarda kemik iliği ve periferik kanda MRD negatiflik oranı sırasıyla %40,7 ve%59,3'dür. Venetoklaks ile kombinasyon halinde IMBRUVICA ile tedavi edilen hastalarda TLS bildirilmemiştir. Daha önce en az bir tedavi almış KLL hastalar Tek ajan IMBRUVICA'nın KLL hastalarındaki güvenliliği ve etkililiği bir kontrolsüz ve bir randomize, kontrollü çalışmada değerlendirilmiştir. Açık etiketli, çok merkezli çalışma(PCYC-1102-CA) günde bir kere 420 mg kullanan relaps ya da refrakter 51 KLL hastasınıkapsamıştır. IMBRUVICA hastalık progresyonu veya kabul edilemez toksisite meydanagelene kadar uygulanmıştır. Hastaların medyan yaşı 68 (37 - 82 yaş aralığı) idi; tanıdan sonrageçen medyan süre 80 ay ve daha önce alınan medyan tedavi sayısı 4 (aralık: 1 - 12 tedavi)idi; hastaların %92,2'si geçmişte bir nükleozid analoğu, %98'i rituksimab, %86,3'ü biralkilleyici, %39,2'si bendamustin ve %19,6'sı ofatumumab kullanmıştır. Başlangıçta,hastaların %39,2'sinde Rai Evre IV hastalık, %45,1'inde kitlesel (bulky) hastalık (5 cm veüzerinde); %35,3'ü 17p delesyonu ve %31,4'ünde 11q delesyonu vardı. ORR, 2008 KLL Uluslararası Çalışması (IWCLL) kriterlerine göre araştırmacılar ve IRC tarafından değerlendirilmiştir. Medyan 16,4 aylık bir takip sürecinde tümü kısmi yanıtlı olan,relaps ya da refrakter 51 hastanın IRC değerlendirmesinde ORR %64,7 (%95 GA; %50,1,%77,6) idi. Lenfositozlu PR dahil ORR %70,6 idi. Yanıta kadar geçen medyan süre 1,9 aydır.DOR 3,9 ile 24,2+ay arasında değişmiştir. Medyan DOR'ye erişilmemiştir. Relaps ya da refrakter KLL hastalarında ofatumumaba karşı IMBRUVICA ile randomize, çok merkezli, açık etiketli bir Faz 3 çalışması yürütülmüştür (PCYC-1112-CA). Hastalar (n=391)hastalık progresyonu ya da kabul edilemez toksisiteye kadar günde 420 mg IMBRUVICA yada 12 doza kadar ofatumumaba 1:1 oranında randomize edilmiştir (300/2.000 mg).Ofatumumaba randomize edilen 57 hasta progresyonu takiben IMBRUVICA'ya geçişyapmıştır. Medyan yaş 67 (aralık: 30 ile 88 yaş arası) olup, hastaların %68'i erkek, %90'ıbeyaz ırktandı. Tüm hastalarda 0 ya da 1 değerinde bir başlangıç ECOG performans durumugözlenmiştir. Teşhisten itibaren geçen medyan süre 91 ay ve geçirilen önceki tedavi sayısı 2idi (aralık: 1 ile 13 arası). Başlangıçta hastaların %58'inde 5 cm ve üzerinde en az bir tümörsaptanmıştır. Hastaların %32'sinde 17p delesyonu (hastaların %50'si 17p delesyonu/TP53mutasyonuna sahip) ve %24'ünde 11q delesyonu ve %47'sinde mutasyonsuz IGHVmevcuttu. IRC tarafından IWCLL kriterlerine göre değerlendirilen PFS IMBRUVICA kolundaki hastalar için ölüm ya da progresyon riskinde %78 düzeyinde, istatistiksel olarak anlamlı birazalma ortaya koymuştur. OS analizi IMBRUVICA kolundaki hastalar için ölüm riskinde%57 düzeyinde, istatistiksel olarak anlamlı bir azalma ortaya koymuştur. PCYC-1112-CAçalışması için etkililik bulguları Tablo 13'de sunulmuştur. Şekil 7: E1912 Çalışmasında OS'nin Kaplan-Meier Eğrisi (ITT Popülasyonu)I I ! 11 ¦
