Valamor 200 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA URUN BILGISI¡BU1. BEŞERI TIBBİ ÜRÜNÜN ADIVALAMOR 200 mg film kaplı tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Ribosiklib süksinat 254,4 mg (200 mg ribosiklibe karşılık gelmektedir) Yardımcı maddeler:Soya lesitin 0,344 mg Yardımcı maddelerin tam listesi için Bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tablet Açık grimsi mor, çentiksiz, yuvarlak, eğimli kenarlı, bir tarafında RIC ve diğer tarafında NVR işlemeli. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarVALAMOR, NSAİ (nonsteroidal aromataz inhibitörü) ile kombine kullanımı halinde Östrojen reseptörü (ER) en az %10 pozitif, insan epidermal büyüme faktörü 2 (HER2) negatifpre/peri/postmenopozal metastatik meme kanseri olan, daha önce metastatik hastalık içinendokrin tedavi almamış hastalar ile birlikte adjuvan NSAİ tedavisinin tamamlanmasından 12ay sonra relaps yapmış veya adjuvan tamoksifen tedavisi sırasında veya sonrasında relapsyapmış hastalarda endikedir. VALAMOR Östrojen reseptörü (ER) en az %10 pozitif ve HER2 (-) olan pre/peri/postmenopozal metastatik meme kanserli daha önce fulvestrant kullanmamışhastalarda: 1- Metastatik hastalık tedavisi için bir sıra ve en az 6 ay aromataz inhibitörü alırken klinikve/veya radyolojik hastalık progresyonu görülenlerde fulvestrant ile birlikte kullanılır. 2- Adjuvan aromataz inhibitörü tedavisini en az 12 ay süreyle kullandıktan sonra ya daadjuvan aromataz inhibitörü tedavisi tamamlandıktan sonraki 12 ay içinde nüks/metastazgelişen hastalarda fulvestrant ile birlikte kullanılır. Adjuvan aromataz inhibitörü alırken ilk 12ay içinde relaps görülen hastalarda veya metastatik hastalık nedeniyle bir sıradan daha fazlaaromataz inhibitörü tedavisi almış olan hastalarda kullanılamaz. Pre/perimenopozal kadınlarda endokrin tedavisi, luteinize edici hormon salgılatıcı hormon (LHRH) agonisti ile birleştirilmelidir. 4.2 Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:VALAMOR ile tedavi, anti-kanser tedavilerin kullanımında deneyimli bir hekim tarafından başlatılmalıdır. Pozoloji:VALAMOR'un önerilen dozu, 28 günlük bir siklusu tamamlayacak şekilde arka arkaya 21 gün süreyle günde bir kez oral yoldan alınan 600 mg (üç adet 200 mg film kaplı tablet) vebunu izleyen tedavisiz 7 gün şeklindedir. Tedavi, hasta tedaviden klinik fayda gördüğü süreceveya kabul edilemez toksisite meydana gelene kadar sürdürülmelidir. VALAMOR, 2,5 mg letrozol ya da 500 mg fulvestrant ile birlikte kullanılmalıdır. VALAMOR letrozol ile birlikte kullanıldığında letrozol, 28 günlük siklus boyunca günde bir kez alınmalıdır. Lütfen ilave bilgiler için letrozolün Kısa Ürün Bilgisine başvurunuz. VALAMOR fulvestrant ile birlikte kullanıldığında fulvestrant, intramüsküler olarak 1., 15. ve 29. günlerde ve daha sonra ayda bir kere uygulanır. Lütfen ilave bilgiler için fulvestrantınKısa Ürün Bilgisine başvurunuz. VALAMOR aç veya tok karnına alınabilir (bkz. Bölüm 4.5). Hastalar dozlarını her gün yaklaşık olarak aynı saatte, tercihen sabah saatlerinde almaya teşvik edilmelidir. Hasta dozualdıktan sonra kusarsa veya bir dozu atlarsa, o gün ilave bir doz alınmamalıdır. Bir sonrakireçete edilmiş doz normal zamanında alınmalıdır. Doz modifikasyonlarıAğır veya tolere edilemeyen advers ilaç reaksiyonlarının kontrol edilebilmesi için dozlara geçici olarak ara vermek, dozu azaltmak veya VALAMOR tedavisini kesmek gerekebilir.Dozda azaltma gerekiyorsa önerilen doz azaltımı kılavuzları Tablo 1'de verilmektedir. Tablo 1 Önerilen doz modifikasyonu kılavuzları
Tablo 2, 3, 4 ve 5'te belirli advers ilaç reaksiyonlarının kontrolü için dozlara ara verme, doz azaltma ve tedavinin kesilmesi ile ilgili öneriler özetlenmektedir. Her hastanın tedavi planına,hasta bazında yapılan fayda/risk değerlendirmesine dayalı olarak tedaviden sorumlu hekiminklinik değerlendirmesi yön vermelidir (bkz. Bölüm 4.4). VALAMOR ile tedaviye başlanmadan önce tam kan sayımı yapılmalıdır. VALAMOR ile tedaviye başlandıktan sonra tam kan sayımı izlemi ilk 2 siklus süresince iki haftada bir,sonraki her 4 siklusun başında ve sonrasında klinik durum gerektirdikçe yapılmalıdır.
VALAMOR ile tedaviye başlanmadan karaciğer fonksiyon testleri yapılmalıdır. VALAMOR ile tedaviye başlandıktan sonra karaciğer fonksiyon testleri izlemi ilk 2 döngü süresince ikihaftada bir, sonraki her 4 siklusun başında ve sonrasında klinik durum gerektirdikçeyapılmalıdır. Eğer derece >2 anomaliler görülürse daha sık izlem yapılması önerilir.
Tedaviye başlanmadan önce EKG değerlendirmesi yapılmalıdır. VALAMOR ile tedaviye başlandıktan sonra EKG, ilk siklusun yaklaşık 14. gününde ve ikinci siklusun başında,sonrasında klinik durum gerektirdikçe tekrarlanmalıdır. Tedavi sırasında QTcF uzaması olursadaha sık EKG izlemi önerilir.
Doz modifikasyonu kılavuzları ve toksisite durumunda diğer ilgili güvenlilik bilgileri için birlikte uygulanan letrozolün, fulvestrantın veya gonadotropin salgılatıcı hormonun (LHRH)agonistinin KÜB'ünü inceleyiniz. VALAMOR 'un güçlü CYP3A4 inhibitörleri ile birlikte kullanımı için doz modifikasyonuGüçlü CYP3A4 inhibitörlerinin eş zamanlı kullanımından kaçınılmalıdır ve daha düşük CYP3A4 inhibisyonu potansiyeli bulunan alternatif bir eş zamanlı tıbbi ürün düşünülmelidir.Eğer hastaların ribosiklib ile aynı zamanda güçlü bir CYP3A4 inhibitörü ile tedavi edilmesişart ise VALAMOR dozu günde bir kez 400 mg'a düşürülmelidir (bkz. Bölüm 4.5). Dozları günlük 400 mg ribosiklibe azaltılmış hastalarda ve bir güçlü CYP3A4 inhibitörünün birlikte uygulanmaya başlatılmasının kaçınılamadığı hastalarda doz 200 mg'a düşürülmelidir. lAH/Pnömonit
CTCAE Versiyon 4.03'e göre derecelendirme. *VALAMOR tedavisi tekrar başlanacaksa bireysel yarar/risk analizi yapılmalıdır. IAH= Interstisyel Akciğer Hastalığı_Dozları günlük 200 mg ribosiklibe azaltılmış hastalarda ve bir güçlü CYP3A4 inhibitörünün birlikte uygulanmaya başlatılmasının kaçınılamadığı hastalarda VALAMOR tedavisikesilmelidir. Hasta bazında değişkenlik nedeniyle, önerilen doz ayarlamaları tüm hastalarda optimum olmayabilir, bu nedenle toksisite belirtilerinin yakın takibi önerilmektedir. Eğer güçlüinhibitör kesilirse, güçlü CYP3A4 inhibitörünün en az 5 yarılanma ömrü geçtikten sonraVALAMOR dozu, güçlü CYP3A4 inhibitörü ile tedaviye başlanmadan önce kullanılmaktaolan doza değiştirilmelidir (bkz. Bölüm 4.4, 4.5 ve 5.2). Uygulama şekli:Oral kullanım içindir. VALAMOR günde bir defa oral yolla, aç veya tok karnına alınmalıdır. Tabletler bütün olarak yutulmalıdır ve yutulmadan önce çiğnenmemeli, ezilmemeli ya da bölünmemelidir. Kırık,çatlak veya başka bir şekilde bütünlüğü bozulmuş hiçbir tablet alınmamalıdır. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Hafif veya orta dereceli böbrek bozukluğu olan hastalarda herhangi bir doz ayarlaması gerekli değildir (bkz. Bölüm 5.2). Şiddetli böbrek bozukluğu olan hastalarda 200 mg'lık bir başlangıçdozu önerilir (bkz. Bölüm 5.2). VALAMOR, şiddetli böbrek bozukluğu olan meme kanserihastalarında çalışılmamıştır. (bkz. Bölüm 4.4,5.1 ve 5.2). Karaciğer yetmezliği:Hafif karaciğer bozukluğu (Child-Pugh sınıf A) olan hastalarda herhangi bir doz ayarlaması gerekli değildir. Orta şiddetli (Child-Pugh sınıf B) ve şiddetli karaciğer bozukluğu (Child-Pugh sınıf C) olan hastalarda ribosiklib maruziyeti artabilir (2 kattan daha az) ve bu hastalardagünde bir kez 400 mg başlangıç dozu önerilir (bkz. Bölüm 5.2). Pediyatrik popülasyon:VALAMOR'un 18 yaşın altındaki çocuklarda ve adölesanlarda güvenliliği ve etkililiği belirlenmemiştir. Veri bulunmamaktadır. Geriyatrik popülasyon:65 yaşın üzerindeki hastalarda herhangi bir doz ayarlaması gerekli değildir (bkz. Bölüm 5.2). 4.3 KontrendikasyonlarEtkin maddeye ya da yer fıstığına, soyaya ya da Bölüm 6.1'de listelenen yardımcı maddelerin herhangi birine karşı aşırı duyarlılık. 