Admiraz 0,25 Mg Film Kaplı Tablet Kısa Ürün BilgisiKISA URUN BILGISI¡ Bu tıbbi ürün ek izleme tabidir. Bu, yeni güvenlilik bilgilerinin hızla tanımlanmasına olanak sağlayacaktır. Sağlık mesleği mensuplarından tüm şüpheli advers reaksiyonları bildirmeleri ricaedilmektedir. Advers reaksiyonların nasıl bildirileceği ile ilgili bilgi için bölüm 4.8'e bakınız. 1. BEŞERI TIBBI URUNUN ADIADMİRAZ 0,25 mg film kaplı tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Siponimod fumarik asit 0,278 mg (0,25 mg siponimoda karşılık gelmektedir) Yardımcı maddeler:Laktoz monohidrat (sığır kaynaklı) 62,197 mg Soya lesitin 0,092 mg Yardımcı maddelerin tam listesi için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tablet Bir yüzünde logo ve diğer yüzünde T baskısı bulunan, yaklaşık 6,1 mm çapında, soluk kırmızı, yuvarlak, bikonveks, kenarları eğimli film kaplı tablet. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarADMİRAZ, erişkinlerde, radyolojik veya klinik olarak aktif sekonder progresif multipl skleroz hastalarının tedavisi için endikedir. 4.2 Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:Siponimod tedavisi, multipl skleroz tedavisinde deneyimli bir doktor tarafından başlatılmalı ve denetlenmelidir. Tedaviye başlamadan önce hastalar, CYP2C9 metabolizör durumlarını belirlemek için CYP2C9 için genotiplenmelidir (bkz. Bölüm 4.4, 4.5 ve 5.2). CYP2C9*3*3 genotipi olan hastalarda siponimod kullanılmamalıdır (bkz. Bölüm 4.3, 4.4 ve 5.2). CYP2C9*2*3 veya *1*3 genotipi olan hastalarda, önerilen idame dozu günde bir kez alınan 1 mg'dır (0,25 mg'lık dört tablet) (bkz. Bölüm 4.4 ve 5.2). Diğer tüm CYP2C9 genotip hastalarında önerilen siponimod idame dozu 2 mg'dır. ADMİRAZ, günde bir defa alınır. Pozoloji:Tedaviye başlamaTedaviye 5 gün süren titrasyon paketi ile başlanmalıdır. Tedavi, 1. ve 2. günlerde günde bir kez 0,25 mg ile başlar, ardından 3. günde bir kez 0,5 mg, 4. günde 0,75 mg ve 5. günde 1,25 mgdozlarıyla devam edilir, ardından 6. günde başlayarak hastanın reçete edilen siponimod idamedozuna ulaşılır (bakınız Tablo 1). Tedaviye başlandıktan sonraki ilk 6 günü boyunca, önerilen günlük doz günde bir kez sabah, aç veya tok karnına alınmalıdır. Tablo 1
1 YP2C9*2*3 veya *1*3 genotipi olan hastalarda, önerilen idame dozu günde bir kez (4 x 0,25 mg) alınan 1 mg'dır (yukarıya ve bölüm 4.4 ve 5.2'ye bakınız). 5. günde 0,25 mg'lık ek maruziyet, hasta güvenliğini tehlikeye atmaz. Tedaviye başlama sırasında kaçırılan doz/dozlarTedavinin ilk 6 günü boyunca, bir titrasyon dozunun bir gün için kaçırılması durumunda, tedavinin yeni bir titrasyon paketi ile yeniden başlatılması gerekir. 6. günden sonra kaçırılan dozBir doz kaçırılırsa, reçete edilen doz bir sonraki planlanan zamanda alınmalıdır; bir sonraki doz iki katına çıkarılmamalıdır. Tedaviye ara verildikten sonra idame tedavisinin yeniden başlatılmasıİdame tedavisine 4 veya daha fazla günlük doz süresiyle ara verilirse, siponimodun yeni bir titrasyon paketi ile yeniden başlatılması gerekir. Uygulama şekli:Ağız yolu ile alınır. Siponimod aç veya tok karına alınır. Film kaplı tabletler suyla birlikte bütün halde yutulmalıdır. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Klinik farmakoloji çalışmalarına dayanarak, böbrek yetmezliği olan hastalarda doz ayarlamasına gerek yoktur (bkz. Bölüm 5.2). Karaciğer yetmezliği:Siponimod, ciddi karaciğer yetmezliği olan hastalarda (Child-Pugh sınıf C) kullanılmamalıdır (bkz. Bölüm 4.3). Hafif veya orta şiddette karaciğer yetmezliği olan hastalarda doz ayarlamasınagerek olmamakla birlikte, bu hastalarda tedaviye başlarken dikkatli olunmalıdır (bkz. Bölüm4.4 ve 5.2). Pediyatrik popülasyon:0 ila 18 yaş arası çocuklarda ve adölesanlarda siponimodun güvenliliği ve etkililiği henüz belirlenmemiştir. Veri bulunmamaktadır. Geriyatrik popülasyon:Siponimod 65 yaş ve üstü hastalarda çalışılmamıştır. Klinik çalışmalar 61 yaşına kadar olan hastaları içermektedir. Siponimod, güvenlilik ve etkililik hakkında yeterli veri bulunmadığındanyaşlılarda dikkatle kullanılmalıdır (bkz. Bölüm 5.2). 4.3 Kontrendikasyonlar Etkin maddeye veya yer fıstığına, soyaya veya bölüm 6.1'de listelenen yardımcımaddelerden herhangi birine karşı aşırı duyarlılık İmmün yetmezlik sendromu Progresif multifokal lökoensefalopati veya kriptokokal menenjit öyküsü Aktif maligniteler Şiddetli karaciğer yetmezliği (Child-Pugh sınıf C) Önceki 6 ayda miyokard enfarktüsü (MI), unstabil angina pektoris, inme/geçici iskemikatak (TIA), dekompanse kalp yetmezliği (yatarak tedavi gerektiren) veya New YorkKalp Derneği (NYHA) sınıf III/IV kalp yetmezliği (bkz. bölüm 4.4) olan hastalar Kalp pili kullanmıyorlarsa ikinci derece Mobitz tip II atriyoventriküler (AV) blok,üçüncü derece AV blok, sino-atriyal kalp bloğu veya hasta sinüs sendromu öyküsü olanhastalar (bkz. Bölüm 4.4) CYP2C9*3 (CYP2C9*3*3) genotipi (zayıf metabolize edici) için homozigot olanhastalar Gebelik sırasında ve etkili doğum kontrolü kullanmayan çocuk