co oIMBRUVICA+rituximab-IIHtM44HHI-IHIIIH4İIM İII44fludarabin,siklofosfamid+rituximab
HR = tehlike oranı; GA = güven aralığı; ORR = genel yanıt oranı; OS = genel sağkalım; PFS = progresyonsuz sağkalım; PR = kısmi yanıta Medyan OS'ye iki kolda da erişilmemiştir. OS için p <0,005 b Ofatumumaba randomize edilen-hastalar uygun olduğunda IMBRUVICA'ya başlarken sansürlenmiştir. c Ofatumumab kolundan çapraz geçiş yapan hastaların ilk IMBRUVICA dozu tarihinde sansürlenmediği sensitivite analizi d IRC'ye göre. Yanıtı teyit etmek için tekrarlayan BT grafileri gereklidir. e Tüm PR'lere ulaşılmıştır; ORR için p < 0,0001. Çalışmadaki medyan takip süresi=9 ay Etkililik, daha önceden belirlenen bir tabakalandırma faktörü olan 17p delesyonunun görüldüğü ve görülmediği hastalar da dahil olmak üzere, incelenen tüm alt gruplardabenzerlik göstermiştir (Tablo 14). Tablo 14: PFS (PCYC-1112-CA Çalışması) İçin Alt Grup Analizi
PFS için Kaplan-Meier eğrileri sırasıyla Şekil 9'da gösterilmektedir.
65 aylık takip dönemindeki final analiz PCYC-ni2-CA Çalışmasında 65 aylık medyan takip süresi ile IMBRUVICA kolundaki hastalar için, araştırmacı değerlendirmesine göre ölüm veya progresyon riskinde %85oranında bir azalma gözlenmiştir. IWCLL kriterlerine göre araştırmacı tarafındandeğerlendirilen medyan PFS IMBRUVICA kolunda 44,1 ay [%95 GA (38,47, 56,18)] veofatumumab kolunda 8,1 ay [%95 GA (7,79, 8,25)] olmuştur (HR=0,15 [%95 GA (0,11,0,2)]). Güncellenmiş Kaplan-Meier PFS eğrisi Şekil 10'da gösterilmektedir. Araştırmacıtarafından değerlendirilen ORR, ofatumumab kolunda %22,4'e kıyasla IMBRUVICAkolunda %87,7 olmuştur. Final analiz döneminde , başlangıçta ofatumumab tedavi kolunarandomize edilen 196 hastanın 133'ü (%67,9) ibrutinib tedavisine geçiş yapmıştır. IWCLLkriterlerine göre araştırmacı tarafından değerlendirilen medyan PFS2 (randomizasyondan PFSolayına kadar ilk ardışık anti-neoplastik tedaviden sonra geçen süre) sırasıyla IMBRUVICAkolunda 65,4 ay [%95 GA (51,61; tahmin edilemez)] ve ofatumumab kolunda 38,5 aydı [%95GA (19,98; 47,24)]; HR=0,54 [%95 GA (0,41, 0,71]. Medyan OS, IMBRUVICA kolunda67,7 aydır [%95 GA (61; tahmin edilemez)]. PCYC-1112-CA çalışmasında ibrutinib ile görülen tedavi etkisi, del17p/TP53 mutasyonu, 11q delesyonu ve/veya mutasyonsuz IGHV olan yüksek riskli hastalar genelinde tutarlılıkgöstermiştir. Şekil 10: 65 Aylık izlem Yapılan Final Analizde PCYC-1112-CA Çalışmasında PFS İçin Kaplan-Meier Eğrisi (ITT Popülasyonu)
Risk altındaki gönüllüler