4.4 Özel kullanım uyarıları ve önlemleriKritik visseral hastalıkRibosiklibin etkililiği ve güvenliliği kritik visseral hastalığı olan hastalarda araştırılmamıştır. NötropeniNötropeninin şiddetine bağlı olarak Tablo 2'de tarif edildiği gibi VALAMOR ile tedaviye ara verilmesi, dozun azaltılması veya tedavinin kesilmesi gerekebilir (bkz. Bölüm 4.2 ve 4.8). Hepatobiliyer toksisiteVALAMOR ile tedaviye başlanmadan karaciğer fonksiyon testleri yapılmalıdır. VALAMOR ile tedaviye başlandıktan sonra karaciğer fonksiyon testleri izlenmelidir (bkz. Bölüm 4.2 ve4.8). Transaminaz yükselmelerinin şiddetine bağlı olarak Tablo 3'te tarif edildiği gibi VALAMOR ile tedaviye ara verilmesi, dozun azaltılması veya tedavinin kesilmesi gerekebilir (bkz. Bölüm4.2 ve 4.8). Başlangıçta AST/ALT derece > 3 yükselmeleri olan hastalar için önerilerbelirlenmemiştir. QT aralığında uzamaÇalışma E2301'de (MONALEESA-7) ribosiklib artı tamoksifen alan hastaların 14/87'sinde (%16,1) ve ribosiklib artı bir nonsteroidal aromataz inhibitörü (NSAI) alan hastaların18/245'inde (%7,3), çalışma başlangıcına kıyasla > 60 milisaniyelik bir QTcF aralığı artışıgözlenmiştir. VALAMOR'un tamoksifen ile kombinasyon halinde kullanımıönerilmemektedir (bkz. Bölüm 4.8 ve 5.1). Tedaviye başlanmadan önce EKG değerlendirmesi yapılmalıdır. VALAMOR ile tedavi sadece QTcF değerleri <450 milisaniye olan hastalarda başlatılmalıdır. EKG, ilk siklusun yaklaşık14. gününde ve ikinci siklusun başında, sonrasında klinik durum gerektirdikçetekrarlanmalıdır (bkz. Bölüm 4.2 ve 4.8). Tedaviye başlanmadan önce, ilk altı siklusun başında ve sonrasında klinik durum gerektirdikçe serum elektrolitlerinin (potasyum, kalsiyum, fosfor ve magnezyum dahil) izlemiyapılmalıdır. Anormallik varsa VALAMOR ile tedaviye başlanmadan önce ve VALAMOR iletedavi sırasında düzeltilmelidir. Halihazırda QTc uzaması olan ya da QTc uzaması açısından önemli riske sahip hastalarda VALAMOR kullanımından kaçınılmalıdır. Bu hastalar: Uzun QT sendromu olan hastalar. Yakın zamanda geçirilmiş miyokard enfarktüsü, konjestif kalp yetmezliği, stabil olmayan anjina ve bradiaritmiler dahil kontrol edilmemiş veya önemli kalp hastalığı olan hastalar. Elektrolit anormallikleri olan hastalardır. QTcF aralığında klinik olarak anlamlı uzamaya yol açabileceğinden VALAMOR'un QTc aralığını uzattığı bilinen ilaçlar ve/veya güçlü CYP3A4 inhibitörleri ile birlikte kullanımındankaçınılmalıdır (bkz. Bölüm 4.2, 4.5 ve 5.1). Güçlü bir CYP3A4 inhibitörü ile tedavindenkaçınılamazsa, doz günde bir kez 400 mg'a düşürülmelidir (bkz. Bölüm 4.2 ve 4.5). Tedavi sırasında gözlenen QT uzamasına bağlı olarak Tablo 4'te tarif edildiği gibi VALAMOR ile tedaviye ara verilmesi, dozun azaltılması veya tedavinin kesilmesi gerekebilir(bkz. Bölüm 4.2, 4.8 ve 5.2). Şiddetli kutanöz reaksiyonlarVALAMOR tedavisi ile toksik epidermal nekroliz (TEN) bildirilmiştir. Şiddetli kutanöz reaksiyonları düşündüren işaret ve semptomlar (örn., sıklıkla kabarcıklar veya mukozallezyonlar ile birlikte progresif geniş çaplı deri döküntüsü) ortaya çıkarsa, VALAMOR acilenbırakılmalıdır. İnterstisyel Akciğer Hastalığı/PnömonitVALAMOR ve diğer siklin-bağımlı kinaz 4/6 (CDK4/6) inhibitörleriyle tedavi edilen hastalarda şiddetli, yaşamı tehdit eden veya ölümcül interstisiyel akciğer hastalığı (İAH)ve/veya pnömonit oluşabilir. Üç Faz-III klinik çalışmada (MONALEESA-2, MONALEESA-3, MONALEESA-7) ribosiklib tedavisi alan hastaların %1,6'sında herhangi bir dereceden İAH/pnömonit,%0,4'ünde Derece 3 ya da Derece 4, %0,1'inde ölüm rapor edilmiştir. Ruhsatlandırma sonrasında ek İAH/Pnömonit olgularına rastlanmış olup, ölümlü vakalar bildirilmiştir. (bkz. Bölüm 4.8) Hastalar, İAH/Pnömonit düşündüren akciğer semptomları (örn., hipoksi, öksürük, dispne) açısından takip edilmelidir. İAH/Pnömonit kuşkusu yaratacak yeni veya kötüleşen solunumsemptomları olan hastalarda hemen VALAMOR kullanımı durdurularak hastadeğerlendirmeye tabi tutulmalıdır. Tekrarlayan semptomatik veya şiddetli İAH veya pnömonitgözlenen hastalarda VALAMOR kullanımı kalıcı olarak bırakılmalıdır. (bkz. Bölüm 4.2) Kan kreatinin yükselmesiRibosiklib, proksimal tübüllerden kreatinin aktif salgılanmasında rol oynayan renal taşıyıcılar organik katyon taşıyıcı 2 (OCT2) ve çoklu ilaç ve toksin ekstrüzyon proteini 1'in (MATE1)bir inhibitörü olarak kan kreatinin artışına neden olabilir (bkz. Bölüm 4.5). Tedavi sırasındakan kreatinin artışı durumunda, böbrek yetmezliğini dışlamak için böbrek fonksiyonunun dahafazla değerlendirilmesi önerilir. CYP3A4 substratlarıRibosiklib 600 mg dozda güçlü bir CYP3A4 inhibitörü ve 400 mg dozda orta güçte bir CYP3A4 inhibitörüdür. Bu nedenle, ribosiklib CYP3A4 ile metabolize edilen tıbbi ürünlerleetkileşime girebilir ki bu da CYP3A4 substratlarının serum konsantrasyonlarında artışa yolaçabilir (bkz. Bölüm 4.5). Dar terapötik indekse sahip duyarlı CYP3A4 substratları ile eşzamanlı kullanım durumunda dikkat önerilmektedir ve CYP3A4 inhibitörleri ile birlikteuygulamaya ilişkin öneriler için diğer ürünün KÜB'üne danışılmalıdır. Böbrek yetmezliğiŞiddetli böbrek yetmezliği olan hastalar için önerilen 200 mg'lık başlangıç dozunun, normal böbrek fonksiyonu olan hastalardaki standart başlangıç dozuna kıyasla yaklaşık %45 dahadüşük maruziyet ile sonuçlandığı tahmin edilmektedir. Bu başlangıç dozundaki etkinlikaraştırılmamıştır. Şiddetli böbrek yetmezliği olan hastalar, toksisite belirtileri açısındanyakından takip edilerek dikkatli olunmalıdır (bkz. Bölüm 4.2 ve 5.2). Çocuk doğurma potansiyeli olan kadınlarÇocuk doğurma potansiyeli olan kadınların, VALAMOR'u kullanırken ve son dozdan en az 21 gün sonrasına kadar etkili bir doğum kontrol yöntemi kullanmaları önerilmelidir (bkz.Bölüm 4.6). Soya lesitiniVALAMOR soya lesitini içerir. Yer fıstığına veya soyaya aşırı duyarlılığı olan hastalar VALAMOR kullanmamalıdır (bkz. Bölüm 4.3). 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriRibosiklibin plazma konsantrasyonlarını yükseltebilen maddelerRibosiklib başlıca CYP3A4 ile metabolize olur. Bu nedenle, CYP3A4 enzimi aktivitesini etkileyebilen tıbbi ürünler, ribosiklibin farmakokinetiğini etkileyebilir. Güçlü CYP3A4inhibitörü ritonavirin (14 gün boyunca günde iki kez 100 mg) 400 mg'lık tekli ribosiklib dozuile birlikte uygulanması sağlıklı gönüllülerde ribosiklib maruziyetini (EAAinf) ve pikkonsantrasyonunu (Cmaks) tek başına verildiğinde 400 mg'lık tekli ribosiklib dozuna kıyaslasırasıyla 3,2 ve 1,7 kat artırmıştır. LEQ803 (ana maruziyetin %10'undan azını oluşturanbaskın ribosiklib metaboliti) için Cmaks ve EAAlast sırasıyla %96 ve %98 azalmıştır. Bunlarla sınırlı olmamak üzere klaritromisin, indinavir, itrakonazol, ketokonazol, lopinavir, ritonavir, nefazodon, nelfinavir, posakonazol, sakuinavir, telaprevir, telitromisin verapamil vevorikonazol dahil güçlü CYP3A4 inhibitörlerinin eş zamanlı kullanımından kaçınılmalıdır(bkz. Bölüm 4.4). CYP3A4'yı inhibe etme potansiyeli daha düşük alternatif eş zamanlı tıbbiürünler göz önünde bulundurulmalı ve hastalar ribosiklib ile ilişkili advers olaylar açısındanizlenmelidir (bkz. Bölüm 4.2, 4.4 ve 5.2). Eğer VALAMOR'un güçlü bir CYP3A4 inhibitörü ile bir arada kullanımından kaçınılamıyorsa, VALAMOR'un dozu bölüm 4.2'de tanımlandığı şekilde azaltılmalıdır. Öteyandan, bu doz ayarlaması ile ilgili klinik veri bulunmamaktadır. Hastalar arasındakideğişkenlik nedeniyle önerilen doz ayarlamaları tüm hastalarda optimal olmayabilir ve bunedenle ribosiklib ile ilişkili advers olaylar için yakın izlem önerilir. VALAMOR ile ilişkilitoksisite görülmesi durumunda doz ayarlanmanlı veya toksisite düzelene kadar tedaviye araverilmelidir (bkz. Bölüm 4.2 ve 5.2). Güçlü CYP3A4 inhibitörü bırakılırsa CYP3A4inhibitörünün en az 5 yarılanma ömründen sonra (söz konusu CYP3A4 inhibitörününKÜB'ünü inceleyiniz), VALAMOR, güçlü CYP3A4 inhibitörünün başlatılmasından öncekullanılanla aynı dozda yeniden başlatılmalıdır. Fizyoloji temelli farmakokinetik simülasyonları, 600 mg'lık ribosiklib dozunda orta güçte bir CYP3A4 inhibitörünün (eritromisin) ribosiklib kararlı durum Cmaks ve EAA değerini sırasıyla1,2 kat ve 1,3 kat artırabileceğini düşündürmüştür. Ribosiklib dozları günde bir kez 400 mg'aazaltılmış hastalar için kararlı durum Cmaks ve EAA değerindeki artışın sırasıyla tahminen 1,4ve 2,1 kat olduğu hesaplanmıştır. 200 mg'lık günlük dozdaki etkinin sırasıyla 1,7 ve 2,8 katartması öngörülmüştür. Hafif veya orta şiddette CYP3A4 inhibitörleri ile tedavininbaşlatılmasında ribosiklib dozunda ayarlama gerekli değildir. Bununla birlikte, ribosiklib ileilişkili advers olayların izlenmesi önerilmektedir. Hastalara greyfurt veya greyfurt suyundan uzak durmaları söylenmelidir. Bunların sitokrom CYP3A4 enzimlerini inhibe ettiği bilinmektedir ve ribosiklibe maruziyeti artırabilirler. Ribosiklibin plazma konsantrasyonlarını düşürebilen maddelerGüçlü CYP3A4 indükleyici rifampisinin (14 gün boyunca günde 600 mg) 600 mg'lık tekli ribosiklib dozu ile birlikte uygulanması ribosiklib EAAinf ve Cmaks değerlerini sağlıklıgönüllülerde tek başına verilen tekli 600 mg ribosiklib dozuna kıyasla sırasıyla %89 ve %81azaltmıştır. LEQ803 Cmaks değeri 1,7 kat artarken EAAinf değeri %27 azalmıştır. Böylecegüçlü CYP3A4 indükleyicilerinin eş zamanlı kullanımı azalmış maruziyete ve sonuçtaetkisizlik riskine yol açabilir. Güçlü CYP3A4 indükleyicilerinin eş zamanlı kullanımındankaçınılmalıdır; bunlar fenitoin, rifampisin, karbamazepin ve sarı kantaronu (Hypericumperforatum)içermekle beraber bunlarla sınırlı değildir. CYP3A4'ü indükleme potansiyeliolmayan ya da minimum potansiyele sahip alternatif bir eş zamanlı tıbbi ürün düşünülmelidir.Orta güçte CYP3A4 indükleyicisinin ribosiklib maruziyeti üzerindeki etkisi araştırılmamıştır. Fizyoloji bazlı farmakokinetik simülasyonları orta güçte CYP3A4 indükleyicisinin (efavirenz)kararlı durum ribosiklib Cmaks ve EAA değerini sırasıyla %51 ve %70 azaltabileceğinidüşündürmüştür. Böylece orta güçte CYP3A4 indükleyicilerinin eş zamanlı kullanımı azalmışmaruziyete ve sonuç olarak özellikle günde bir kez 400 mg veya 200 mg'da ribosiklib iletedavi edilen hastalarda yetersiz etkililik riskine yol açabilir. Plazma konsantrasyonları VALAMOR ile değişebilen maddelerRibosiklib orta güç ila güçlü bir CYP3A4 inhibitörü olup, CYP3A4 ile metabolize edilen tıbbi substratlarla etkileşime girebilir ki bu da eş zamanlı kullanılan tıbbi ürünün serumkonsantrasyonlarında artışa yol açabilir. Sağlıklı gönüllülerde tek başına midazolam (CYP3A4 substratı) uygulaması ile karşılaştırıldığında midazolamın çoklu ribosiklib (400 mg) dozları ile birlikte uygulanması,midazolam maruziyetini %280 yükseltmiştir (3,8 kat). Fizyolojik esaslı farmakokinetiğin(FEFK) kullanıldığı simülasyonlar, klinik olarak anlamlı 600 mg dozunda verilenVALAMOR'un midazolam EAA değerini 5,2 kat artırmasının beklendiğini göstermiştir. Bunedenle, genel olarak, VALAMOR başka bir tıbbi ürün ile birlikte uygulandığında, CYP3A4inhibitörleri ile birlikte kullanıma ilişkin öneriler için diğer ürünün KÜB'üne başvurulmalıdır.VALAMOR, dar terapötik indekse sahip CYP3A4 substratları ile birlikte uygulanırkendikkatli olunması önerilir (bkz. Bölüm 4.4). Bunlarla sınırlı olmamak üzere alfentanil,siklosporin, everolimus, fentanil, sirolimus ve takrolimus dahil dar terapötik indekse sahipduyarlı CYP3A4 substratlarının dozunun azaltılması gerekebilir çünkü ribosiklib bunlarınmaruziyetini artırabilir. 