doğurma potansiyeliolan kadınlar (bkz. Bölüm 4.4 ve 4.6) 4.4 Özel kullanım uyarıları ve önlemleriEnfeksiyonlarEnfeksiyon riskiSiponimodun temel farmakodinamik etkisi, periferik lenfosit sayısını doza bağlı olarak başlangıç değerinin %20-30'una azaltmaktır. Bu, lenfoid dokulardaki lenfositlerin geri dönüşlüsekestrasyonundan kaynaklanmaktadır (bkz. Bölüm 5.1). Siponimodun bağışıklık sistemi üzerindeki etkileri enfeksiyon riskini artırabilir (bkz. Bölüm 4.8). Tedaviye başlamadan önce, yeni bir tam kan sayımı (CBC) (yani son 6 ay içinde veya önceki tedavinin kesilmesinden sonra) mevcut olmalıdır. CBC değerlendirmeleri tedavi başlangıcından3 ila 4 ay sonra ve sonrasında en az yılda bir kez ve ayrıca enfeksiyon belirtileri olmasıdurumunda da önerilir. Klinik çalışmalarda mutlak lenfosit sayısı <0,2 x 109/l olan hastalardasiponimod dozu azaltıldığından, mutlak lenfosit sayılarının <0,2 x 109/l olarak ölçülmesi halinde,siponimod dozu 1 mg'a düşürülmelidir. Halihazırda siponimod 1 mg alan bir hastadadoğrulanmış mutlak lenfosit sayısı <0,2 x 109/l ise 0,6 x 109/l'ye ulaşıncaya kadar siponimodtedavisi kesilmelidir. Lenfosit sayısı bu değere ulaştığında tedaviye tekrar başlanmasıdüşünülebilir. Şiddetli aktif enfeksiyonu olan hastalarda tedavinin başlatılması, bu durum düzelene kadar ertelenmelidir. Periferik lenfosit sayısı üzerindeki azaltıcı etkiler gibi rezidüel farmakodinamiketkiler, ilaç kesildikten sonra 3 ila 4 haftaya kadar sürebileceğinden, bu süre zarfında enfeksiyoniçin dikkatli olmaya devam edilmelidir (bkz. Aşağıda Siponimod tedavisinin durdurulmasıbölümü). Hastalara enfeksiyon belirtilerini derhal doktorlarına bildirmeleri söylenmelidir. Tedavi sırasında enfeksiyon semptomları olan hastalarda etkili tanı ve tedavi stratejileri uygulanmalıdır.Bir hastada ciddi bir enfeksiyon gelişirse, siponimod ile tedavinin askıya alınmasıdüşünülmelidir. Siponimod için kriptokokal menenjit (CM) vakaları bildirilmiştir. CM ile uyumlu semptom ve bulguları olan hastalara derhal tanısal değerlendirme yapılmalıdır. CM ekarte edilene kadarsiponimod tedavisi askıya alınmalıdır. CM teşhisi konulursa, uygun tedavi başlatılmalıdır. Siponimod dahil olmak üzere S1P reseptör modülatörleri ve diğer multipl skleroz tedavileri için progresif multifokal lökoensefalopati (PML) vakaları bildirilmiştir (bkz. Bölüm 4.8). Doktorlar,PML'yi düşündürecek klinik semptomlar veya manyetik rezonans görüntüleme (MRG)bulguları konusunda dikkatli olmalıdır. PML'den şüpheleniliyorsa, PML ekarte edilinceyekadar siponimod tedavisi askıya alınmalıdır. PML doğrulanırsa siponimod tedavisi kesilmelidir. Herpes viral enfeksiyonuTedavi sırasında herhangi bir zamanda siponimod ile herpes viral enfeksiyonu vakaları (varicella zoster virüslerinin [VZV] neden olduğu menenjit veya meningoensefalit vakalarıdahil) meydana gelmiştir. Herpes menenjit veya meningoensefalit meydana gelirse, siponimodkesilmeli ve ilgili enfeksiyon için uygun tedavi uygulanmalıdır. Hekim tarafından onaylanmışsuçiçeği öyküsü olmayan veya VZV'ye karşı tam bir aşılama süreci belgelenmemiş hastalarsiponimod başlamadan önce VZV antikorları açısından test edilmelidir (aşağıda Aşılamabölümüne bakınız). AşılamaSiponimod ile tedaviye başlamadan önce antikor negatif hastalar için tam kür suçiçeği aşılaması önerilir; bunun ardından aşılamanın tam etkisinin ortaya çıkmasını beklemek üzere tedavininbaşlatılması 1 ay ertelenmelidir (bkz. Bölüm 4.8). Hastalar siponimod alırken ve tedaviyi bıraktıktan sonra 4 hafta boyunca canlı zayıflatılmış aşıların kullanımından kaçınılmalıdır (bkz. Bölüm 4.5). Siponimod tedavisi sırasında aşılar daha az etkili olabilir. Planlı aşılamadan 1 hafta önce başlayarak 4 hafta sonrasına kadar tedavinin kesilmesi önerilir. Siponimod tedavisini aşılamaiçin durdururken, hastalık aktivitesinin olası geri dönüşü göz önünde bulundurulmalıdır (aşağıdaSiponimod tedavisinin durdurulması bölümüne bakınız). Anti-neoplastik, immünomodülatör veya immünosüpresif tedavilerle birlikte tedaviBu tür bir tedavi sırasında edinsel bağışıklık sistemi etkileri riski nedeniyle anti-neoplastik, immünomodülatör veya immünosüpresif tedaviler (kortikosteroidler dahil) bir arada dikkatle uygulanmalıdır (bkz. Bölüm 4.5). Makula ödemiFaz III klinik çalışmada görme semptomlarının eşlik ettiği veya etmediği makula ödemi, siponimod tedavisinde (%1,8) plaseboya (%0,2) kıyasla daha sık bildirilmiştir (bkz. Bölüm 4.8).Olguların çoğu tedavinin ilk 3-4 ayında meydana gelmiştir. Bu nedenle, tedavinin başlamasından3-4 ay sonra oftalmolojik bir değerlendirme önerilir. Makula ödemi vakaları daha uzun sürelitedavide de meydana geldiğinden, hastalar siponimod tedavisi sırasında herhangi bir zamandaortaya çıkabilecek görme bozukluklarını bildirmelidir ve makula da dahil olmak üzere fundusundeğerlendirilmesi önerilir. Makula ödemi olan hastalarda, bu durum düzelinceye kadar siponimod tedavisi başlatılmamalıdır. Siponimod, makula ödemi riskinde potansiyel bir