Ay Kombinasyon tedavisi IMBRUVICA'nın daha önce KLL için tedavi edilen hastalardaki güvenlilik ve etkililiği, IMBRUVICA + BR kombinasyon tedavisinin plasebo + BR'ye karşı araştırıldığı randomize,çok merkezli, çift kör Faz 3 çalışmada (CLL3001 Çalışması) daha fazla değerlendirilmiştir.Hastalar (n=578), hastalık progresyonu veya kabul edilemez toksisiteye kadar günde 420 mgIMBRUVICA ya da plasebo + BR kombinasyonunu almak üzere 1:1 oranında randomizeedilmiştir. Tüm hastalar en fazla 6 adet 28 günlük kür boyunca BR almıştır. Bendamustin, enfazla 6 kür için 1. kürün 2 ve 3. günlerinde ve 2-6. kürlerin 1 ve 2. günlerinde 70 mg/m2 dozda30 dakika boyunca IV infüzyon yoluyla uygulanmıştır. Rituksimab, ilk kürün 1. gününde 375mg/m2 dozda ve 2-6. kürlerin 1. gününde 500 mg/m2 dozda uygulanmıştır. Plasebo + BRtedavisine randomize edilen 90 hasta, IRC tarafından doğrulanan progresyonu takibenIMBRUVICA grubuna geçiş yapmıştır. Hastaların medyan hasta yaşı 64 (aralık: 31 - 86),%66'sı erkek ve %91'i beyaz ırktandı. Tüm hastaların başlangıç ECOG performans durumu 0ya da 1'di. Tanıdan itibaren geçen medyan süre 6 yıl ve daha önce alınan tedavilerin medyansayısı 2'ydi (aralık: 1-11 tedavi). Başlangıçta, hastaların %56'sında >5 cm'lik en az birtümör, %26'sında del11q mevcuttu. Progresyonsuz sağkalım (PFS) IRC tarafından IWCLL kriterlerine göre değerlendirilmiştir. CLL3001 Çalışması için etkililik sonuçları Tablo 15'de sunulmuştur. Tablo 15: KLL Hastalarında Etkililik Sonuçları (CLL3001 Çalışması)
GA=güven aralığı; HR=tehlike oranı; ORR=genel yanıt oranı; OS=genel sağkalım; PFS=progresyonsuz sağkalıma IRC tarafından değerlendirilen. b IRC tarafından değerlendirilen, ORR (tam yanıt, kemik iliğinde tam düzelme olmaksızın tam yanıt, nodüler kısmi yanıt, kısmi yanıt).c İki kol için de medyan OS'ye ulaşılmadı. WM Tek ajan IMBRUVICA'nın WM'deki (IgM salgılayan lenfoplazmasitik lenfoma) güvenliliği ve etkililiği, açık etiketli, çok merkezli ve daha önce tedavi görmüş olan 63 hastadan oluşan tekkollu bir çalışmada incelenmiştir. Medyan yaş 63'tür (aralık: 44 ila 86), hastaların %76'sıerkek, %95'i beyazdır. Tüm hastaların bazal ECOG performans statüsü 0 veya 1'dir. Tanıdanberi gelen medyan süre 74 aydır ve medyan geçmiş tedavi sayısı 2'dir (aralık: 1 ila 11 tedavi).Başlangıçta, medyan serum IgM değeri 3,5 g/dL'dir ve hastaların %60'ı anemiktir(hemoglobin <11 g/dL veya 6,8 mmol/L). Hastalık progresyonu veya kabul edilemez toksisiteye kadar oral yoldan günde bir kez 420 mg IMBRUVICA uygulanmıştır. Bu çalışmanın primer bitiş noktası, araştırmacıdeğerlendirmesine göre ORR'dir. ORR ve DOR, Üçüncü Uluslararası WaldenströmMakroglobulinemisi Çalıştayı'ndan uyarlanan kriterler kullanılarak değerlendirilmiştir.IMBRUVICA'ya verilen yanıtlar Tablo 16'da gösterilmektedir. Tablo 16: WM Hastalarında Genel Yanıt Oranı (ORR) ve Yanıt Süresi (DOR)