600 mg dozda ribosiklibin şu CYP3A4 substratları ile eş zamanlı uygulanmasından kaçınılmalıdır: Alfuzosin, amiodaron, sisaprid, pimozid, kinidin, ergotamin, dihiroergotamin,ketiapin, lovastatin, simvastatin, sildenafil, midazolam, triazolam. Sağlıklı gönüllülerde tek başına kafein (CYP1A2 substratı) alınması ile karşılaştırıldığında kafeinin çoklu ribosiklib (400 mg) dozları ile birlikte alınması kafein maruziyetini %20 (1,20kat) artırmıştır. Klinik olarak anlamlı 600 mg dozunda FEFK'nin kullanıldığı simülasyonlar,ribosiklibin CYP1A2 substratları üzerinde sadece çok zayıf inhibitör etkisini öngörmüştür(EAA değerinde <2 kat artış). Taşıyıcıların substratları olan maddelerİn vitrodeğerlendirmeler, ribosiklibin ilaç taşıyıcıları P-gp, BCRP, OATP1B1/B3, OCT1, OCT2, MATE1 ve BSEP'in aktivitelerini inhibe etme yönünde potansiyele sahip olduğunugöstermiştir. Digoksin, pitavastatin, pravastatin, rosuvastatin ve metformini içeren ancakbunlarla sınırlı olmayan dar terapötik indeks sergileyen bu taşıyıcıların duyarlı substratları ileeş zamanlı tedavisi sırasında toksisite açısından dikkat ve izleme önerilir.İlaç-besin etkileşimleriVALAMOR aç veya tok karnına uygulanabilir (bkz. Bölüm 4.2 ve 5.2). Gastrik pH değerini yükselten tıbbi ürünlerRibosiklib 4,5 pH değerinde veya bu değerin altında ve biyo-uyumlu ortamlarda (pH 5 ve 6,5) yüksek çözünürlüğe sahiptir. Ribosiklibin gastrik pH'i yükselten tıbbi ürünler ile birlikteuygulanması klinik bir çalışmada değerlendirilmemiştir; ancak popülasyon farmakokinetiği vekompartımanlı olmayan farmakokinetik analizlerinde değişmiş ribosiklib emilimigözlenmemiştir. Ribosiklib ve letrozol arasındaki ilaç-ilaç etkileşimiMeme kanseri olan hastalarda yürütülen klinik çalışmadan ve popülasyon farmakokinetiği analizinden veriler bu tıbbi ürünlerin birlikte uygulanmasından sonra ribosiklib ve letrozolarasında ilaç etkileşimi olmadığını göstermiştir. Ribosiklib ile anastrozol arasındaki ilaç-ilaç etkileşimiMeme kanseri olan hastalardaki bir klinik çalışmaya ait veriler, ribosiklib ile anastrozolün bir arada uygulanmasından sonra bu tıbbi ürünler arasında klinik olarak anlamlı bir ilaçetkileşimine işaret etmemiştir. Ribosiklib ve fulvestrant arasındaki ilaç-ilaç etkileşimiMeme kanserli hastalarda yürütülen klinik çalışmadan elde edilen veri, bu iki tıbbi ürünün birlikte uygulanmasını takiben fulvestrantın ribosiklib maruziyeti üzerinde klinik olarakilişkili etkisinin olmadığını göstermiştir. Ribosiklib ile tamoksifen arasındaki ilaç-ilaç etkileşimiMeme kanseri olan hastalardaki bir klinik çalışmaya ait veriler, ribosiklib ile tamoksifenin bir arada uygulanmasından sonra tamoksifen maruziyetinin yaklaşık 2 kat arttığını göstermiştir. Ribosiklib ile oral kontraseptifler arasındaki ilaç-ilaç etkileşimiRibosiklib ile oral kontraseptifler arasında ilaç-ilaç etkileşimi çalışmaları gerçekleştirilmemiştir (bkz. Bölüm 4.6). Beklenen etkileşimlerQTaralığını uzatabilen anti-aritmik tıbbi ürünlervediğer tıbbi ürünler:VALAMOR'un anti-aritmik tıbbi ürünler gibi QT aralığını uzatma potansiyeli olduğu bilinen tıbbi ürünler (bunlarla sınırlı olmamak üzere amiodaron, disopiramid, prokainamid, kinidin vesotalol) ve QT aralığını uzattığı bilinen diğer tıbbi ürünler (bunlarla sınırlı olmamak üzereklorokuin, halofantrin, klaritromisin, siprofloksazin, levofloksasin, azitromisin, haloperidol,metadon, moksifloksasin, bepridil, pimozid ve intravenöz ondansetron) ile birlikteuygulanmasından kaçınılmalıdır (bkz. Bölüm 4.4). Ayrıca, VALAMOR'un tamoksifen ilekombinasyon halinde kullanılması önerilmemektedir (bkz. Bölüm 4.1, 4.4 ve 5.1). Özel popülasyonlara ilişkin ek bilgiler:Etkileşim açısından özel popülasyonlara ilişkin veri bulunmamaktadır. Pediyatrik popülasyon:Etkileşim açısından özel popülasyonlara ilişkin veri bulunmamaktadır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)VALAMOR ile tedaviye başlanmadan önce gebelik durumu teyit edilmelidir. VALAMOR kullanmakta olan çocuk doğurma potansiyeline sahip kadınların tedavi sırasında ve VALAMOR ile tedaviyi bıraktıktan sonra en az 21 gün süreyle etkili doğum kontrolü (örn.çift bariyer kontrasepsiyon) uygulamaları gerekmektedir. Gebelik dönemiGebe kadınlarda yeterli ve iyi kontrollü çalışmalar bulunmamaktadır. Hayvanlar üzerinde yapılan araştırmaların sonuçlarına ve etki mekanizmasına göre, ribosiklib hamile kadınlarauygulandığında fetal zarara neden olabilir (bkz. Bölüm 5.3). VALAMOR'un hamileliksırasında ve çocuk doğurma potansiyeli olup kontrasepsiyon yöntemi uygulamayan kadınlardauygulanması önerilmemektedir. Ribosiklibin gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. Bölüm 5.3).İnsanlara yönelik potansiyel risk bilinmemektedir. VALAMOR, yarar-risk değerlendirmesine göre gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Gebe kadınlara fetüse yönelik potansiyel risk konusunda tavsiyelerdebulunulmalıdır. Laktasyon dönemiRibosiklibin insan sütüyle atılıp atılmadığı bilinmemektedir. Ribosiklibin anne sütü ile beslenen bebek ve süt üretimi üzerine etkisini gösteren veri bulunmamaktadır. Ribosiklib vemetabolitleri sıçan sütüne hızla geçmiştir. Hayvanlar üzerinde yapılan çalışmalar, ribosiklibinsütle atıldığını göstermektedir. Emzirmenin durdurulup durdurulmayacağına ya daVALAMOR tedavisinin durdurulup durdurulmayacağına/ tedaviden kaçınılıpkaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası veVALAMOR tedavisinin emziren anne açısından faydası dikkate alınmalıdır. VALAMORkullanan ve emziren kadınların, VALAMOR'un anne sütü ile beslenen bebeklerde ciddiadvers reaksiyon oluşturma potansiyeli sebebiyle son VALAMOR dozundan en az 21 günsonrasına kadar emzirmemeleri önerilir. Üreme yeteneği/FertiliteRibosiklibin fertilite üzerine etkilerini gösteren klinik veri bulunmamaktadır. Hayvanlarda yapılan çalışmalara göre, ribosiklib üreme potansiyeli bulunan erkeklerde fertiliteye zararverebilir (bkz. Bölüm 5.3). 4.7 Araç ve makine kullanımı üzerindeki etkilerVALAMOR, araç ve makine kullanma becerisi üzerinde minör etkiye sahip olabilir. Hastalara, VALAMOR ile tedavileri sırasında yorgunluk, baş dönmesi veya vertigodeneyimlemeleri halinde araç ve makine kullanırken dikkatli olmaları söylenmelidir (bkz.Bölüm 4.8). 4.8 İstenmeyen etkilerGüvenlilik profilinin özetiBirleştirilmiş veri kümesinde ribosiklib artı herhangi bir kombinasyon için sıklığın plasebo artı herhangi bir kombinasyon için sıklıktan fazla olduğu en yaygın advers reaksiyonlar (>%20 sıklıkla bildirilen) nötropeni, enfeksiyonlar, bulantı, yorgunluk, diyare, lökopeni, kusma,baş ağrısı, konstipasyon, alopesi, öksürük, döküntü, sırt ağrısı, anemi ve anormal karaciğerfonksiyonu testleridir. Birleştirilmiş veri kümesinde ribosiklib artı herhangi bir kombinasyon için sıklığın plasebo artı herhangi bir kombinasyon için sıklıktan fazla olduğu en yaygın derece 3/4 adversreaksiyonlar (>2 sıklıkla bildirilen) nötropeni, lökopeni, anormal karaciğer fonksiyonu testleri,lenfopeni, enfeksiyonlar, sırt ağrısı, anemi, yorgunluk, hipofosfatemi ve kusmadır. Nedensel ilişkiye bakılmaksızın, faz III çalışmalarında kombinasyona bakmaksızın ribosiklib alan hastalarda advers olaylara bağlı doz azaltımı hastaların % 39,5'inde; faz IIIçalışmalarında herhangi bir kombinasyon ve ribosiklib alan hastalarda tedavinin kalıcı olarakdurdurulması hastaların % 8,7'sinde bildirilmiştir. Advers reaksiyonların tablo halinde özetiRibosiklibin genel güvenlilik profili, ribosiklibi endokrin tedavisi (N=582, bir aromataz inhibitörü ile kombinasyon halinde ve N=483, fulvestrant ile kombinasyon halinde) ilekombinasyon halinde alan ve HR-pozitif, HER2-negatif ileri veya metastatik meme kanseriüzerine randomize, çift kör, plasebo kontrollü faz III klinik çalışmalara (MONALEESA-2,MONALEESA-7 NSAI alt grubu ve MONALEESA-3) katılan 1065 hastanın birleştirilmişveri kümesine dayanmaktadır. Pazarlama sonrasında, ilave advers reaksiyonlar görülmüştür. Birleştirilmiş faz III çalışmaların veri kümesi genelinde ribosiklib tedavisine medyan maruziyet süresi 19,2 ay olup hastaların %61,7'sinde maruziyet >12 aydır. Faz III çalışmalardan bildirilen advers reaksiyonlar (Tablo 6) MedDRA sistem organ sınıfına göre listelenmektedir. Her bir sistem organ sınıfı içinde advers reaksiyonlar sıklığa göre, ensık reaksiyonlar ilk belirtilerek sıralanmaktadır. Her bir sıklık gruplandırması içinde adversreaksiyonlar azalan ciddilik derecelerine göre sunulmaktadır. Ayrıca, her advers reaksiyoniçin karşılık gelen sıklık kategorisi şu sisteme dayanmaktadır (CIOMS III): Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerdenhareketle tahmin edilemiyor). Tablo 7 Üç Faz III klinik çalışmada ve pazarlama sonrası deneyim sırasında gözlenen advers reaksiyonlar