artış nedeniyle diyabet, üveit veya altta yatan/birlikte var olan retina hastalığı öyküsü olan hastalarda dikkatli kullanılmalıdır (bkz.Bölüm 4.8). Makula ödemini tespit etmek için bu hastaların tedaviye başlamadan önce vesiponimod tedavisi sırasında düzenli olarak oftalmolojik bir değerlendirmeye tabi tutulmasıönerilir. Makula ödemi olan hastalarda siponimod tedavisinin devamı değerlendirilmemiştir. Bir hastada makula ödemi gelişirse siponimodun kesilmesi önerilir. Durumun düzelmesinin ardındansiponimodun yeniden başlatılması gerekip gerekmediğine dair kararda, her hastadaki potansiyelfaydalar ve riskler dikkate alınmalıdır. BradiaritmiKalp atım hızında azalmaSiponimod tedavisinin başlatılması kalp atım hızında geçici bir azalmaya neden olur (bkz. Bölüm 4.8 ve 5.1) ve bu nedenle tedavinin başlangıcında, 6. günde idame dozuna ulaşmak üzere, birtitrasyon şeması uygulanır (bakınız bölüm 4.2). İlk titrasyon dozundan sonra, kalp atım hızının azalması bir saat içinde başlar ve 1. gündeki düşüş yaklaşık 3 ila 4 saatte maksimum seviyeye ulaşmıştır. Artırmalı titrasyon devam ettikçe, sonrakigünlerde kalp atım hızındaki düşüşlerin daha fazla olduğu görülür ve 1. gündeki (başlangıç)değerden maksimum düşüşe 5 ila 6. günde ulaşılır. Mutlak saatlik ortalama kalp atım hızında enyüksek günlük doz sonrası azalma 1. günde görülür, nabız dakikada ortalama 5-6 vuruş (bpm)azalır. Sonraki günlerde doz sonrası düşüşler daha az belirgindir. Dozlara devam edildikçe kalpatım hızı 6. günden sonra artmaya başlar ve tedavinin başlamasından sonraki 10 gün içindeplasebo düzeylerine ulaşır. Dakikada 40 bpm'nin altındaki kalp atım hızları nadiren gözlenmiştir. Bradikardi yaşayan hastalar genellikle asemptomatik olmuştur. Birkaç hastada baş dönmesi ve kardiyak olmayangöğüs ağrısı dahil olmak üzere hafif ve orta şiddette semptomlar görülmüştür; bunlar müdahaleolmadan 24 saat içinde düzelmiştir (bkz. Bölüm 4.8). Gerekirse, siponimodun neden olduğu kalpatım hızındaki azalma, parenteral dozlarda atropin veya izoprenalin ile tersine çevrilebilir. Atriyoventriküler iletimSiponimod tedavisinin başlatılması, doz titrasyonu sırasında kalp atım hızında gözlenen azalmaya benzer geçici bir patern izleyen geçici atriyoventriküler iletim gecikmeleri ileilişkilendirilmiştir. Atriyoventriküler iletim gecikmeleri çoğu durumda birinci dereceatriyoventriküler (AV) bloklar (elektrokardiyogramda uzamış PR aralığı) olarak kendinigöstermiştir. Klinik çalışmalarda, tedavinin başlaması sırasında hastaların %1,7'sinden azında,genellikle Mobitz tip I (Wenckebach) olmak üzere ikinci derece AV blokları gözlenmiştir. İletimanormallikleri tipik olarak geçici ve asemptomatik olmuş, 24 saat içinde düzelmiş ve tedavininkesilmesini gerektirmemiştir. Önceden belirli kardiyak rahatsızlıkları olan hastalarda tedaviye başlama önerisiÖnlem olarak, aşağıdaki kardiyak rahatsızlıkları olan hastalar, ilk siponimod dozundan sonra 6saat boyunca bradikardi belirtileri ve semptomları açısından izlenmelidir (ayrıca bkz. Bölüm4.3):- sinüs bradikardisi (kalp atım hızı <55 bpm/dakika), - birinci veya ikinci derece [Mobitz tip I] AV bloğu geçmişi, - miyokard enfarktüsü öyküsü - kalp yetmezliği öyküsü (NYHA sınıf I ve II hastaları). Bu hastalarda, dozlamadan önce ve gözlem süresinin sonunda elektrokardiyogram (EKG) alınması önerilir. Doz sonrası bradiaritmi veya iletimle ilgili semptomlar ortaya çıkarsa veya dozsonrası 6 saat sonra EKG yeni başlangıç ikinci derece veya daha yüksek AV bloğu veya QTc>500 msn gösterirse, uygun tedavi protokolü başlatılmalı ve semptomlar/bulgular düzelinceyekadar gözleme devam edilmelidir. Farmakolojik tedavi gerekiyorsa, gözlem gece boyuncasürdürülmeli ve ikinci dozdan sonra 6 saatlik gözlem tekrarlanmalıdır. Ciddi kardiyak ritim bozuklukları veya önemli bradikardi riski nedeniyle, siponimod aşağıdaki hastalarda kullanılmamalıdır: - semptomatik bradikardi veya tekrarlayan senkop öyküsü, - kontrolsüz hipertansiyon veya - tedavi edilmemiş ciddi uyku apnesi. Bu tür hastalarda, siponimod ile tedavi, ancak beklenen faydalar potansiyel risklere ağır bastığı takdirde düşünülmeli ve en uygun gözlem stratejisini belirlemek için tedaviye başlamadan öncebir kardiyologdan tavsiye alınmalıdır. Kapsamlı bir QT çalışması, önemli bir doğrudan QT uzatma etkisi göstermemiştir ve siponimod, QT uzaması ile ilişkili bir aritmojenik potansiyel ile ilişkili değildir. Tedavinin başlatılması,titrasyon aşamasında kalp atım hızının düşmesine ve QT aralığının dolaylı uzamasına nedenolabilir. Siponimod, önemli QT uzaması (QTc> 500 msn) olan veya QT uzamasına neden olantıbbi ürünlerle tedavi edilen hastalarda çalışılmamıştır. Önceden belirgin QT uzaması olan veyabilinen aritmojenik özelliklere sahip QT uzamasına neden olan tıbbi ürünlerle tedavi edilenhastalarda siponimod ile tedavi düşünülüyorsa, tedavinin başlatılması sırasında uygun gözlemstratejisine karar vermek üzere tedaviyi başlatmadan önce bir kardiyologdan tavsiye