GA=Güven Aralığı; DOR = yanıt süresi; NR = ulaşılamamıştır; MR = minör yanıt; PR = kısmi yanıt; VGPR = çok iyi kısmi yanıt; ORR = MR + PR + VGPRÇalışmadaki medyan izlem süresi: 14,8 ay Yanıta kadar geçen medyan süre 1 aydır (aralık: 0,7-13,4 ay). Bir Bağımsız Araştırma Komitesi (IRC) tarafından etkililik sonuçları da incelenmiştir ve %83 ORR, %11 VGPR ve %51 PR oranı görülmüştür. Kombinasyon tedavisi IMBRUVICA'nın daha önce tedavi görmemiş veya daha önce tedavi görmüş WM hastalarındaki güvenliliği ve etkililiği, rituksimabla kombinasyon halinde IMBRUVICA'nın,rituksimabla kombinasyon halinde plaseboyla karşılaştırıldığı randomize, çok merkezli, çiftkör Faz 3 çalışmasında (PCYC-1127-CA) daha ayrıntılı biçimde incelenmiştir. Hastalar(n=150), hastalık progresyonu ya da kabul edilemez toksisiteye kadar rituksimab ilekombinasyon halinde günde 420 mg IMBRUVICA veya rituksimab ile kombinasyon halindeplasebo almak üzere 1:1 oranında randomize edilmiştir. Rituksimab 4 hafta boyunca haftalık375 mg/m2 dozunda uygulanmıştır (1. - 4. haftalar) ve ardından 4 hafta boyunca haftalıkrituksimab uygulaması yapılmıştır (17. - 20. haftalar). Medyan yaş 69'dur (aralık, 36 ila 89), %66'sı erkektir ve %79'u beyazdır. Hastaların %93'ünün bazal ECOG performans statüsü 0 ve 1, %7'sinin bazal ECOG performans statüsü2'dir. Hastaların %45'i tedavi görmemiştir %55'i daha önce tedavi görmüştür. Tanıdan berigeçen medyan süre 52,6 aydır (tedavi görmemiş hastalarda=6,5 ay ve daha önce tedavigörmüş hastalarda=94,3 ay). Daha önce tedavi görmüş hastalarda, geçmiş tedavi medyansayısı 2'dir (aralık, 1 ila 6 tedavi). Başlangıçta, medyan serum IgM değeri 3,2 g/dL'dir (aralık,0,6 ila 8,3 g/dL), hastaların %63'ü anemiktir (hemoglobin <11 g/dL veya 6,8 mmol/L)MYD88 L265P mutasyonları %77'sinde mevcuttur, %13'ünde yoktur ve hastaların %9'ununmutasyon durumu için değerlendirilebilir değildir. 26,5 aylık medyan takip ile birincil analizde, IRC tarafından değerlendirilen PFS tehlike oranı 0,2 [%95 GA (0,11, 0,38)] idi. Daha önce tedavi görmemiş hastalar, daha önce tedavi görmüşhastalar ve MYD88 L265P mutasyonları olan veya olmayan hastalar için PFS tehlike oranları,ITT popülasyonu için PFS tehlike oranı ile tutarlıydı. IMBRUVICA+rituksimab ile tedavi edilen hastaların %1'inde ve plasebo+rituksimab ile tedavi edilen hastaların %16'sında derece 3 veya 4 infüzyonla ilişkili reaksiyonlargözlenmiştir. IMBRUVICA+rituksimab kolundaki deneklerin %8'inde ve plasebo+rituksimab kolundaki deneklerin %46,7'sinde IgM artışı şeklinde tümör parlaması meydana geldi. 63 aylık takipte Nihai Analiz 63 aylık genel bir takip ile, PCYC-1127-CA için son analiz sırasında bir IRC tarafından değerlendirilen etkinlik sonuçları Tablo 17'de ve PFS için Kaplan-Meier eğrisi Şekil 11'degösterilmektedir. Daha önce tedavi görmemiş hastalar (0.31 [%95 GA (0,14, 0,69)]) ve dahaönce tedavi görmüş hastalar (0,22 [%95 GA (0,11, 0,43)]) için tehlike oranları, ITTpopülasyonu için PFS tehlike oranı ile tutarlıydı. Tablo 17: PCYC-1127-CA (Final Analiz*) Çalışmasındaki Etkililik Sonuçları