Kardiyak hastalıklar
Senkop
Yaygın Solunum, göğüs bozuklukları ve mediastinal hastalıklar
Dispne, öksürük
Çok yaygın
Interstisyel akciğer hastalığı (lAH)/pnömonit
Yaygın Gastrointestinal hastalıklarBulantı, diyare, kusma, konstipasyon, karın ağrısı2 , stomatit, dispepsi Disguzi Çok yaygın Yaygın Hepatobiliyer hastalıklar
Hepatotoksisite ¦
Yaygın Deri ve deri altı doku hastalıkları
Alopesi, döküntü 4, prurit Eritem, cilt kuruluğu, vitiligoToksik epidermal nekroliz (TEN)*
Çok yaygın Yaygın Bilinmiyor Kas-iskelet bozukluklar, bağ doku ve kemik hastalıkları
Sırt ağrısı
Çok yaygın Genel bozukluklar ve uygulama bölgesine ilişkin durumlarYorgunluk, periferik ödem, pireksi, asteni Orofarengeal ağrı, ağız kuruluğu Çok yaygın Yaygın AraştırmalarAnormal karaciğer fonksiyonu testleri 5 Kan kreatinin düzeyinde artış, elektrokardiyogramda QT uzaması Çok yaygın Yaygın Pazarlama sonrası deneyim sırasında bildirilen advers reaksiyonlar. 1 Enfeksiyonlar: İdrar yolu enfeksiyonları, solunum yolu enfeksiyonları, gastroenterit,sepsis (<%1). 2 Karın ağrısı: Karın ağrısı, üst karın ağrısı. 3 Hepatotoksisite: Hepatik sitoliz, ilaç kaynaklı karaciğer hasarı (<%1), hepatotoksisite,karaciğer yetmezliği, otoimmün hepatit (tek bir vaka). 4 Döküntü: Döküntü, makülo-papüler döküntü, kaşıntılı doküntü. 5 Anormal karaciğer fonksiyonu testleri: ALT düzeyinde yükselme, AST düzeyindeyükselme, kan bilirubin düzeyinde yükselme. Seçilen advers reaksiyonların tanımıNötropeniNötropeni en sık bildirilen advers reaksiyon (%75,4) olup, faz III çalışmalarında ribosiklib ve herhangi bir kombinasyon kullanan hastaların %62'sinde nötrofil sayımlarında derece 3 veya4 azalma (laboratuvar bulgularına dayanarak) bildirilmiştir. Derece 2, 3 veya 4 nötropeni görülen hastalar arasında, bir olay yaşamış hastalar için başlangıca kadar geçen medyan süre 17 gündür. Derece >3 düzelmeye (normalizasyona veyaderece <3'e) kadar geçen medyan süre ribosiklib artı herhangi bir kombinasyon tedavisikollarında tedavide ara vermeyi ve/veya azaltmayı ve/veya tedavinin bırakılmasını takiben 12gündür. Febril nötropeni faz III çalışmalarında ribosiklibe maruz kalmış hastaların yaklaşık%1,7'sinde bildirilmiştir. Hastalara herhangi bir ateş durumunu derhal bildirmelerisöylenmelidir. Nötropeni, şiddet derecesine dayalı olarak laboratuvar izlemi, dozlara ara verme ve/veya doz modifikasyonu ile kontrol edilmiştir. Nötropeniye bağlı tedavinin bırakılması düşük orandagerçekleşmiştir (%0,8) (bkz. Bölüm 4.2 ve 4.4). Hepatobiliyer toksisiteFaz III klinik çalışmalarda, hepatobiliyer toksisite durumu ribosiklib artı herhangi bir kombinasyon kolundaki hastalarda, plasebo artı herhangi bir kombinasyon kolundaki hastalarakıyasla daha yüksek bir oranda görülmüş (sırasıyla, %27,3'e karşılık %19,6), ribosiklib artıherhangi bir kombinasyon ile tedavi edilen hastalar arasında daha fazla derece 3/4 advers olaybildirilmiştir (sırasıyla, %13,2'ye kıyasla %6,1). Transaminazlarda yükselme gözlemlenmiştir.Ribosiklib ve plasebo kollarında sırasıyla ALT (%11,2'ye karşılık %1,7) ve AST'de (%7,8'ekarşılık %2,1) derece 3 veya 4 artışlar bildirilmiştir. Kolestaz yokluğunda alkalen fosfataznormal iken ALT veya AST değerlerinde normalin üst sınırının üç katından büyük ve toplambilirubin değerinde normalin üst sınırının iki katından büyük eş zamanlı yükselmeler, 6hastada görülmüştür (Çalışma A2301'de [MONALEESA-2] 4 hasta; ribosiklib tedavisibırakıldıktan sonra düzeyleri 154 gün içinde normale dönmüştür ve Çalışma F2301'de[MONALEESA-3] 2 hasta; ribosiklib tedavisi bırakıldıktan sonra düzeyleri sırasıyla 121 ve532 gün içinde normale dönmüştür). Çalışma E2301'de (MONALEESA-7) bu gibi vakalarraporlanmamıştır. Hepatobiliyer toksisite olayları nedeniyle dozlara ara verme ve/veya doz ayarlamaları, ribosiklib artı herhangi bir kombinasyon ile tedavi edilen hastaların %12,3'ünde, ağırlıklıolarak ALT düzeyinde yükselme (%7,9) ve/veya AST düzeyinde yükselme (%7,3) nedeniylebildirilmiştir. Ribosiklib artı herhangi bir kombinasyon tedavisinin, anormal karaciğerfonksiyonu testleri veya hepatotoksisite nedeniyle kesilmesi, hastaların sırasıyla %2,4 ve%0,3'ünde söz konusu olmuştur (bkz. Bölüm 4.2 ve 4.4). Faz III çalışmalarda, derece 3 veya 4 ALT veya AST artışı olaylarının %70,9'u (90/127) tedavinin ilk 6 ayı içinde meydana gelmiştir. Derece 3 veya 4 ALT/AST artışı görülen hastalararasında başlangıca kadar geçen medyan süre ribosiklib artı herhangi bir kombinasyon koluiçin 92 gündür. Ribosiklib artı herhangi bir kombinasyon kolunda düzelmeye (normalizasyonaveya derece <2'ye) kadar geçen medyan süre 21 gündür. QT uzamasıÇalışma E2301'de (MONALEESA-7) çalışma başlangıcına göre gözlenen ortalama QTcF artışı, NSAI artı plasebo alt grubu ile karşılaştırıldığında tamoksifen artı plasebo alt grubundayaklaşık 10 milisaniye daha yüksek olarak tek başına tamoksifenin, ribosiklib artı tamoksifengrubunda gözlenen QTcF değerlerine katkıda bulunmuş olabilecek bir QTcF uzatma etkisininolduğunu düşündürmüştür. Plasebo kolunda, çalışma başlangıcına göre >60 milisaniyelikQTcF aralığı artışı tamoksifen alan 6/90 (%6,7) hastada görülürken, NSAI alan hiçbir hastadagörülmemiştir (bkz. Bölüm 5.2). Ribosiklib artı tamoksifen alan hastaların 14/87'sinde(%16,1) ve ribosiklib artı bir NSAI alan hastaların 18/245'inde (%7,3), çalışma başlangıcınakıyasla > 60 milisaniyelik bir QTcF aralığı artışı gözlenmiştir. Ribosiklibin tamoksifen ilekombinasyon halinde kullanımı önerilmemektedir (bkz. Bölüm 5.1). Faz III klinik çalışmalarda, ribosiklib artı aromataz inhibitörü veya fulvestrant kollarında hastaların %9,3'ü ve plasebo artı aromataz inhibitörü veya fulvestrant kollarında hastaların%3,5'i en az bir QT aralığı uzaması olayı deneyimlemiştir (EKG'de QT uzaması ve senkopdahil). EKG verilerinde inceleme, 15 hastanın (%1,4) >500 milisaniye başlangıç sonrası QTcFdeğerine sahip olduğunu ve 61 hastanın (%5,8) QTcF aralıklarında başlangıca göre >60milisaniye artış yaşadığını göstermiştir. Torsade de pointes vakaları bildirilmemiştir.Elektrokardiyogramda QT uzaması ve senkop nedeniyle dozlara ara verme/doz ayarlamaları,ribosiklib artı aromataz inhibitörü veya fulvestrant ile tedavi edilen hastaların %2,9'undabildirilmiştir. EKG verilerinin analizi, ribosiklib artı aromataz inhibitörü veya fulvestrant kollarında ve plasebo artı aromataz inhibitörü veya fulvestrant kollarında sırasıyla 55 hasta (%5,2) ve 12hastada (%1,5) en az bir >480 milisaniye başlangıç sonrası QTcF sonucu göstermiştir. >480milisaniye QTcF uzaması olan hastalar arasında olayın başlangıcına kadar geçen medyan sürekombinasyona bakılmaksızın 15 gün olmuş ve dozlara ara verildiğinde ve/veya dozazaltıldığında bu değişikliklerin geri dönüşlü olduğu görülmüştür (bkz. Bölüm 4.2, 4.4 ve 5.2). Böbrek yetmezliği olan hastalarÜç önemli çalışmada, hafif böbrek yetmezliği olan 341 hasta ve orta derecede böbrek yetmezliği olan 97 hasta ribosiklib ile tedavi edilmiştir. Şiddetli böbrek yetmezliği olanhastalar çalışmaya dahil edilmemiştir (bkz. Bölüm 5.1). Tedavi sırasında başlangıçtaki böbrekyetmezliğinin derecesi ile kan kreatinin değerleri arasında bir korelasyon bulunmaktadır. Hafifveya orta derecede böbrek yetmezliği olan hastalarda QT uzaması ve trombositopenioranlarında hafif artış gözlenmiştir. Bu toksisiteler durumunda izleme ve doz ayarlamaönerileri için Bölüm 4.2 ve 4.4 bakınız. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr[email protected]: tel: 0 800 314 00 08; faks: 0 312 218 35 99)4.9 Doz aşımı ve tedavisiRaporlanmış VALAMOR ile doz aşımı vakalarına ait sınırlı deneyim bulunmaktadır. Bir doz aşımı olayında, bulantı ve kusma gibi semptomlar meydana gelebilir. Buna ilaveten,hematolojik (örn., nötropeni, trombositopeni) toksisite ve olası QTc uzaması meydanagelebilir. Tüm doz aşımı durumlarında gerekli genel semptomatik ve destekleyici tedavibaşlatılmalıdır. 