alınmalıdır. Siponimod, sınıf la (örn. kinidin, prokainamid) veya sınıf III (örn. amiodaron, sotalol) antiaritmik tıbbi ürünler ile tedavi gerektiren aritmili hastalarda araştırılmamıştır. Sınıf la vesınıf III antiaritmik tıbbi ürünler, bradikardi hastalarında Torsades de Pointes vakalarıylailişkilendirilmiştir. Tedaviye başlanması kalp atım hızının düşmesine neden olduğundan,siponimod, tedavinin başlaması sırasında bu tıbbi ürünlerle birlikte kullanılmamalıdır. Klinik çalışmalarda bu tıbbi ürünler, siponimod alan hastalarda incelenmediği için kalp atım hızını düşüren kalsiyum kanal blokerleri (verapamil veya diltiazem gibi) veya kalp atım hızınıdüşürebilen diğer maddeler (örn. ivabradin veya digoksin) ile eş zamanlı tedavi alan hastalardadeneyim sınırlıdır. Tedaviye başlama sırasında bu maddelerin birlikte kullanılması ciddibradikardi ve kalp bloğu ile ilişkili olabilir. Kalp atım hızı üzerindeki potansiyel katkısınedeniyle, siponimod tedavisi genellikle halihazırda bu maddelerle tedavi edilen hastalardabaşlatılmamalıdır (bkz. Bölüm 4.5). Bu gibi hastalarda, sadece beklenen faydalar potansiyelrisklerden daha ağır basarsa siponimod ile tedavi düşünülmelidir. Siponimod ile tedavinin başlatılması sırasında yukarıdaki maddelerden biriyle birlikte tedavi düşünülürse, kalp atım hızını düşürmeyen bir tıbbi ürüne geçiş veya tedavinin başlatılması içinuygun gözlem ile ilgili olarak bir kardiyologdan tavsiye alınmalıdır. Beta-bloker tedavisine siponimod eklendiğinde bradiaritmik etkiler daha belirgindir. Stabil bir beta bloker dozu alan hastalar için, tedaviye başlamadan önce istirahat kalp atım hızı dikkatealınmalıdır. Kronik beta-bloker tedavisi altında istirahat kalp atım hızı >50 bpm ise, siponimoduygulanabilir. İstirahat kalp atım hızı <50 bpm ise, taban kalp atım hızı >50 bpm olana kadarbeta-bloker tedavisi kesilmelidir. Daha sonra siponimod ile tedavi başlatılabilir ve siponimodhedef idame dozuna yükseltildikten sonra beta bloker ile tedavi yeniden başlatılabilir (bkz.Bölüm 4.5). Karaciğer fonksiyonuSiponimod ile tedaviye başlamadan önce yakın tarihli (yani son 6 ay içinde) transaminaz ve bilirubin düzeyleri mevcut olmalıdır. Faz III klinik çalışmada, plasebo alan hastalarda %1,5'i ile karşılaştırıldığında siponimod 2 mg ile tedavi edilen hastaların %5,6'sında normalin üst sınırının (ULN) üç katı alaninaminotransferaz (ALT) veya aspartat aminotransferaz (AST) değerleri gözlenmiştir (bölüm4.8). Klinik çalışmalarda, ALT ve AST değerlerindeki yükseklik 3 katlık bir artışı aştığı takdirdeve hasta karaciğer fonksiyonuyla ilgili semptomlar gösterirse veya yükseklik 5 katlık bir artışıaştığı takdirde tedavi kesilmiştir. Faz III klinik çalışmada, tüm tedavi kesme olaylarının %1'ibu kriterlerden birini karşılamıştır. Karaciğer fonksiyon bozukluğunu düşündüren semptomlar geliştiren hastalarda karaciğer enzimleri kontrol edilmeli ve anlamlı karaciğer hasarı doğrulanırsa siponimod kesilmelidir.Tedavinin yeniden başlatılması, karaciğer hasarının başka bir nedeni olduğunun belirlenipbelirlenmemesine ve karaciğer fonksiyon bozukluğunun tekrarlama riskine karşı hastanıntedaviye devam etmesinin yararlarına bağlı olacaktır. Önceden karaciğer hastalığı olan hastalarda siponimod alırken yüksek karaciğer fonksiyon testi değerlerinin ortaya çıkma riskinin daha yüksek olduğunu gösteren herhangi bir veri olmamasına rağmen, önemli karaciğer hastalığı geçmişi olan hastalarda dikkatli olunmalıdır. Kutanöz neoplazmalarSiponimod alan hastalarda, özellikle tedavi süresi daha uzun olan hastalarda, bazal hücreli karsinom (BCC) ve skuamöz hücreli karsinom (SCC) dahil olmak üzere diğer kutanözneoplazmalar bildirilmiştir (bkz. bölüm 4.8). Tüm hastalar için tedavi başlangıcında ve klinik değerlendirmeye göre her 6 ila 12 ayda bir cilt muayenesi önerilmektedir. Daha uzun tedavi sürelerinde dikkatli deri muayeneleri yapılmalıdır.Hastalara, tüm şüpheli cilt lezyonlarını vakit kaybetmeden hekimlerine bildirmelerisöylenmelidir. Siponimod ile tedavi edilen hastalar, korunmadan güneş ışığına maruz kalmayakarşı uyarılmalıdır. Bu hastalar eş zamanlı olarak UV-B radyasyonu ile fototerapi veya PUVA-fotokemoterapi almamalıdır. Beklenmedik nörolojik veya psikiyatrik semptomlar/bulgularBaşka bir sfingozin 1-fosfat (S1P) reseptör modülatörü için seyrek sıklıkla posterior reversibl ensefalopati sendromu (PRES) olguları bildirilmiştir. Geliştirme programında siponimod içinbu tür olaylar rapor edilmemiştir. Bununla birlikte, siponimod tedavisi gören bir hastadabeklenmedik nörolojik veya psikiyatrik semptomlar/bulgular ortaya çıkar (örn. bilişselbozukluklar, davranış değişiklikleri, kortikal görme bozuklukları veya diğer nörolojik kortikalsemptomlar/bulgular veya intrakraniyal basınçta bir artışa işaret eden herhangi birsemptom/bulgu) veya hızlandırılmış nörolojik bozulma gelişirse, derhal tam bir fiziksel venörolojik muayene programlanmalı ve MRG düşünülmelidir. İmmünosüpresif veya immünomodülatör