GA=Güven Aralığı; CR = tam yanıt; HR = tehlike oranı; MR = minör yanıt; PR = kısmi yanıt; R = rituksimab; VGPR = çok iyi kısmi yanıt* Çalışmada medyan takip süresi = 49,7 ay.a IRC tarafından değerlendirilmiştir. b 4 yıllık PFS tahminleri, IMBRUVICA + R kolunda %70,6 [%95 GA (58,1, 80)] iken plasebo + R kolunda %25,3 [%95 GA (15,3, 36,6)] olmuştur.c Yanıt oranıyla ilişkili p değeri <0,0001 d IMBRUVICA + R koluna karşı plasebo + R kolu için yanıt oranı, daha önce tedavi görmemiş hastalarda sırasıyla %76'ya karşı %41 ve daha önce tedavi görmüş hastalarda%76'ya karşı %22 idi. e Başlangıç değerinden bağımsız olarak başlangıç değerine göre >2 g/dL'lik artış veya başlangıç değeri <11 g/dL ise >0,5 g/dL'lik bir iyileşme ile >11 g/dL'ye artış olaraktanımlanır. Şekil 11: PCYC-1127-CA (Final Analiz) Çalışmasında PFS İçin Kaplan-Meier Eğrisi (ITT Popülasyonu)İbr+RPbo+R
L40Vlll-IH- - - IhI3020
Ay PCYC-1127-CA çalışmasının, daha önce tedavi görmüş ve rituksimab içeren tedavisi başarısız olmuş olan ve tek ajanlı IMBRUVICA verilen 31 hastadan oluşan ayrı birmonoterapi kolu vardır. Medyan yaş 67'dir (aralık, 47 ila 90). Hastaların %81'inin bazalECOG performans statüsü 0 ve 1, %19'unun bazal ECOG performans statüsü 2'dir. Geçmiştedavi medyan sayısı 4'tür (aralık, 1 ila 7 tedavi). 61 aylık genel bir takiple, IRC'ye göremonoterapi kolunda PCYC-1127-CA çalışmasında gözlemlenen yanıt oranı %77'dir (%0 CR;%29 VGPR, %48 PR). Medyan yanıt süresi 33 aydı (aralık, 2,4 ila 60,2+ ay). IRC'ye göremonoterapi kolunda gözlemlenen toplam yanıt oranı %87'dir (%0 CR, %29 VGPR, %48 PR,%10 MR). Genel yanıtın medyan süresi 39 aydı (aralık, 2,07 ila 60,2+ ay). Pediatrik popülasyonIMBRUVICA'nın güvenliliği, etkililiği ve farmakokinetiği, nükseden veya refrakter matür B hücreli non-Hodgkin lenfoma olan pediyatrik ve genç erişkin hastalarda, IMBRUVICA'nınrituksimab, ifosfamid, karboplatin, etoposid ve deksametazon (RICE) rejimi veya rituksimab,vinkristin, ifosfamid, karboplatin, idarubisin ve deksametazon (RVICI) rejimi ilekombinasyon halinde arka plan tedavisi olarak iki bölümlü, çok merkezli, açık etiketli bir Faz3 çalışmasında (LYM3003) değerlendirilmiştir. Çalışmanın 1. kısmı (3 ila 17 yaş arası 21 hasta), 2. kısımdaki (3 ila 19 yaş arası 51 hasta) kullanılacak dozu değerlendirmiştir (bkz. bölüm 5.2). 2. kısımda hastalar, arka plan tedavisi ile günde 440 mg/m2 (12 yaş altı) veya 329 mg/m2 (12 yaş ve üzeri) şeklinde IMBRUVICA almak veya 3 döngü tedavi, transplantasyon, hastalıkilerlemesi veya kabul edilemez toksisite tamamlanana kadar tek başına arka plan tedavisialmak üzere 2:1 oranında randomize edilmiştir. 5.2 Farmakokinetik özelliklerGenel özellikler Emilim:İbrutinib oral uygulama sonrasında 1 ila 2 saatlik medyan Tmaks değeri ile hızla emilir. Aç koşullarda (n=8) mutlak biyoyararlanım %2,9'dur (%90 GA = 2,1 - 3,9) ve bir öğün ilebirlikte kullanıldığında iki katına çıkar. İbrutinibin farmakokinetiği farklı B hücrelimaligniteleri olan hastalarda anlamlı bir farklılık göstermez. İbrutinib maruziyeti 840 mg'akadar olan dozlarda artar. Dozun 560 mg olarak uygulandığı hastalarda gözlenen kararlıdurum EAA değeri (ortalama ± standart sapma) 953 ± 705 ng sa/mL'dir. İbrutinibin açkarnına alınması ile yüksek oranda yağ içeren bir kahvaltıdan 30 dakika önce, 30 dakika sonra(tok koşullar) ya da yüksek yağlı bir kahvaltıdan 2 saat sonrasına kıyasla maruziyetin yaklaşık%60'ına (EAAson) neden olmuştur. İbrutinib pH'a bağlı çözünürlük sergiler ve daha yüksek pH değerinde daha düşük çözünürlüğe sahiptir. 