5. FARMAKOLOJIK ÖZELLIKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grubu: Antineoplastik ve immünomodülatör ajanlar, protein kinaz inhibitörleri, sikline bağımlı kinaz (CDK) inhibitörleri ATC kodu: L01EF02 Etki mekanizması:Ribosiklib seçici sikline bağımlı kinaz (CDK) 4 ve 6 inhibitörü olup, biyokimyasal analizlerde sırasıyla 0,01 (4,3 ng/ml) ve 0,039 ^M'lik (16,9 ng/ml) %50 inhibisyon (IC50) değerlerinisağlamıştır. Bu kinazlar D-sikline bağlanma ile birlikte aktive olurlar ve hücre döngüsüprogresyonu ve hücresel proliferasyona yol alan sinyal yolaklarında önemli bir rol oynarlar.Siklin D-CDK4/6 kompleksi hücre döngüsü progresyonunu, retinoblastoma proteininin (pRb)fosforilasyonu yoluyla regüle eder. İn vitroortamda ribosiklib pRb fosforilasyonunu azaltarak hücre döngüsünün G1 fazında durmaya ve meme kanseri hücre dizilerinde hücre proliferasyonunda azalmaya yol açmıştır. İnvivo ortamda tek ajan ribosiklib ile tedavi, iyi tolere edilen dozlarda pRb fosforilasyonununinhibisyonu ile korelasyon gösteren tümör regresyonuna yol açmıştır.Hastadan elde edilmiş östrojen reseptör-pozitif meme kanseri ksenogreft modelinin kullanıldığı in vivo çalışmalarda, ribosiklib ve antiöstrojenlerin (örn. letrozol) kombinasyonu,ajanların tek başına uygulandığı durumla karşılaştırıldığında, kalıcı tümör regresyonu ve dozuygulaması bırakıldıktan sonra yeni tümör büyümesinde gecikme ile birlikte daha iyi birtümör büyümesi inhibisyonu sağlamıştır. Ek olarak, ZR751 ER+ insan meme kanseriksenogreftlerini taşıyan ve bağışıklık yetmezliği bulunan farelerde fulvestrant ilekombinasyon halindeki ribosiklibin in vivo anti-tümör aktivitesi değerlendirilmiştir vefulvestrant ile kombinasyon, tam tümör büyümesi inhibisyonu ile sonuçlanmıştır. Bilinen ER durumuna sahip meme kanseri hücre hatları paneli test edildiğinde, ribosiklibin ER- olanlara kıyasla ER+ meme kanseri hücre hatlarında daha etkili olduğu gösterilmiştir.Şimdiye kadar test edilen klinik öncesi modellerde, ribosiklib aktivitesi için intakt pRbgerekmiştir. Kardiyak elektrofızyoloH:İleri evre kanser hastalarında ribosiklibin QTc aralığı üzerindeki etkisini değerlendirmek için tek dozdan sonra ve kararlı durumda seri, üçlü EKG'ler kaydedilmiştir. Bir farmakokinetik-farmakodinamik analizde, 50-1200 mg aralığında dozlarla ribosiklib ile tedavi edilen 997hasta yer almıştır. Bu analiz, ribosiklibin QTc aralığında konsantrasyona bağımlı artışlaraneden olduğunu düşündürmüştür. NSAI veya fulvestrant ile kombinasyon halindeki 600 mgribosiklib için başlangıça göre tahmini ortalama QTcF değişimi, tamoksifen ile kombinasyonhalinde 34,7 milisaniye (%90 GA: 31,64; 37,78) ile karşılaştırıldığında, sırasıyla kararlıdurumdaki geometrik ortalamada Cmaks'ta sırasıyla 22 milisaniye (%90 GA: 20,56; 23,44) ve23,7 milisaniye (%90 GA: 22,31; 25,08) olmuştur (bkz. Bölüm 4.4). Klinik etkililik ve güvenlilik: Çalışma CLEE011A2301 (MONALEESA-2)Hormon reseptörü pozitif, HER2 negatif ileri evre meme kanseri olan, ileri evre hastalık için daha önce bir tedavi almamış postmenopozal kadınlarda tek başına letrozole kıyaslaribosiklibin letrozol ile kombine tedavisi randomize, çift kör, plasebo kontrollü, çok merkezlibir faz III klinik çalışmada değerlendirilmiştir. Toplam 668 hasta, karaciğer ve/veya akciğer metastazlarının varlığına (Var [n=292 (44%)]) kıyasla Yok [n=376 (56%)]) göre katmanlandırılarak 1:1 oranında ribosiklib 600 mg veletrozol (n=334) veya plasebo ve letrozol (n=334) almak üzere randomize edilmiştir.Demografik özellikler ve başlangıç hastalık karakteristikleri çalışma kolları arasında dengelive karşılaştırılabilir nitelikte olmuştur. Ribosiklib, 28 gün süreyle günde bir kez 2,5 mgletrozol ile kombinasyon halinde oral yolla günde bir kez 600 mg dozunda arka arkaya 21 günverilmiş, ardından 7 gün tedavisiz ara bırakılmıştır. Çalışma süresince veya hastalıkprogresyonundan sonra hastaların plasebodan ribosiklibe geçiş yapmalarına izin verilmemiştir. Bu çalışmaya alınan hastaların medyan yaşı 62'dir (aralık 23 - 91). Hastaların %44,2'si 65 yaşın üzerindedir ve bu hastalarında 69'u 75 yaşın üzerindedir. Dahil edilen hastalar beyaz(%82,2), Asyalı (%7,6) ve siyahtır (%2,5). Tüm hastaların ECOG performans durumu 0 veya1'dir. Ribosiklib kolunda, hastaların %46,6'sı çalışmaya girmeden önce neoadjuvan veyaadjuvan koşullarda kemoterapi görmüştür ve %51,3'ü neoadjuvan veya adjuvan koşullardaanti-hormonal tedavi almıştır. Hastaların %34,1'i de novohastadır. Hastaların %22'sindesadece kemiği tutan hastalık ve %58,8'inde visseral hastalık vardır. Daha önce anastrozolveya letrozol ile (neo)adjuvan tedavi görmüş hastalar çalışma randomizasyonundan en az 12ay önce bu tedaviyi tamamlamış olmalıdır.Birincil analizÇalışma için birincil sonlanım noktası tüm popülasyonda (tüm randomize hastalar) araştırıcı değerlendirmesi temelinde Solid Tümörlerde Yanıt Değerlendirme Kriterleri (RECIST v1.1)kullanılarak hedeflenen progresyonsuz sağkalım (PFS) olaylarının %80'i gözlendikten sonrayürütülmüş planlanan ara analizde karşılanmış ve körlenmiş bağımsız merkezi radyolojikdeğerlendirme ile doğrulanmıştır. Tam analiz setinde etkililik sonuçları, plasebo artı letrozol alan hastalara kıyasla, ribosiklib artı letrozol kombinasyonu ile tedavi edilen hastalar için klinik olarak anlamlı tedavi etkisi ilePFS'de istatistiksel olarak anlamlı düzelme göstermiştir (tehlike oranı [TO] = 0,556, %95 GA:0,429; 0,720, tek yönlü stratifiye edilmiş log sıra testi p değeri 0,00000329). Global sağlık durumu/QoL verileri ribosiklib artı letrozol kolu ve plasebo artı letrozol kolu arasında ilgili bir fark olmadığını göstermiştir. Tablo 8 ve 9'da etkililik verilerine ilişkin daha olgun bir güncelleme (02 Ocak 2017 veri kesme) sunulmaktadır. Medyan PFS ribosiklib artı letrozol ile tedavi edilen hastalar için 25,3 ay (%95 GA: 23; 30,3) iken, plasebo artı letrozol alan hastalar için 16 (%95 GA: 13,4; 18,2) aydır. Ribosiklib artıletrozol alan hastaların %54,7'sinin, plasebo artı letrozol alan hastaların ise %35,9'unun 24.ayda progresyonsuz olduğu öngörüldü. Tablo 8 MONALEESA-2 - Araştırmacının radyolojik değerlendirmesine dayalı etkililik sonuçları (PFS) (02 Ocak 2017 veri kesme)
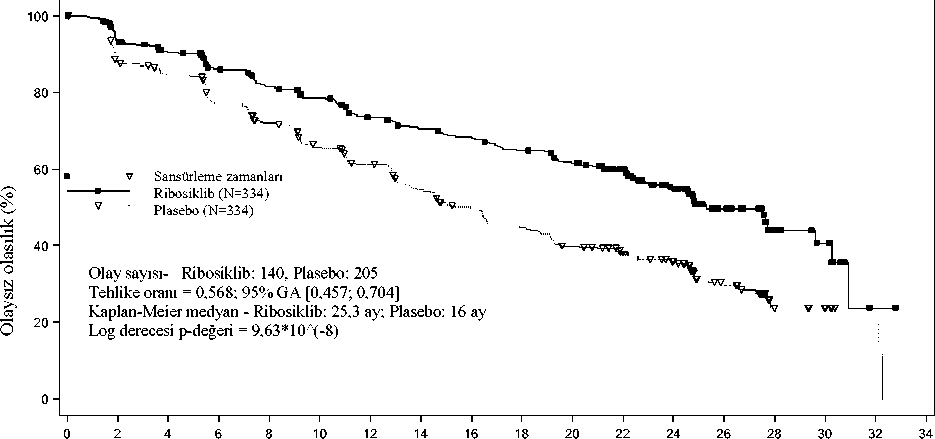
Şekil 1 MONALEESA-2 - Araştırmacının değerlendirmesine dayalı PFS'nin Kaplan-Meier grafiği (02 Ocak 2017 veri kesme)
Halen risk altında olan hasta sayısı Zaman 02468101214161820Ribosiklib 334294277257240227207196188176164Plasebo 33427926523921919617915613812411028171030117Önceden belirlenmiş alt grup PFS analizleri serisi prognostik faktörler ve başlangıç özellikleri temelinde tedavi etkisinin tutarlılığını araştırmak üzere yürütülmüştür. Yaş, ırk, öncekiadjuvan veya neo adjuvan kemoterapi veya hormonal tedaviler, karaciğer ve/veya akciğertutulumu ve sadece kemiğe metastaz yapan hastalığı içeren tüm bireysel hasta alt gruplarındahastalık progresyonu veya ölüm riskinde ribosiklib artı letrozol kolu lehine bir azalmagözlenmiştir. Bu, karaciğer ve/veya akciğer metastazları olan hastalar için (TO 0,561 [%95GA: 0,424; 0,743], medyan progresyonsuz sağkalım [mPFS] ribosiklib artı letrozol için 24,8ay ve tek başına letrozol için 13,4 ay) ya da karaciğer ve/veya akciğer metastazları olmayanhastalar için (TO 0,597 [%95 GA: 0,426; 0,837], mPFS 27,6 aya karşılık 18,2 ay) belirgindir. Genel yanıt ve klinik fayda oranları için güncellenmiş bulgular Tablo 9'da gösterilmektedir. Tablo 9 MONALEESA-2 - Araştırmacı değerlendirmesine dayalı etkililik sonuçları (ORR, CBR) (02 Ocak 2017 veri kesme)
Nihai genel saşkalım analiziGenel çalışma popülasyonunda bu nihai genel sağkalım analizinden bulgular Tablo 10 ve Şekil 2'de sunulmaktadır.Tablo 10 MONALEESA-2- Etkililik bulguları (OS) (10 Haziran 2021 veri kesme)
Şekil 2 MONALEESA-2- Genel popülasyonda OS'nin Kaplan-Meier grafiği (10 Haziran 2021 veri kesme)
Log-sıra testiPH modeli IRT'ye göre karaciğer ve/veya akciğer metastazları durumuna göre sınıflandırılır.Tek taraflı P-değeri sınıflandırılmış log-sıra testinden elde edilir.