tedavilerle önceden tedaviDiğer hastalığı modifiye edici terapilerden geçiş yapılırken, hastalığın yeniden aktivasyon riskini en aza indirirken aynı zamanda ilave bir bağışıklık etkisinden kaçınmak için diğertedavinin yarılanma ömrü ve etki şekli göz önünde bulundurulmalıdır. Önceki tedavinin (yanisitopeni) bağışıklık etkilerinin ortadan kalktığından emin olmak için siponimod başlatılmadanönce periferik bir lenfosit sayımı (CBC) önerilir. Alemtuzumabın, ürün bilgilerinde açıklanan bağışıklık baskılayıcı etkilerinin karakteristikleri ve süresi nedeniyle, alemtuzumabdan sonra siponimod ile tedaviye başlanması önerilmez. Siponimod genellikle beta interferon veya glatiramer asetatın kesilmesinden hemen sonra başlatılabilir. Kan basıncı etkileriTıbbi ürünle kontrol edilemeyen hipertansiyonu olan hastalar klinik çalışmalara dahil edilmemiştir ve kontrolsüz hipertansiyonu olan hastalar siponimod ile tedavi edildiği takdirdeözel dikkat gerekir. SPMS hastalarında faz III klinik çalışmada hipertansiyon, siponimod kullanan hastalarda (%12,6) plasebo verilenlere (%9,0) göre daha sık bildirilmiştir. Siponimod ile tedavi, tedavininbaşlamasından kısa bir süre sonra başlayan ve yaklaşık 6 aylık tedaviden sonra maksimumetkiye ulaşan (sistolik 3 mmHg, diyastolik 1,2 mmHg) ve daha sonra stabil seyreden bir artışlasonuçlanmıştır. Devam eden tedavi ile bu etki de devam etmiştir. Siponimod tedavisi sırasında kan basıncı düzenli olarak izlenmelidir. CYP2C9 genotipiSiponimod ile tedaviye başlamadan önce hastalar CYP2C9 metabolizör durumlarını belirlemek üzere CYP2C9 için genotiplendirilmelidir (bkz. Bölüm 4.2). CYP2C9*3 (CYP2C9*3*3genotipi: popülasyonun yaklaşık %0,3 ila 0,4'ü) için homozigot hastalar siponimod ile tedaviedilmemelidir. Bu hastalarda siponimod kullanımı, önemli ölçüde yüksek siponimod plazmadüzeylerine yol açar. Maruziyet artışını önlemek için CYP2C9*2*3 genotipi (popülasyonun%1,4-1,7'si) olan hastalarda ve CYP2C9*1*3 genotipi (popülasyonun %9-12'si) olanhastalarda önerilen idame dozu günde 1 mg'dır (bkz. bölüm 4.2 ve 5.2). Çocuk doğurma potansiyeli olan kadınlarFetüs ile ilgili risk nedeniyle, siponimod gebelik sırasında ve etkili doğum kontrolü kullanmayan çocuk doğurma potansiyeli olan kadınlarda kontrendikedir. Tedaviye başlamadanönce, çocuk doğurma potansiyeli olan kadınlar fetüs ile ilgili bu risk konusundabilgilendirilmeli, gebelik testi negatif olmalı, tedavi sırasında ve tedavinin kesilmesinden sonraen az 10 gün boyunca etkili bir doğum kontrol yöntemi kullanmalıdır (bkz. Bölüm 4.3 ve 4.6). Siponimod tedavisinin durdurulmasıBaşka bir S1P reseptör modülatörünün kesilmesinden sonra hastalığın geri tepmesi de dahil olmak üzere hastalığın şiddetli alevlenmesi seyrek sıklıkla bildirilmiştir. Siponimod tedavisinidurdurduktan sonra hastalığın şiddetli alevlenmesi olasılığı dikkate alınmalıdır. Hastalar,siponimod kesildikten sonra olası şiddetli alevlenme veya yüksek hastalık aktivitesinin geridönüşü ile ilgili belirtiler açısından gözlemlenmeli ve gerektiği şekilde uygun tedaviuygulanmalıdır. Siponimod tedavisi durdurulduktan sonra, siponimod 10 güne kadar kanda kalır. Bu aralıkta diğer tedavilere başlamak, siponimoda eşzamanlı maruziyet ile sonuçlanacaktır. SPMS hastalarının büyük çoğunluğunda (%90), tedaviyi bıraktıktan sonraki 10 gün içinde lenfosit sayıları normal aralığa döner. Bununla birlikte, periferik lenfosit sayısı üzerindekiazaltıcı etkiler gibi rezidüel farmakodinamik etkiler, son dozdan sonra 3-4 haftaya kadar devamedebilir. Bu süre zarfında immünosüpresanların kullanımı bağışıklık sistemi üzerinde ilave biretkiye yol açabilir ve bu nedenle son dozdan sonra 3 ila 4 hafta boyunca dikkatli olunmalıdır. Hematolojik testler ile etkileşimSiponimod, sekonder lenfoid organlarda yeniden dağılım yoluyla kan lenfosit sayısını azalttığından, siponimod ile tedavi edilen bir hastanın lenfosit alt kümesi durumunudeğerlendirmek için periferik kan lenfosit sayıları kullanılamaz. Dolaşımdaki mononükleerhücrelerin kullanımını içeren laboratuvar testleri, dolaşımdaki lenfositlerin sayısındakiazalmaya bağlı olarak daha büyük kan hacimleri gerektirir. Yardımcı maddelerADMİRAZ soya lesitin içerir. Yer fıstığına ya da soyaya aşırı duyarlılığı olan hastalar ADM İRAZ almamalıdır (bkz. bölüm 4.3).ADM İRAZ tabletler laktoz içerir. Galaktoz intoleransı, total laktoz yetmezliği veya glikoz-galaktoz malabsorpsiyonu gibi nadir kalıtsal problemleri olan hastaların bu ilacı almamalıdır.Antineoplastik, bağışıklık modüle edici veya immünosüpresif tedavilerSiponimod; antineoplastik, immünomodülatör veya immünosüpresif tedavilerle kombinasyon halinde çalışılmamıştır. Bu tür bir tedavi sırasında ve bu tıbbi ürünlerin herhangi birinindurdurulmasından sonraki haftalarda ek bağışıklık etkileri riski nedeniyle, birlikte uygulamasırasında dikkatli olunmalıdır (bkz. Bölüm 4.4). Ürün bilgilerinde açıklanan alemtuzumab bağışıklık baskılayıcı etkilerinin özellikleri ve süresi