5 gün süreyle günde bir kere 40 mg omeprazol aldıktan sonra aç karnına560 mg'lık tek bir ibrutinib uygulanan sağlıklı gönüllülerde, geometrik ortalama oranları(%90 GA) EAA0-24 için %83 (%68-102), EAAson için %92 (%78-110) ve Cmaks için %38(%26-53) olmuştur. Dağılım:İbrutinibin insan plazma proteinlerine in vitrogeri dönüşümlü bağlanması %97,3 olup, 50 ila1.000 ng/mL aralığında hiçbir konsantrasyon bağımlılığı yoktur. Kararlı durumdaki görünürdağılım hacmi (Vd, ss/F) yaklaşık 10.000 L'dir. Biyotransformasyon:İbrutinib primer olarak CYP3A4 ile metabolize olur ve BTK'yı inhibe edici aktivitesi ibrutinibden yaklaşık 15 kat daha düşük olan bir dihidrodiol metaboliti oluşur. CYP2D6ibrutinib metabolizmasında minimal bir rol oynar. Bu nedenle, farklı CYP2D6 genotiplerine sahip hastalarda önlem alınması gerekli değildir. Eliminasyon:Görünür klerens (CL/F) yaklaşık 1000 L/saat'tir. İbrutinibin yarılanma ömrü 4 ila 13 saattir. Sağlıklı deneklerde radyoaktif olarak etiketlenmiş tek doz oral [14C]-ibrutinib uygulamasındansonra radyoaktivitenin yaklaşık %90'ı 168 saat içerisinde atılmış olup, bunun büyük bölümü(%80) feçeste ve %10'undan azı idrarda atılmıştır. Değişmemiş ibrutinib radyoaktif olaraketiketlenmiş boşaltım ürününün feçeste yaklaşık %1'ini oluşturur ve idrarda görülmez. Doğrusallık/doğrusal olmayan durum:İbrutinib maruziyeti doz ile orantılı olarak artar. Hastalardaki karakteristik özelliklerGeriyatik popülasyon:Popülasyon farmakokinetiği yaşın, dolaşımdan ibrutinib klerensini anlamlı düzeyde etkilemediğini göstermiştir. Pediyatrik popülasyon:Farmakokinetik veriler, nükseden veya refrakter matür B-hücreli non-Hodgkin lenfomalı çocuklarda, 12 yaş ve üzeri çocuklarda günlük 329 mg/m2 doz ve 3 yaş ila 12 yaş altıçocuklarda günlük 440 mg/m2 doz uygulanan ibrutinib maruziyetlerinin genellikle günlük 560mg doz uygulanan yetişkin hastalarda gözlenen maruziyet aralığı içinde olduğunugöstermektedir. Cinsiyet:
dolaşımdan ibrutinib klerensini anlamlı düzeyde
Popülasyon farmakokinetiği cinsiyetin, etkilemediğini göstermiştir. Irk:Irkın ibrutinib farmakokinetiği üzerindeki potansiyel etkisini karşılaştırmak için yeterli bir veri yoktur. Vücut ağırlığı:Popülasyon farmakokinetiği verileri vücut ağırlığının (aralık: 41-146 kg; ortalama [standart sapma]: 83 [19 kg]) ibrutinib klerensi üzerinde ihmal edilebilir bir etkiye sahip olduğunugöstermiştir. Böbrek yetmezliği:İbrutinib minimal bir renal klerense sahiptir; metabolitlerin üriner yolla atılan miktarı dozun < %10'udur. Bugüne kadar böbrek fonksiyon bozukluğu olan hastalarda