Çalışma CLEE011E2301 (MONALEESA-7)Ribosiklib, randomize çift kör, plasebo kontrollü, çok merkezli bir faz III klinik çalışmada, hormon reseptörü pozitif, HER2-negatif ilerlemiş meme kanseri olan premenopozal veperimenopozal kadınların tedavisinde, NSAI veya tamoksifen artı goserelin ile kombinasyonhalinde, bir NSAI veya tamoksifen artı goserelin ile kombinasyon halinde plasebo karşısındadeğerlendirilmiştir. MONALEESA-7'deki hastalar ilerlemiş meme kanseri için öncedenendokrin tedavisi görmemişlerdir. Toplam 672 hasta, karaciğer ve/veya akciğer metastazlarının varlığına (Var [n=344 (%51,2)] karşısında Yok [n=328 (%48,8)]), ilerlemiş hastalık için önceki kemoterapi (Var [n=120(%17,9)] karşısında Yok [n=552 (%82,1)]) ve endokrin kombinasyon partneri (NSAI vegoserelin [n=493 (%73,4)] karşısında tamoksifen ve goserelin [n=179 (%26,6)]) görekatmanlandırılarak 1:1 oranında ya ribosiklib 600 mg artı NSAI/tamoksifen artı goserelin(n=335) ya da plasebo artı NSAI/tamoksifen artı goserelin (n=337) almak üzere randomizeedilmiştir. Demografik özellikler ve çalışma başlangıcı hastalık karakteristikleri çalışmakolları arasında dengeli ve birbirine yakın olmuştur. Ribosiklib, 28 gün süreyle günde bir kezoral yolla NSAI (letrozol 2,5 mg veya anastrozol 1 mg) veya tamoksifen (20 mg) ve 28 gündebir subkutan yolla goserelin (3,6 mg) ile kombinasyon halinde, hastalık progresyonuna veyakabul edilemez toksisiteye kadar oral yolla günde bir kez 600 mg dozunda arka arkaya 21 günverilmiş, ardından 7 gün tedavisiz ara bırakılmıştır. Çalışma sırasında veya hastalıkprogresyonundan sonra hastaların plasebodan ribosiklibe geçiş yapmalarına izin verilmemiştir.Endokrin kombinasyon partnerlerinin değiştirilmesine de izin verilmemiştir. Bu çalışmaya kaydedilen hastaların medyan yaşı 44 yıldır (aralık: 25 ila 58) ve hastaların %27,7'si 40 yaşın altındadır. Dahil edilen hastaların çoğu Beyaz (%57,7), Asyalı (%29,5)veya Siyahtır (%2,8) ve neredeyse tüm hastaların (%99) başlangıç ECOG performans durumu0 veya 1'dir. Çalışmaya giriş öncesinde bu 672 hastanın %14'ü önceden metastatik hastalıkiçin kemoterapi görmüş, %32,6'sı adjuvan ve %18'i neoadjuvan koşullarda kemoterapi almış,ve %39,6'sı adjuvan ve %0,7'si neoadjuvan koşullarda endokrin tedavisi almıştır. ÇalışmaE2301'de hastaların %40,2'sinde de novo metastatik hastalık, %23,7'sinde sadece kemiğitutan hastalık ve %56,7'sinde visseral hastalık söz konusudur. Çalışma, tam analiz setinde (randomize edilen tüm hastalar) araştırmacı değerlendirmesine dayanan ve RECIST v1.1 kriterleri kullanılarak 318 progresyonsuz sağkalım (PFS) olayındansonra gerçekleştirilen birincil analizde birincil sonlanım noktasına ulaşmıştır. Birincil etkililiksonuçları, körlenmiş bağımsız merkezi radyolojik değerlendirmeye dayalı PFS sonuçlarıyladesteklenmiştir. Birincil PFS analizi tarihinde medyan takip süresi 19,2 ay olmuştur. Genel çalışma popülasyonunda etkililik sonuçları, plasebo artı NSAI/tamoksifen artı goserelin kullanan hastalar ile karşılaştırıldığında ribosiklib artı NSAI/tamoksifen artı goserelinkullanan hastalarda, klinik olarak anlamlı bir tedavi etkisiyle birlikte PFS'de istatistikselolarak anlamlı bir düzelme göstermiştir (Tehlike Oranı: 0,553, %95 GA: 0,441; 0,694, tekyanlı katmanlandırılmış log sıra testi p değeri 9,83x10-8). Medyan PFS, ribosiklib artıNSAI/tamoksifen artı goserelin ile tedavi edilen hastalarda 23,8 ay (%95 GA: 19,2; TE) veplasebo artı NSAI/tamoksifen artı goserelin verilen hastalarda 13 ay (%95 GA: 11, 16,4)olmuştur. PFS için dağılımı, Şekil 3'te, PFS için Kaplan-Meier eğrisinde özetlenmektedir. PFS'nin araştırmacıŞekil 3 MONALEESA-7 - Genel popülasyonda değerlendirmesine dayalı Kaplan-Meier grafiği
100-Log rank p-değeri = 9,83*10^(-8)30252624221620
1Ö
12
Halen risk altında olan hasta sayısı Zaman (Ay) ö Ribosiklib 335Plasebo 337
2Ö 54313000281126332420132240241221916513906282452071Ö23518323012734204248526423t>Randomize edilen hastaların yaklaşık %40'ından rastgele seçilmiş bir alt grubun körlenmiş bağımsız merkezi radyolojik değerlendirmesine dayalı PFS sonuçları, araştırmacıdeğerlendirmesine dayalı birincil etkililik sonuçlarını destekler nitelikte olmuştur (TehlikeOranı: 0,427; %95 GA: 0,288; 0,633). Birincil PFS analizi tarihinde genel sağkalım verileri hazır olmayıp 89 (%13) ölüm (Tehlike Oranı: 0,916 [%95 GA: 0,601; 1,396]) söz konusu olmuştur. RECIST v1.1'e göre araştırmacı değerlendirmesine dayalı genel yanıt oranı (ORR) ribosiklib kolunda (%40,9; %95 GA: 35,6; 46,2), plasebo kolundan daha yüksek bulunmuştur (%29,7; %95 GA: 24,8; 34,6, p=0,00098). Gözlenen klinik fayda oranı (CBR), plasebo kolu (%69,7; %95 GA: 64,8; 74,6, p=0,002) ile karşılaştırıldığında ribosiklib kolunda (%79,1; %95 GA:74,8; 83,5) daha yüksektir. NSAI artı goserelin ile kombinasyon halinde ribosiklib veya plasebo alan 495 hastanın önceden tanımlanmış alt grup analizinde, medyan PFS ribosiklib artı NSAI alt grubunda 27,5ay (%95 GA: 19,1; TE) ve plasebo artı NSAI alt grubunda 13,8 ay (%95 GA: 12,6; 17,4)bulunmuştur [Tehlike Oranı: 0,569; %95 GA: 0,436; 0,743]. Etkililik sonuçları Tablo 11'deözetlenmekte ve PFS için Kaplan-Meier eğrileri Şekil 4'te verilmektedir. Tablo 11 MONALEESA-7 - NSAI alan hastalarda etkililik sonuçları (PFS)
Şekil 4 MONALEESA-7 - NSAI alan hastalarda değerlendirmesine dayalı Kaplan-Meier grafiği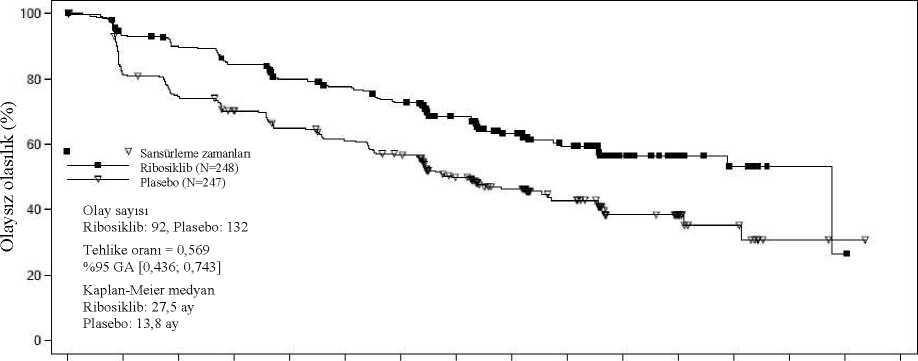
Tablo 12 MONALEESA-7 - NSAI alan hastalarda araştırmacı değerlendirmesine dayalı etkililik sonuçları (ORR, CBR)
Ribosiklib artı NSAI alt grubundaki sonuçlar yaş, ırk, önceki adjuvan/ neoadjuvan kemoterapi veya hormonal tedaviler, karaciğer ve/veya akciğer tutulumu ve sadece kemiği tutanmetastatik hastalığı içeren farklı alt gruplar genelinde tutarlı olmuştur. Genel sağkalım verilerinin daha iyi bir güncellemesi (veri kesme tarihi 30 Kasım 2018) Tablo 13 ve Şekil 5 ve 6'da verilmektedir. İkinci OS analizinde çalışma, OS'de istatistiksel olarak anlamlı bir iyileşme göstererek anahtar ikincil sonlanım noktasını karşılamıştır. Tablo 13 MONALEESA-7 - Etkililik sonuçları (OS) (veri kesme tarihi 30 Kasım 2018)
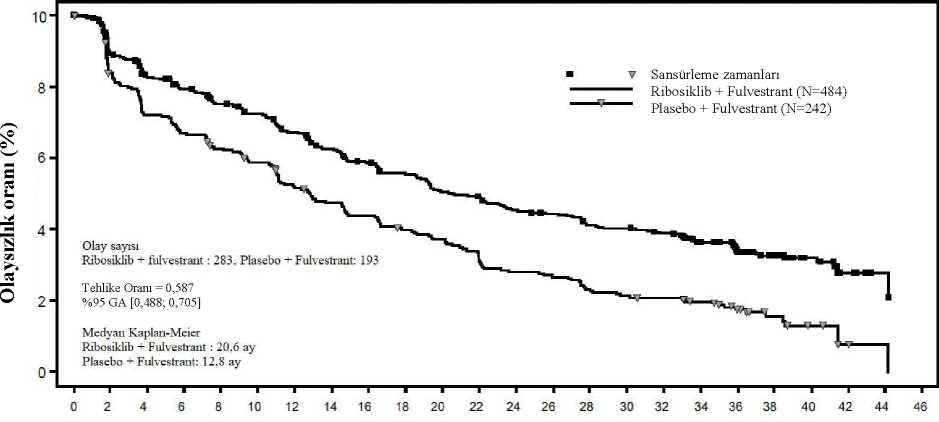
Şekil 5 MONALEESA-7- Son OS analizinin Kaplan Meier grafiği (veri kesme tarihi 30 Kasım 2018)1(W V Sansürleme zamanlan Ribosiklib (N=335)Plasebo (N=337)80 Olay sayısıRibosiklib: 83, Plasebo: 109O M X/;JS Tehlike Oranı = 0,712%95 GA [0,535; 0,948]Kaplan-Meier medyan Ribosiklib: TEPlasebo: 40,9 ayLog sıra p değeri = 0,00973
20
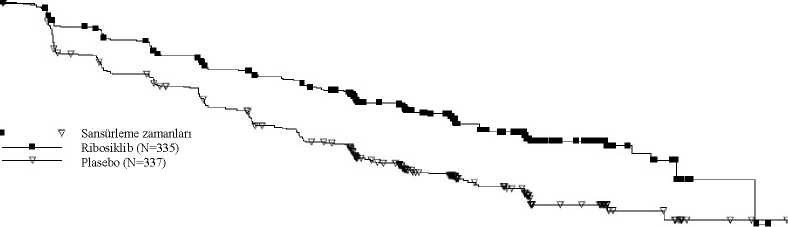
Log-sıra testi ve Cox modeli akciğer ve/veya karaciğer metastazı, ileri evre hastalık için önceki kemoterapi ve IRT başına endokrin kombinasyon partneri ile stratifiye edilmiştir. Şekil 6 MONALEESA-7- NSAI alan hastalarda son OS analizinin Kaplan Meier grafiği (veri kesme tarihi 30 Kasım 2018)¦¦¦¦Ç* V w Vly" *Sansürleme zamanları Ribosiklib (N=248)Plasebo (N=247) M OUOlay sayısıRibosiklib: 61, Plasebo: 80
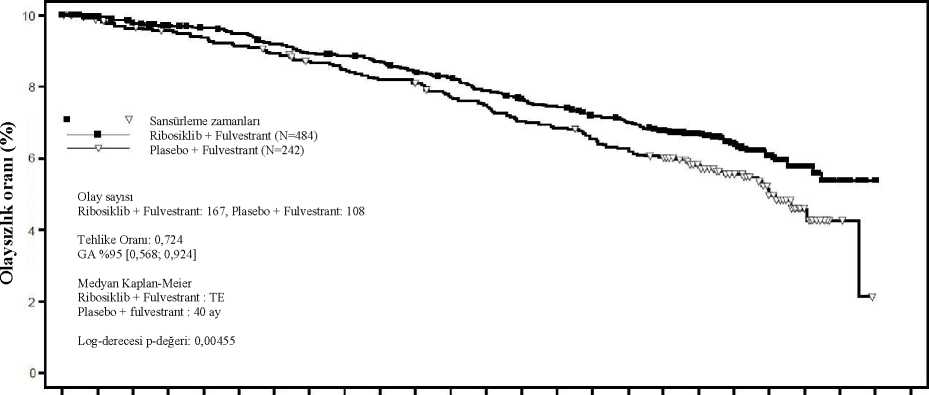
O Tehlike Oranı = 0,699 %95 GA [0,501; 0,976]Kaplan-Meier medyan Ribosiklib: TEPlasebo: 40,7 ayTV V VWAO '20
Tehlike oranı katmanlandırılmamış Cox modeline dayanmaktadır. Ek olarak, genel çalışma popülasyonundaki 0,692 TO değeri (%95 GA: 0,548; 0,875) ile bu çalışmada daha önce ribosiklib alan hastalarda sonraki basamak tedavide progresyon veyaölüm (PFS2) olasılığı, plasebo kolundaki hastalara göre daha düşük olmuştur. Medyan PFS2,plasebo kolunda 32,3 ay bulunmuştur (%95 GA: 27,6; 38,3) ve ribosiklib kolundaulaşılmamıştır (%95 GA: 39,4; TE). NSAI alt grubu için benzer sonuçlar gözlenmiş, TOdeğeri 0,660 (%95 GA: 0,503; 0,868) ile medyan PFS2 plasebo kolunda 32,3 ay olurken (%95GA: 26,9; 38,3), ribosiklib kolunda ulaşılmamıştır (%95 GA: 39,4; TE). Çalışma CLEE011F2301 (MONALEESA-3)Ribosiklib, 2:1 randomize çift kör, plasebo kontrollü, çok merkezli bir faz III klinik çalışmada, önceden endokrin tedavi almamış ya da sadece bir basamak endokrin tedavi almış hormonreseptörü pozitif, HER2-negatif ileri evre meme kanseri olan 726 postmenopozal kadınıntedavisinde, tek başına fulvestrant karşısında fulvestrant ile kombinasyon halindedeğerlendirilmiştir. Bu çalışmaya kaydedilen hastaların medyan yaşı 63'tür (aralık 31 - 89). Hastaların %46,7'si 65 yaş ve üzeri olup bunlar arasında 75 yaş ve üzeri hasta oranı %13,8'dir. Dahil edilenhastalar beyaz (%85,3), Asyalı (%8,7) veya siyahtır (%0,7) ve neredeyse tüm hastaların(%99,7) ECOG performans durumu 0 veya 1'dir. Bu çalışmaya birinci ve ikinci basamakhastalar kaydedilmiştir (hastaların %19,1'inde de novometastatik hastalık söz konusudur).Çalışmaya giriş öncesinde hastaların %42,7'si adjuvan ve %13,1'i neoadjuvan koşullardakemoterapi almış, %58,5'i adjuvan ve %1,4'ü neoadjuvan koşullarda endokrin tedavisi almış,%21'i ise ilerlemiş meme kanseri koşullarında önceden endokrin tedavisi görmüştür. ÇalışmaF2301'de %21,2'sinde sadece kemiği tutan hastalık ve %60,5'inde visseral hastalık sözkonusudur.Birincil analizÇalışma, tam analiz setinde (randomize edilen tüm hastalar, veri kesme tarihi: 3 Kasım 2017) araştırmacı değerlendirmesine dayanan ve RECIST v1.1 kriterleri kullanılarak 361progresyonsuz sağkalım (PFS) olayından sonra gerçekleştirilen birincil analizde birincilsonlanım noktasına ulaşmıştır. Birincil PFS analizi tarihinde medyan takip süresi 20,4 ayolmuştur. Birincil etkililik sonuçları, tam analiz setinde plasebo artı fulvestrant alan hastalar ile karşılaştırıldığında ribosiklib artı fulvestrant alan hastalarda, progresyon veya ölüm bağılriskinde ribosiklib artı fulvestrant kolu lehine tahmini %41'lik azalmayla, PFS'de istatistikselolarak anlamlı bir düzelme göstermiştir (tehlike oranı: 0,593, %95 GA: 0,480; 0,732, tek yanlıkatmanlandırılmış log sıra testi p değeri 4,1x10-7). Birincil etkililik bulguları körlenmiş bir bağımsız merkezi radyolojik değerlendirme ile %40 görüntüleme alt setinin rastgele merkezi denetimi ile desteklenmiştir (tehlike oranı 0,492; %95GA: 0,345; 0,703). İkinci OS ara analizi zamanında PFS için tanımlayıcı bir güncelleme yürütülmüş olup, genel popülasyon ve önceki endokrin tedaviye göre alt gruplarda güncellenmiş PFS bulguları Tablo14'te özetlenmektedir ve Kaplan-Meier eğrisi Şekil 7'de sunulmaktadır.