nedeniyle, tedavinin yararları her bir hasta için risklerden açıkça daha ağır basmadıkça,alemtuzumab sonrası siponimod ile tedaviye başlanması önerilmez (bkz. Bölüm 4.4). Anti-aritmik tıbbi ürünler, QT uzatan tıbbi ürünler, kalp atım hızını düşürebilecek tıbbi ürünlerTedavi başlangıcında sınıf la (örn., kinidin, prokainamid) veya sınıf III (örn. amiodaron, sotalol)anti-aritmik tıbbi ürünler, bilinen aritmojenik özelliklere sahip QT uzatan tıbbi ürünler, kalp atımhızını düşüren kalsiyum kanal blokerleri (verapamil veya diltiazem gibi) veya kalp atım hızınıazaltabilecek diğer maddeler (örn. ivabradin veya digoksin) kullanmakta olan hastalarda, kalpatım hızı üzerindeki potansiyel ilave etkiler nedeniyle, siponimod eşzamanlı olarakkullanılmamalıdır (bkz. bölüm 4.4). Bu tıbbi ürünlerin siponimod ile birlikte kullanımı ile ilgiliveri mevcut değildir. Tedaviye başlama sırasında bu maddelerin birlikte kullanılması ciddibradikardi ve kalp bloğu ile ilişkili olabilir. Kalp atım hızı üzerindeki potansiyel katkı etkisinedeniyle, siponimod tedavisi genellikle bu maddelerle eşzamanlı tedavi edilen hastalardabaşlatılmamalıdır (bkz. Bölüm 4.4). Siponimod ile tedavi düşünülürse, kalp atım hızınıdüşürmeyen tıbbi ürünlere geçiş veya tedavinin başlatılması için uygun izlem ile ilgili olarakbir kardiyologdan tavsiye alınmalıdır.Beta blokerlerBeta bloker alan hastalarda kalp atım hızını düşürme üzerine ilave etkiler nedeniyle siponimod başlatıldığında dikkatli olunmalıdır (bkz. Bölüm 4.4). Stabil dozlarda siponimod alan hastalardabeta-bloker tedavisi başlatılabilir. Siponimod ve propranololün birlikte uygulanmasının olumsuz kronotropik etkisi, özel bir farmakodinamik/güvenlilik çalışmasında değerlendirilmiştir. Siponimod farmakokinetik/farmakodinamik kararlı durum üzerine propranolol eklenmesi, propranololfarmakokinetik/farmakodinamik kararlı durumun üstüne siponimod eklenmesine kıyasla dahaaz belirgin negatif kronotropik etkilere (aditiften daha az) sahip olmuştur (aditif HR etkisi). AşılamaCanlı zayıflatılmış aşıların kullanımı enfeksiyon riski taşıyabilir ve bu nedenle siponimod tedavisi sırasında ve tedaviden sonra 4 haftaya kadar bunlar kullanılmamalıdır (bkz. Bölüm 4.4). Siponimod ile tedavi sırasında ve tedaviden sonra 4 haftaya kadar aşılama daha az etkili olabilir. Siponimod tedavisi aşılamadan 1 hafta öncesinden 4 hafta sonrasına kadar duraklatılırsa,aşılamanın etkililiğinin tehlikeye girmediği kabul edilmektedir (bkz. Bölüm 4.4). Özel bir faz Isağlıklı gönüllü çalışmasında, grip aşılarıyla birlikte siponimod tedavisi veya daha kısa tedaviduraklaması (aşılamadan 10 gün ile 14 gün öncesi arası) plaseboya kıyasla daha düşük yanıtoranlarına (yaklaşık %15 ila %30 daha düşük) neden olmuş, diğer yandan bir PPV 23 aşısınınetkililiği, eşzamanlı siponimod tedavisinden olumsuz etkilenmemiştir (bkz. bölüm 4.4). Diğer tıbbi ürünlerin siponimod farmakokinetiğini etkileme potansiyeliSiponimod esas olarak sitokrom P450 2C9 (CYP2C9) (%79,3) ve daha az ölçüde sitokrom P450 3A4 (CYP3A4) (%18,5) tarafından metabolize edilir. CYP2C9 bir polimorfik enzimdir ve CYP3A veya CYP2C9 inhibitörlerinin veya indükleyicilerinin varlığında ilaç-ilaç etkileşimi(DDI) etkisinin CYP2C9 genotipine bağlı olduğu tahmin edilmektedir. CYP2C9 ve CYP3A4 i^nhibit^^leriSiponimod ile orta derecede CYP2C9 ve orta veya güçlü CYP3A4 inhibisyonuna neden olan tıbbi ürünlerin birlikte kullanılması siponimod maruziyetinde önemli bir artış nedeniyle önerilmez. Bueşzamanlı ilaç rejimi, ayrı bir orta veya güçlü CYP3A4 inhibitörü ile kombinasyon halinde ortaderecede bir CYP2C9/CYP3A4 çift inhibitörünü (örn. flukonazol) veya orta bir CYP2C9inhibitörünü içerebilir. CYP2C9*1*1 genotipi olan sağlıklı gönüllülerde kararlı durumda 200 mg/gün flukonazol (orta CYP2C9/güçlü CYP3A4 inhibitörü) ile tek doz siponimod 4 mg'ın birlikte uygulanması,siponimodun eğri altındaki alanında (EAA) 2 kat artışa neden olmuştur. Fizyolojik temellifarmakokinetik (PBPK) modelleme kullanılarak yapılan bir ilaç etkileşim potansiyelideğerlendirilmesine göre, CYP2C9*2*2 genotipi olan hastalar dışında tüm genotipler genelindeherhangi bir CYP3A4 ve CYP2C9 inhibitörü tipiyle siponimodun EAA'sında maksimum 2 katartış öngörülmektedir. CYP2C9*2*2 hastalarında, orta CYP2C9/CYP3A4 inhibitörlerininvarlığında siponimodun EAA'sında 2,7 kat artış beklenmektedir. CYP2C9 ve CYP3A4 indükleyicil^^iSiponimod, çoğu CYP2C9 ve CYP3A4 indükleyici tipi ile kombine edilebilir. Bununla birlikte, siponimod maruziyetinde beklenen bir azalma nedeniyle, siponimod kombine edildiğindetedavinin uygunluğu ve olası yararı göz önünde bulundurulmalıdır: - genotipten bağımsız olarak tüm hastalarda güçlü CYP3A4/orta CYP2C9 ikili indükleyicileri(örn. karbamazepin) veya ayrı bir güçlü CYP3A4 indükleyicisi ile kombinasyon halindeorta derecede bir CYP2C9 indükleyicisi ile. - CYP2C9*1*3 veya *2*3 genotipli hastalarda orta derecede CYP3A4 indükleyicileri (örn.modafinil) veya güçlü CYP3A4 