spesifik çalışmalaryapılmamıştır. Ağır böbrek yetmezliği olan hastalara veya diyaliz hastalarına ait hiçbir verimevcut değildir (bkz. Bölüm 4.2). Karaciğer yetmezliği:İbrutinib karaciğerde metabolize olur. Kanserli olmayan hastalarda, açlık koşullarında 140 mg IMBRUVICA'nın verildiği bir karaciğer yetmezliği çalışması yürütülmüştür. Hafif (n=6,Child Pugh Sınıf A), orta (n=10, Child Pugh Sınıf B) ve şiddetli (n=8, Child Pugh Sınıf C)karaciğer yetmezliği olan hastalarda bozulmuş karaciğer fonksiyonu bireyler arasında önemliölçüde değişmekte olup, ortalama olarak ibrutinib maruziyetinde (EAAson) sırasıyla 2,7-, 8,2-ve 9,8-kat bir artış gözlenmiştir. Ayrıca ibrutinibin serbest fraksiyonu da yetmezliğin derecesiarttıkça yükselmiştir; bu çalışmada benzer özelliklere sahip sağlıklı kontrollerde plazmadaki%3,3 oranına kıyasla hafif, orta ve şiddetli karaciğer yetmezliği olan gönüllülerde sırasıyla%3, 3,8 ve 4,8 oranları görülmüştür. Bağlı olmayan ibrutinib maruziyetindeki (EAAbağlı olmayan,son) ilgili artışın hafif, orta ve şiddetli karaciğer yetmezliği olan gönüllülerde sırasıyla 4,1-,9,8- ve 13-kat olduğu hesaplanmaktadır (bkz. Bölüm 4.2). CYP substratları ile eş zamanlı kullanım:In vitroin vitroortamda en zayıf CYP450 izoenzim indükleyicidir. Ibrutinib'in hassas bir CYP3A4 substratıolması sebebiyle, kendi maruziyeti üzerinde klinik olarak ilişkili bir etkisi bulunmamaktadır.Transport substratları/inhibitörleri ile eş zamanlı kullanım:In vitroin vitro5.3 Klinik öncesi güvenlilik verileriAşağıdaki advers etkiler sıçanlar ve köpeklerde yapılan 13 haftalık çalışmalarda görülmüştür. İbrutinibin her iki türde de 30 mg/kg/gün'lük bir Yan Etki Gözlenmeyen Seviye (NoObserved Adverse Effect Level; NOAEL) dozunda sıçanlarda ve köpeklerde gastrointestinaletkileri (yumuşak feçes/diyare ve/veya enflamasyon) ve lenfoid tükenmesini indüklediğisaptanmıştır. 560 mg/gün klinik dozundaki ortalama maruziyete dayalı olarak, EAA oranlarıNOAEL'de erkek ve dişi sıçanlarda sırasıyla 2,6 ve 21 ile erkek ve dişi köpeklerde sırasıyla0,4 ve 1,8 saptanmıştır. Gözlenen En Düşük Etki Seviyesi (Lowest Observed Effect Level;LOEL) (60 mg/kg/gün) marjinleri köpeklerde 3,6 (erkekler) ve 2,3 mislidir (dişi). Sıçanlarda,orta dereceli pankreatik asiner hücre atrofisi (advers kabul edilir) erkek sıçanlarda 100 mg/kgve üzerindeki dozlarda (EAA maruziyet marjini: 2,6 kat) gözlenirken, 300 mg/kg/gün'e kadardozlarda dişi sıçanlarda görülmemiştir (EAA maruziyet marjini: 21,3 kat). > 100 mg/kg/gün(EAA maruziyet marjini: 20,3 kat) verilen dişi sıçanlarda hafif düzeyde azalmış trabeküler vekortikal kemik görülmüştür. Tüm gastrointestinal, lenfoid ve kemik bulguları 6 ila 13 haftalıksüreçler sonunda iyileşmiştir. Pankreatik bulgular benzer geri dönüşüm süreçlerinde kısmendüzelmiştir. Juvenil toksisite çalışmaları yapılmamıştır. Karsinojenisite/genotoksisiteTransjenik farelerde (Tg.rasH2) 2000 mg/kg/gün'e