Şekil 7 MONALEESA-3 (F2301)- PFS için Araştırmacı değerlendirmesine dayalı Kaplan-Meier grafiği (Veri kesme tarihi: 3 Haziran 2019)
Genel yanıt oranı (ORR) ve RECIST v1.1'e dayalı araştırmacı değerlendirmesine göre klinik fayda oranı (CBR) için etkililik sonuçları Tablo 15'te verilmektedir. Tablo 15 MONALEESA-3 - Araştırmacı değerlendirmesine dayalı etkililik sonuçları (ORR, CBR) (3 Kasım 2017 veri kesme)
Ribosiklib artı fulvestrant ile tedavi edilen hastaların önceden tanımlanmış alt grup analizine dayalı tehlike oranları, yaş, önceki tedavi (erken veya ilerlemiş), önceki adjuvan/neoadjuvankemoterapi veya hormonal tedaviler, karaciğer ve/veya akciğer tutulumu ve sadece kemiğitutan metastatik hastalığı içeren farklı alt gruplar genelinde tutarlı fayda göstermiştir. Genel Saşkalım Analiziİkinci genel sağkalım analizinde, çalışma ikincil sonlanım noktasını sağlayarak, genel sağkalımda istatistiksel olarak anlamlı bir iyileşme göstermiştir. Genel çalışma popülasyonunda ve alt gruplar analizinde bu nihai genel sağkalım analizinden elde edilen bulgular Tablo 16 ve Şekil 8'de sunulmaktadır. Tablo 16 MONALEESA-3 (F2301) - Etkililik bulguları (OS) (Veri kesme tarihi: 03-
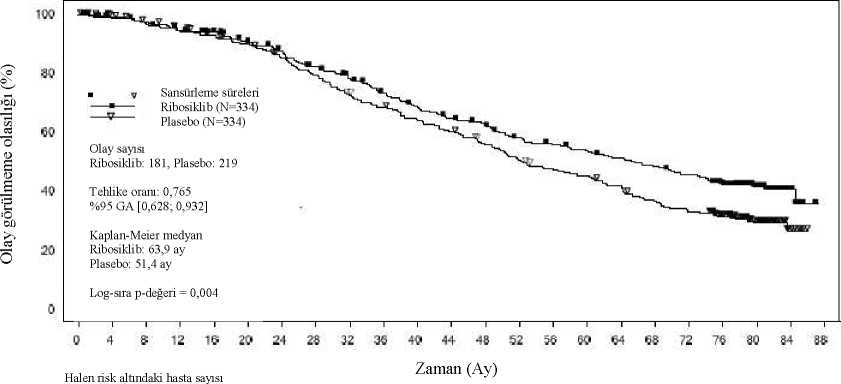
b Tek taraflı P-değeri IRT'ye göre önceki endokrin tedavisi, akciğer ve/veya karaciğer metastazına göre tabakalandırılmış log-sıra testinden elde edilir. P-değeri tek taraflıdırve 0,025'lik bir genel anlam düzeyi için Lan-DeMets (O'Brien-Fleming) alfa-harcamafonksiyonu ile tayin edildiği üzere 0,01129'luk bir eşiğe karşı kıyaslanır. c Tehlike oranı tabakalandırılmamış Cox PH modelinden elde edilir._Şekil 8 MONALEESA-3 (F2301) - OS Kaplan-Meier grafiği (tam analiz seti [FAS]) (Veri kesme tarihi: 3 Haziran 2019)
Log-sıra testimodeli akciğer ve/veya karaciğer metastazları, ileri hastalık için önceki kemoterapi ve IRT'ye göre endokrin kombinasyonu eşine göre tabakalandırılır.Ribosiklib kolundaki hastalarda bir sonraki basamak tedavide progresyona veya ölüme (PFS2) kadar geçen süre, genel çalışma popülasyonunda plasebo kolundaki hastalara kıyasla dahauzundur (TO: 0,670 [%95 GA: 0,542; 0,830]). Medyan PFS2, ribosiklib kolu için 39,8 ay(%95 GA: 32,5; TE), plasebo kolunda 29,4 aydır (%95 GA: 24,1; 33,1). Yaşlı hastalarMONALEESA-2 ve MONALEESA-3 çalışmalarında ribosiklib alan tüm hastaların temsili oranları >65 yaş ve >75 yaş arasındadır (bkz. Bölüm 5.1). Bu hastalar ile daha genç hastalararasında ribosiklibin güvenliliği ve etkililiği bakımından genel farklılıklar gözlenmemiştir(bkz. Bölüm 4.2). Böbrek yetmezliği olan hastalarÜç pivot çalışmada (MONALEESA-2, MONALEESA-3 ve MONALEESA-7), normal böbrek fonksiyonu olan 510 (% 53,8) hasta, hafif böbrek yetmezliği olan 341 (%36) hasta veorta derecede böbrek yetmezliği olan 97 (% 10,2) hasta ribosiklib ile tedavi edilmiştir. Şiddetliböbrek yetmezliği olan hastalar, çalışmalara dahil edilmemiştir. Normal böbrek fonksiyonuolanlara kıyasla, 600 mg başlangıç dozunda ribosiklib alan hafif ve orta derecede böbrekyetmezliği olan hastalarda progresyonsuz sağkalım sonuçları tutarlıdır. Güvenlilik profili,renal kohortlar arasında genel olarak tutarlıdır (bkz. Bölüm 4.8). 5.2 Farmakokinetik özelliklerGenel özelliklerRibosiklibin farmakokinetiği, ilerlemiş meme kanseri olan hastalarda 50 mg ila 1200 mg'lık günlük oral dozlarından sonra incelenmiştir. Sağlıklı gönüllülere 400 mg ile 600 mg arasındatek oral dozlar veya tekrarlı 400 mg dozları (8 gün) verilmiştir. Emilim:Ribosiklibin mutlak biyoyararlanımı bilinmemektedir. Ribosiklib oral kullanımın ardından Cmaks'a ulaşma zamanı (Tmaks) 1-4 saat arasında olmuştur. Ribosiklib, test edilen doz aralığında (50 - 1200 mg) maruziyette (Cmaks ve EAA) oransaldeğerin biraz üzerinde artışlar sergilemiştir. Tekrarlanan günlük tek doz uygulamalarısonrasında kararlı durumuma genellikle 8 gün sonra ulaşılmıştır ve ribosiklib, 2,51 geometrikortalama birikim oranı (aralık: 0,97-6,4) ile birikmiştir. Besin etkisiAçlık durumu ile karşılaştırıldığında, 600 mg tek doz ribosiklib film kaplı tabletin yüksek oranda yağ içeren yüksek kalorili bir öğün ile birlikte oral yolla uygulanmasının, ribosiklibinemilim hızı ve oranı üzerinde herhangi bir etkisi olmamıştır. Dağılım:Ribosiklibin insan plazma proteinlerine bağlanması in vitroortamda yaklaşık %70 bulunmuştur ve konsantrasyondan bağımsız olmuştur (10 - 10000 ng/ml). Ribosiklib kırmızıkan hücreleri ile plazma arasında eşit dağılmış, in vivo ortamda kan-plazma oranı 1,04bulunmuştur. Popülasyon farmakokinetik analizine dayanılarak kararlı durumda görünürdağılım hacmi (Vss/F) 1090 litredir.Biyotransformasyon:In vitrove in vivo çalışmalar ribosiklibin insanda ağırlıklı olarak CYP3A4 aracılığıyla olmak üzere başlıca hepatik metabolizma ile eliminasyona uğradığını göstermiştir. [14C] ribosiklibin600 mg'lık tek dozunun insanlara oral uygulanmasından sonra ribosiklib için ana metabolikyolaklar, oksidasyonu (dealkilasyon, C ve/veya N-oksijenasyon, oksidasyon (-2H)) vebunların kombinasyonlarını içermiştir. Ribosiklib faz 1 metabolitlerinin faz II konjügatları N-asetilasyon, sülfasyon, sistein konjügasyonu, glikozilasyon ve glukuronidasyonu içermiştir.Ribosiklib, plazmadaki başlıca dolaşan ilaç kaynaklı öğedir. Dolaşımdaki başlıca metabolitler,metabolit M13 (CCI284, N-hidroksilasyon), M4 (LEQ803, N-demetilasyon) ve M1'i (ikincilglukuronid) içermiştir. Ribosiklibin klinik aktivitesi (farmakolojik ve güvenlilik) başta anailaç kaynaklı olmuş, dolaşımdaki metabolitlerin göz ardı edilebilir katkısı olduğu görülmüştür.Ribosiklib yaygın olarak metabolize olmuş olup, değişmemiş ilaç feçes ve idrarda sırasıyla dozun %17,3 ve %12,1'ine karşılık gelmiştir. Metabolit LEQ803 dışkıda önemli bir metabolitolmuş ve feçes ve idrarda sırasıyla uygulanan dozun yaklaşık %13,9 ve %3,74'ünü temsiletmiştir. Gerek feçes gerekse idrarda çok sayıda başka metabolit de minör miktarlarda tespitedilmiştir (uygulanan dozun < %2,78'i). Eliminasyon:İlerlemiş kanseri olan hastalarda 600 mg'da kararlı durumda geometrik ortalama plazma efektif yarılanma ömrü (birikim oranına dayalı) 32 saat (%63 CV) ve geometrik ortalamagörünür oral klirens (CL/F) 25,5 l/saat (%66 CV) bulunmuştur. Sağlıklı gönüllülerdekiçalışmalarda 600 mg dozda ribosiklibin geometrik ortalama görünür plazma terminalyarılanma ömrü (T1/2) 29,7 ile 54,7 saat, ribosiklibin geometrik ortalama CL/F değeri ise 39,9ila 77,5 l/saat aralığında olmuştur. Ribosiklib ve metabolitleri renal yolağın küçük bir katkısı ile ağırlıklı olarak feçes ile elimine olur. 6 sağlıklı erkek gönüllüde, tek bir oral [14C] ribosiklib dozunun uygulanmasından sonrauygulanan toplam radyoaktivitenin %91,7'si 22 gün içinde belirlenmiştir; feçes ana atılımyolu olup (%69,1) dozun %22,6'sı idrarda elde edilmiştir. Doğrusallık/doğrusal olmayan durum:Ribosiklib, test edilen 50 ila 1200 mg doz aralığında hem tek hem de tekrarlı dozlardan sonra maruziyette (Cmaks ve EAA) oransal değerin biraz üzerinde artışlar sergilemiştir. Bu analiz dozgruplarının çoğu için küçük örneklem büyüklükleri ile sınırlıdır ve verilerin çoğu 600 mg dozgrubundan gelmektedir. Hastalardaki karakteristik özelliklerÖzel popülasyonlar Böbrek bozukluğuBöbrek fonksiyonunun ribosiklib farmakokinetiği üzerindeki etkisi 400 mg'lık tek bir ribosiklib dozunda, normal böbrek fonksiyonuna sahip (mutlak GFR [aGFR] >90ml/dakika)14 sağlıklı gönüllü ile hafif böbrek yetmezliği (aGFR 60 ila <90 ml/dakika) olan 8,orta derece böbrek yetmezliği olan (aGFR 30 ile <60 ml/dakika) 6, şiddetli böbrek yetmezliğiolan (aGFR 15 ila <30 ml/dakika) 7 gönüllünün ve son dönem böbrek hastalığı (aGFR <15ml/dakika) olan (SDBY) 3 hastanın dahil edildiği bir böbrek yetmezliği çalışmasındadeğerlendirilmiştir. Normal böbrek fonksiyonu olan gönüllülerde maruz kalmaya göre hafif, orta ve şiddetli böbrek yetmezliği olan hastalarda EAAinf 1,6 kat, 1,9 kat ve 2,7 kat ve Cmaks 1,8 kat, 1,8 kat ve 2,3 kat artmıştır. Ribosiklibin etkililik ve güvenlilik çalışmaları, hafif böbrek yetmezliği olanhastaların büyük bir kısmını kapsadığı için (bkz. Bölüm 5.1), böbrek yetmezliği çalışmasındaorta veya şiddetli böbrek yetmezliği olan gönüllülerden elde edilen veriler, normal böbrekfonksiyonu olan ve hafif renal yetmezliği olan gönüllülere ait birleştirilmiş verilerlekarşılaştırılmıştır. Normal böbrek fonksiyonu ve hafif böbrek yetmezliği olan gönüllülere aitbirleştirilmiş verilerle karşılaştırıldığında, orta ve şiddetli böbrek yetmezliği olan hastalardaEAAinf sırasıyla 1,6 kat ve 2,2 kat ve Cmaks ise 1,5 kat ve 1,9 kat artmıştır. Az sayıda deneknedeniyle SDBY'li denekler için bir kat farkı hesaplanmamıştır, ancak sonuçlar şiddetliböbrek yetmezliği olan deneklere kıyasla ribosiklib maruziyetinde benzer veya biraz dahabüyük bir artışa işaret etmektedir. Böbrek fonksiyonunun ribosiklib farmakokinetiği üzerindeki etkisi, hastalara 600 mg başlangıç dozunun verildiği etkinlik ve güvenlik çalışmalarına dahil edilen kanser hastalarındada değerlendirilmiştir (bkz. Bölüm 5.1). 