indükleyicileri ile. PBPK modellemesi kullanılarak ilaç etkileşim potansiyeli üzerinde yapılan bir değerlendirmeye göre, siponimod maruziyetinde önemli bir azalma (sırasıyla %76 ve %51'e kadar)beklenmektedir. Günde 2 mg siponimodun rifampinin (güçlü CYP3A4 ve orta CYP2C9indükleyicisi) günlük 600 mg dozları varlığında uygulanması, CY2C9*1*1 olgularındasiponimod EAAtau, ss ve Cmaks, ss değerlerini sırasıyla %57 ve %45 azaltmıştır. Oral kontraseptiflerSiponimod ile birlikte uygulama, kombine etinilöstradiol ve levonorgestrel oral kontraseptifin farmakokinetiği ve farmakodinamiği üzerinde klinik olarak anlamlı etkiler ortaya koymamıştır.Dolayısıyla, siponimod tedavisi sırasında araştırılan oral kontraseptifin etkililiği korunmuştur. Diğer progestajenleri içeren oral kontraseptiflerle herhangi bir etkileşim çalışması yapılmamıştır, ancak siponimodun oral kontraseptiflerin etkinliği üzerinde bir etkisi olması beklenmemektedir. Özel popülasyonlara ilişkin ek bilgiler Böbrek yetmezliği:Böbrek yetmezliği olan hastalarla ilgili bir etkileşim çalışması yapılmamıştır. Karaciğer yetmezliği:Karaciğer yetmezliği olan hastalarla ilgili bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon:Pediyatrik hastalarla ilgili bir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: D Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Siponimod, etkili doğum kontrol yöntemi kullanmayan çocuk doğurma potansiyeli olan kadınlarda kontrendikedir (bkz. Bölüm 4.3). Bu nedenle, çocuk doğurma potansiyeli olankadınlarda tedaviye başlamadan önce gebelik testi sonucu negatif çıkmalı ve fetüs için ciddi riskkonusunda danışmanlık sağlanmalıdır. Çocuk doğurma potansiyeli olan kadınlar, tedavi sırasındave son siponimod dozunu takip eden en az on gün boyunca etkili bir doğum kontrol yöntemikullanmalıdır (bkz. Bölüm 4.4). Ayrıca Doktor Eğitim Paketinde özel önlemler de yer almaktadır. Siponimod kadın hastalara reçete edilmeden önce ve tedavi sırasında bu önlemler uygulanmalıdır. Gebeliği planlamak için siponimod tedavisini durdururken, hastalık aktivitesinde olası bir geri dönüş göz önünde bulundurulmalıdır (bkz. Bölüm 4.4). Gebelik dönemiGebe kadınlarda siponimod kullanımıyla ilgili veri bulunmamaktadır ya da sınırlı veri mevcuttur. Hayvan çalışmaları, günlük 2 mg dozda insan maruziyeti ile karşılaştırılabilirmaruziyet düzeylerinde embriyo-fetal ölümler ve iskelet veya visseral malformasyonlar dahilolmak üzere sıçanlarda ve tavşanlarda siponimod kaynaklı embriyotoksisite ve fetotoksisite vesıçanlarda teratojenisite göstermiştir (bkz. Bölüm 5.3). Ek olarak, başka bir sfingozin-1-fosfatreseptör modülatörü ile sahip olunan klinik deneyim, gebelik sırasında uygulandığında genelpopülasyonda gözlenen orana kıyasla 2 kat daha fazla büyük konjenital malformasyon riskigöstermiştir. Sonuç olarak, siponimod gebelik sırasında kontrendikedir (bkz. Bölüm 4.3). Siponimod, gebelik planlanmadan en az 10 gün önce durdurulmalıdır (bkz. Bölüm 4.4). Bir kadın tedavi sırasındagebe kalırsa, siponimod kesilmelidir. Tedaviyle ilişkili fetüse zararlı etki riski konusunda tıbbitavsiye verilmeli ve ultrasonografi muayeneleri yapılmalıdır. Laktasyon dönemiSiponimod veya ana metabolitlerinin anne sütüne geçip geçmediği bilinmemektedir. Siponimod ve metabolitleri sıçanların sütüne geçer. Siponimod emzirme döneminde kullanılmamalıdır. Üreme yeteneği/FertiliteSiponimodun insan fertilitesi üzerindeki etkisi değerlendirilmemiştir. Siponimodun sıçanlarda ve maymunlarda erkek üreme organları veya sıçanlarda fertilite parametreleri üzerinde hiçbiretkisi olmamıştır. 4.7 Araç ve makine kullanımı üzerindeki etkilerSiponimodun araç ve makine kullanımı üzerinde hiçbir etkisi yoktur veya etkileri göz ardı edilebilir düzeydedir. Bununla birlikte, siponimod ile tedaviye başlarken bazen baş dönmesimeydana gelebilir. Bu nedenle, hastalar, siponimod tedavisine başlanan ilk gün boyunca araçveya makine kullanmamalıdır (bkz. Bölüm 4.4). 4.8 İstenmeyen etkilerGüvenlilik profilinin özetiEn yaygın advers ilaç reaksiyonları baş ağrısı (%15) ve hipertansiyondur (%12,6). Advers ilaç reaksiyonlarının tablo halinde özetiHer sistem organ sınıfı içinde, advers ilaç reaksiyonları, en sık reaksiyonlar önce olacak şekilde sıklığa göre sıralanmaktadır. Ek olarak, her advers ilaç reaksiyonu için karşılık gelen sıklıkkategorisi aşağıdaki sisteme dayanmaktadır: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10);yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000),bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor).
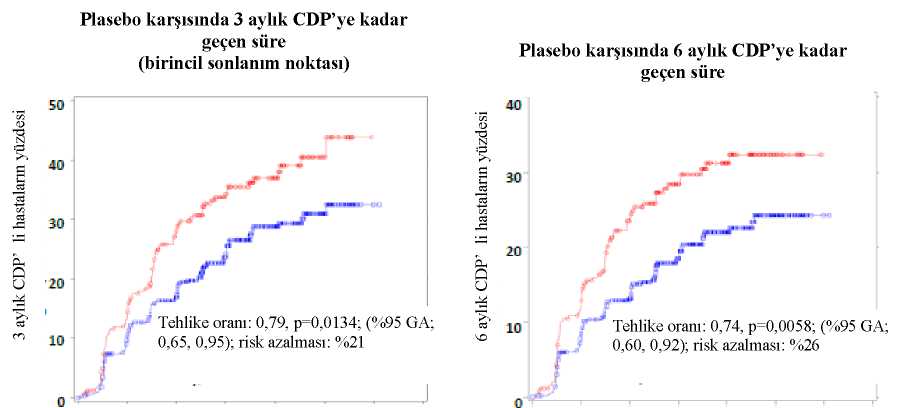