kadar oral dozlarla yürütülen 6 aylık bir çalışmada, ibrutinib karsinojenisite göstermemiştir. Bu, insanlarda 560 mg/gün olarakuygulanan doz ile karşılaştırıldığında, EAA bakımından erkeklerde yaklaşık 23 kat,kadınlarda ise yaklaşık 37 kat ibrutinib maruziyetine karşılık gelmektedir. İbrutinib bakterilerde, memeli hücrelerinde ya da farelerde test edildiğinde genotoksik özellikler sergilememiştir. Üreme toksisitesiİbrutinib gebe sıçanlara 80 mg/kg/gün dozunda verildiğinde, günde 560 mg doz uygulanan hastalardaki maruziyetin (EAA) yaklaşık 14 katı bir maruziyet marjini ile visseralmalformasyonlar (kalp ve ana damarlar) ve iskeletsel değişimler ile artmış bir post-implantasyon kaybıyla ilişkilendirilmiştir. Hayvanlarda uygulanan > 40 mg/kg/gün dozunda,ibrutinib (günde 560 mg doz uygulanan hastalara kıyasla, EAA oranı > 5,6) azalmış fetusağırlıklarına neden olmuştur. Sonuçta, fötal NOAEL 10 mg/kg/gün olarak belirlenmiştir(günde 560 mg dozuna kıyasla yaklaşık 1,3 katı EAA) (bkz. Bölüm 4.6). Hamile tavşanlarda 15 mg/kg/gün ve üzerinde uygulanan ibrutinib iskeletsel malformasyonlar (kaynaşmış sternebra) ve 45 mg/kg/gün dozda uygulanan ibrutinib de post-implantasyonkaybıyla ile ilişkilendirilmiştir. İbrutinib 15 mg/kg/gün (günlük 560 mg ibrutinib uygulananMHL hastalarında maruziyetin (EAA) yaklaşık 2 katı ve günlük 420 mg ibrutinib uygulananKLL ve WM hastalarında maruziyetin (EAA) yaklaşık 2,8 katı) uygulanan tavşanlardamalformasyonlara sebep olmuştur. Sonuçta, fötal NOAEL 5 mg/kg/gün olarak belirlenmiştir(günde 560 mg dozuna kıyasla yaklaşık 0,7 katı EAA) (bkz. Bölüm 4.6). FertiliteErkek ya da dişi sıçanlarda test edilen maksimum doza kadar (100 mg/kg/gün - insana eşdeğer doz 16 mg/kg/gün) fertilite ya da üreme kapasitesine bir etki görülmemiştir. 6. FARMASÖTİK ÖZELLİKLER6.1 Yardımcı maddelerin listesiTablet çekirdeğiKolloidal susuz silikaKroskarmelloz sodyumLaktoz monohidrat (sığır sütü kaynaklı)Magnezyum stearat Mikrokristalin selülozPovidon Sodyum lauril sülfat (E487) Film kaplamaMakrogolPolivinil alkolTitanyum dioksit (E171)Siyah demir oksit (E172) Sarı demir oksit (E172) Talk 6.2 GeçimsizliklerGeçerli değil. 6.3 Raf ömrü24 ay. 6.4 Saklamaya yönelik özel tedbirler30 °C altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriğiHer bir karton cüzdanda poliklorotrifloroetilen (PCTFE) ile lamine edilmiş polivinil klorür (PVC)/alüminyum blister ile 10 film kaplı tablet içerir. Her kartonda (30 film kaplı tablet) 3cüzdan bulunur. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİ Johnson and Johnson Sıhhi Mal. San. ve Tic. Ltd. Şti. Kavacık/Beykoz/İstanbul 8. RUHSAT NUMARASI 2022/479 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİ İlk ruhsat tarihi: 25.08.2022 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ |
İlaç BilgileriImbruvica 420 Mg Film Kaplı TabletEtken Maddesi: İbrutinib Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.