600 mg ribosiklibin tek doz veya tekrarlı dozlarhalinde oral uygulamasını takiben kanser hastalarında yapılan çalışmalardan elde edilenfarmakokinetik verilerin bir alt grup analizinde, hafif (n=57) veya orta (n=14) böbrekyetmezliği olan hastalarda ribosiklibin EAAinf ve Cmaks değerleri, normal böbrek fonksiyonuolan hastalardaki (n=86) EAAinf ve Cmaks ile karşılaştırılabilir olup, hafif veya orta derecedeböbrek yetmezliğinin ribosiklib maruziyeti üzerinde klinik olarak anlamlı bir etkisi olmadığınıdüşündürmektedir. Karaciğer bozukluğuKaraciğer bozukluğu olan kanser hastalığı olmayan gönüllülerde yürütülen bir farmakokinetik çalışmaya dayanılarak, hafif karaciğer bozukluğunun ribosiklib maruziyetine herhangi biretkisi olmamıştır (bkz. Bölüm 4.2). Ribosiklib için ortalama maruziyet, orta şiddetli(geometrik ortalama oran [GMR]: Cmaks için 1,44; EAAinf için 1,28) ve şiddetli (GMR: Cmaksiçin 1,32; EAAinf için 1,29) karaciğer bozukluğu olan hastalarda iki kattan az artış göstermiştir(bkz. Bölüm 4.2). Normal karaciğer fonksiyonu olan 160 meme kanserli hasta ve hafif karaciğer bozukluğu olan 47 hastayı içeren bir popülasyon farmakokinetiği analizi, spesifik karaciğer bozukluğuçalışmasının bulgularını destekleyerek, hafif karaciğer bozukluğunun ribosiklib maruziyetineherhangi bir etkisinin olmadığını göstermiştir. Ribosiklib orta şiddette veya şiddetli hepatikbozukluğu olan meme kanseri hastalarında araştırılmamıştır. Yaş, ağırlık, cinsiyet ve ırkın etkisiPopülasyon farmakokinetik analizi yaş, vücut ağırlığı veya cinsiyetin ribosiklibin sistemik maruziyetinde doz ayarlaması gerektirebilecek bir etkisinin olmadığını göstermiştir.Farmakokinetikte ırka bağlı değişikliklere ilişkin veriler, sonuca varabilmek için çok sınırlıdır. In vitroetkileşimlerRibosiklibin sitokrom P450 enzimleri üzerindeki etkisiİn vitro,İn vitrodeğerlendirmeler ribosiklibin klinik açıdan ilgili konsantrasyonlarda CYP2A6, CYP2B6,CYP2C8, CYP2C9, CYP2C19 ve CYP2D6'nın aktivitelerini inhibe etme potansiyeline sahipolmadığını göstermiştir. Ribosiklib CYP1A2, CYP2C9 ve CYP2D6'nın zamana bağımlıinhibisyonu için potansiyele sahip değildir.İn vitroİn vitroveriler CARaracılığıyla CYP2B6'yı indükleme potansiyelini dışlamak için yeterli değildir.Taşıyıcıların ribosiklib üzerindeki etkisiRibosiklib, in vitroin vitroortamda, karaciğer alım taşıyıcıları OATP1B1, OATP1B3 veyaOCT-1'nin bir substratı değildir.Ribosiklibin taşıyıcılar üzerindeki etkisi:İn vitroin vitroortamda klinik açıdan uygun konsantrasyonlarda OAT1, OAT3veya MRP2'yi inhibe etmemiştir.5.3 Klinik öncesi güvenlilik verileriGüvenlilik farmakolojisiKöpeklerdeki in vivokardiyak güvenlilik çalışmalarında, önerilen 600 mg dozundan sonra hastalarda elde edilmesi beklenecek maruziyette doz ve konsantrasyon ile ilişkili QTc aralığıuzaması gösterilmiştir. Artmış maruziyetlerde (beklenen klinik Cmaks'ın yaklaşık 5 katı) prematür ventriküler kontraksiyon (PVC'ler) olaylarını tetikleme potansiyeli debulunmaktadır. Tekrarlı doz toksisitesiSıçanlarda 27 haftaya ve köpeklerde 39 haftaya varan tekrarlı doz toksisitesi çalışmaları (3 haftalık tedavi/1 haftalık tedavisiz tedavi planı), ribosiklib toksisitesinin ana hedef organıolarak hepatobiliyer sistemi göstermiştir (proliferatif değişiklikler, kolestaz, kum benzeri safrataşı ve koyu safra). Tekrarlı doz çalışmalarında ribosiklibin farmakolojik etkisi ile ilişkilihedef organlar kemik iliğini (hiposellülerite), lenfoid sistem (lenfoid deplesyonu), intestinalmukoza (atrofi), deri (atrofi), kemik (kemik oluşumunda azalma), böbrek (tübüler epitelhücrelerde eş zamanlı dejenerasyon ve rejenerasyon) ve testisi (atrofi) içermiştir. Testistegörülen geri dönüşlülük eğilimi göstermiş atrofik değişikliklerin yanı sıra diğer tümdeğişiklikler 4 haftalık tedavisiz periyottan sonra tamamen geri dönüşlü olmuştur. Bu etkiler,testiküler germ hücreler üzerinde, seminiferöz tübüllerde atrofi ile sonuçlanan doğrudan anti-proliferatif etki ile bağlantılı olabilir. Toksisite çalışmalarında hayvanlarda ribosiklibemaruziyet genellikle 600 mg/gün (EAA bazında) çoklu dozları alan hastalarda gözlenendenküçük veya eşit olmuştur. Üreme toksisitesi/FertiliteRibosiklib, sıçanda veya tavşanda maternal toksisite göstermeyen dozlarda fetotoksisite ve teratojenite göstermiştir. Prenatal maruziyetten sonra EAA bazında en yüksek önerilen dozolan 600 mg/gün dozunda, sırasıyla insandaki maruziyetten daha düşük düzeylerde veyainsandaki maruziyetin 1,5 katında ribosiklib ile sıçanlarda implantasyon sonrası kayıpinsidanslarında artış ve fetal ağırlıkta azalma gözlenmiştir ve ribosiklib tavşanlardateratojeniktir. Sıçanlarda, geçici olarak değerlendirilmiş ve/veya düşük fetal ağırlıkla ilişkilendirilmiş iskelet değişikliklerinin eşlik ettiği fetal ağırlıkta azalma belirlenmiştir. Tavşanlarda, embriyofetalgelişim üzerine, fetal anomaliler (malformasyonlar ve dış, iç organ ve iskelet değişiklikleri) vefetal büyüme (daha düşük fetal ağırlıklar) insidansında artış ile kanıtlanmış olan advers etkilerolmuştur. Bu bulgular azalmış/küçük akciğer loblarını ve aort arkında ek damarı ve diyaframfıtığını, aksesuar lobun olmamasını veya (kısmen) kaynaşmış akciğer loblarını veazalmış/küçük aksesuar akciğer lobunu (30 ve 60 mg/kg), ekstra/ rudimenter onüçüncükaburgayı ve şekilsiz hiyoid kemiğini ve polekste azalmış parmak kemiği sayısını içermiştir.Herhangi bir embriyofetal mortalite bulgusu saptanmamıştır. Dişi sıçanlardaki bir fertilite çalışmasında ribosiklib, 300 mg/kg/güne kadar dozlarda (olasılıkla, EAA bazında en yüksek önerilen doz olan 600 mg/gün dozunda hastalardaki klinikmaruziyetten daha düşük veya eşit bir maruziyette) üreme fonksiyonunu, fertiliteyi ya daerken dönem embriyonik gelişimi etkilememiştir. Ribosiklib erkek fertilite çalışmalarında değerlendirilmemiştir. Bununla birlikte, sıçan ve köpek toksisite çalışmalarında, EAA bazında en yüksek önerilen doz olan 600 mg/gündozdaki maruziyetin altında veya bu maruziyete eşit düzeylerde, testislerde atrofikdeğişiklikler bildirilmiştir. Bu etkiler, testiküler üreme hücreleri üzerinde, seminiferöz tübüllerde atrofi ile sonuçlanan doğrudan anti-proliferatif etki ile bağlantılı olabilir. Ribosiklib ve metabolitleri sıçan sütüne hızla geçmiştir. Ribosiklib maruziyeti plazmaya göre sütte daha yüksek bulunmuştur. GenotoksisiteBakteriyel in vitroin vivoin vitrosistemlerde metabolik aktivasyon içeren veya içermeyen genotoksisite çalışmaları, ribosiklibin genotoksik potansiyeline ilişkinherhangi bir kanıt ortaya koymamıştır.KarsinojenezRibosiklib, sıçanlarda yapılan 2 yıllık bir çalışmada karsinojenite açısından değerlendirilmiştir. Ribosiklibin 2 yıl boyunca oral yoldan >300 mg/kg/gün dozlarda verilmesi, dişi sıçanların uterusunda/serviksinde endometriyal epitelyal tümörler ve glandüler ve skuamöz hiperplaziinsidansında artışa ve 50 mg/kg/gün dozunda verilmesi ise erkek sıçanların tiroid bezindefoliküler tümörlerin insidansında artışa neden olmuştur. Neoplastik değişikliklerin görüldüğüdişi ve erkek sıçanlarda kararlı durumda (EAA0-24 saat) ortalama maruziyet, hastalarda önerilen600 mg/gün dozunda elde edilenin sırasıyla 1,2 ve 1,4 katıdır. Neoplastik değişiklikleringörüldüğü dişi ve erkek sıçanlarda kararlı durumda ortalama maruziyet (EAA0-24 saat),hastalarda 400 mg/gün dozunda elde edilenin sırasıyla 2,2 ve 2,5 katı olmuştur. Ek neoplastik olmayan proliferatif değişiklikler, erkek sıçanlarda sırasıyla >5 mg/kg/gün ve 50 mg/kg/gün dozlarında karaciğerde değişikliğe uğramış odaklarda artış (bazofilik ve berrakhücreli) ve testiküler interstisyel (Leydig) hücre hiperplazisinden oluşmuştur. Erkek sıçanlardaki tiroid bulgularının mekanizması muhtemelen karaciğerde kemirgenlere özgü bir mikrozomal enzim indüksiyonunu içermektedir ve bunun insanlarla hiçbir ilişkisiolmadığı düşünülmektedir. Rahim/serviks ve testiküler interstisyel (Leydig) hücrelerüzerindeki etkiler, hipofiz bezinde laktotrofik hücre fonksiyonunun CDK4 inhibisyonunabağlı olarak hipotalamus-hipofiz-gonadal ekseni değiştiren uzun süreli hipoprolaktinemi ileilişkilidir. İnsanlarda bu mekanizma yoluyla östrojen/progesteron oranındaki herhangi bir potansiyel artış, eş zamanlı anti-östrojen tedavisinin östrojen sentezi üzerindeki inhibitör etkisi ilekompanse edilecektir. Bu sebeple, VALAMOR östrojen seviyesini azaltan tedavilerlekombinasyon halinde endikedir. Prolaktin sentezi ve rolü açısından kemirgenler ve insanlar arasındaki önemli farklılıklar göz önüne alındığında, bu etki biçiminin insanlarda sonuçları olması beklenmemektedir. 6. FARMASOTIK BİLGİLER6.1 Yardımcı maddelerin listesiTablet bileşenleri:Mikrokristalin selüloz Krospovidon (Tip A) Düşük sübstitüe hidroksipropilselüloz Magnezyum stearatKolloidal silikon dioksit Film kaplama bileşenleri:Siyah demir oksit (E172) Kırmızı demir oksit (E172) Soya lesitin Polivinil alkol (kısmen hidrolize) Talk Titanyum dioksit (E171) Ksantan sakızı 6.2 GeçimsizliklerGeçerli değildir. 6.3 Raf ömrü36 ay 6.4 Saklamaya yönelik özel tedbirler30oC'nin altında oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriği21 film kaplı tablet içeren PCTFE/PVC blisterler Ambalaj büyüklüğü: 63 film kaplı tablet 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİFarmanova Sağlık Hizmetleri Limited Şirketi 8. RUHSAT NUMARASI:2020/107 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİ:11.05.2020 / - 10. KÜB'ÜN YENİLENME TARİHİ |
İlaç BilgileriValamor 200 Mg Film Kaplı TabletEtken Maddesi: Ribosiklib Süksinat Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.