Seçilmiş advers reaksiyonların tanımıEnfeksiyonlarSPMS hastalarındaki Faz III klinik çalışmada, toplam enfeksiyon oranı, siponimod hastaları ile plasebo hastaları arasında karşılaştırılabilir olmuştur (sırasıyla %49,0'a karşılık %49,1).Bununla birlikte, plasebo (%0,7) ile karşılaştırıldığında siponimod (%2,5) tedavisinde herpeszoster enfeksiyonu oranında bir artış bildirilmiştir. Tedavi sırasında herhangi bir zamanda siponimod ile varicella zoster virüslerinin neden olduğu menenjit veya meningoensefalit vakaları ortaya çıkmıştır. Siponimod için kriptokokal menenjit(CM) vakaları da bildirilmiştir (bkz. bölüm 4.4). Makula ödemiMakula ödemi siponimod alan hastalarda (%1,8) plasebo verilenlerden (%0,2) daha sık bildirilmiştir. Olguların çoğunluğu siponimodun başlamasından sonraki 3 ila 4 ay içindemeydana gelmiş olmakla birlikte, siponimod ile tedavi edilen hastalarda 6 aydan daha uzun birsüreyle vakalar bildirilmiştir (bkz. Bölüm 4.4). Bazı hastalar bulanık görme veya görmekeskinliğinde azalma ile başvururken, bazıları asemptomatik olmuş ve rutin oftalmolojikmuayenesinde tanı konmuştur. Tedavinin kesilmesinden sonra genellikle makula ödemindedüzelme olmuş veya kendiliğinden kaybolmuştur. Yeniden tedavi ile birlikte tekrarlama riskideğerlendirilmemiştir. BradiaritmiSiponimod tedavisinin başlatılması, kalp atım hızında geçici bir düşüşe neden olur ve ayrıca atriyoventriküler iletim gecikmeleriyle de ilişkili olabilir (bkz. Bölüm 4.4). Siponimod ile tedaviedilen hastaların %6,2'sinde bradikardi bildirilirken bu oran plasebo ile %3,1 olmuştur; AVblok ise siponimod ile tedavi edilen hastaların %1,7'sinde ve plasebo uygulanan hastaların%0,7'sinde bildirilmiştir (bkz. Bölüm 4.4). Kalp atım hızındaki maksimum düşüş, dozdan sonraki ilk 6 saatte görülür. İlk dozlama aşamasında geçici, doza bağlı bir düşüş gözlenmiştir ve > 5 mg'lık dozlarda bu etki plato yapmıştır. Bradiaritmik olaylar (AV blokları ve sinüs duraklamaları) siponimod tedavisialtında plaseboya kıyasla daha yüksek bir insidansla tespit edilmiştir. Çoğu AV bloğu ve sinüs duraklaması, 2 mg'lık terapötik dozun üzerinde meydana gelmiştir ve doz titrasyonu yapılmamış koşullar altında doz titrasyon koşullarına kıyasla önemli ölçüde dahayüksek insidans göstermiştir. Siponimodun neden olduğu kalp atım hızındaki azalma, atropin veya izoprenalin ile tersine çevrilebilmektedir. Karaciğer _ fonksiyon testleriSiponimod ile tedavi edilen MS hastalarında hepatik enzimlerde artış (çoğunlukla ALT yüksekliği) bildirilmiştir. SPMS hastalarında yapılan faz III çalışmada, özellikle karaciğertransaminaz (ALT/AST) ve GGT yükselmelerine bağlı olarak, plasebo (%3,1) hastaları ilekarşılaştırıldığında siponimod hastalarında (%11,3) daha sık karaciğer fonksiyon testi artışlarıgözlenmiştir. Yüksekliklerin çoğu tedaviye başladıktan sonraki 6 ay içinde meydana gelmiştir.Siponimod kesildikten sonra yaklaşık 1 ay içinde ALT düzeyleri normale dönmüştür (bkz. Bölüm4.4). Kan basıncıSPMS hastalarındaki faz III klinik çalışmada hipertansiyon, plasebo uygulanan hastalar (%9,0) ile karşılaştırıldığında siponimod kullanan hastalarda (%12,6) daha sık bildirilmiştir. Siponimodile tedavi, tedavinin başlamasından kısa bir sonra başlayarak sistolik ve diyastolik kanbasıncında bir artışla sonuçlanmış, bu etki yaklaşık 6 aylık tedaviden sonra maksimum düzeyeulaşmış (sistolik 3 mmHg, diyastolik 1,2 mmHg) ve sonrasında stabil kalmıştır. Devam edentedavi ile bu etki de devam etmiştir. NöbetlerSPMS hastalarındaki faz III klinik çalışmada plasebodaki %0,4'lük oran ile karşılaştırıldığında, siponimod ile tedavi edilen hastaların %1,7'sinde nöbetler bildirilmiştir. Solunum etkileriSiponimod tedavisi ile 1 saniyede zorlu ekspiratuvar hacimde (FEV1) ve akciğerin karbon monoksit (DLCO) değerleri için difüzyon kapasitesinde küçük düşüşler gözlenmiştir. SPMShastalarındaki faz III klinik çalışmada tedavinin 3. ve 6. aylarında, siponimod grubundaFEV1'de başlangıçtan ortalama değişiklikler, her bir zaman noktasında -0,1 L iken plasebogrubunda değişiklik olmamıştır. Bu gözlemler, siponimod ile tedavi edilen ve kronik obstruktifakciğer hastalığı (KOAH) veya astım gibi solunum bozuklukları olan hastalarda biraz dahayüksek olmuştur (FEV1'de başlangıçtan yaklaşık 0,15 L ortalama değişiklik). Kronik tedavidebu azalma klinik olarak anlamlı advers olaylara dönüşmemiştir ve öksürük veya dispneraporlarındaki artışla ilişkili değildir (bkz. Bölüm 5.1). Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar / risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr[email protected];4.9 Doz aşımı ve tedavisiSağlıklı olgularda 75 mg'lık tek dozlardan sonra semptomatik bradikardi oluşumuna bağlı olarak maksimum tolere edilen tek dozun 25 mg olduğu belirlenmiştir. Birkaç olgu, 3 ila 4 günboyunca kasıtsız olarak günde 200 mg'a kadar dozlar almıştır ve karaciğer fonksiyon testlerindeasemptomatik hafif ila orta derecede geçici artışlar yaşamışlardır. 84 mg siponimod alan bir hastada (depresyon öyküsü olan) karaciğer transaminazlarında hafif bir yükselme görülmüştür. Doz aşımı eğer siponimoda ilk maruziyet ise veya siponimodun doz titrasyon fazı sırasında meydana gelirse, gece boyunca gözlemi de içerebilecek şekilde bradikardinin belirti vesemptomlarını gözlemlemek önemlidir. Kalp atım hızı ve kan basıncının düzenli olarakölçülmesi gerekir ve elektrokardiyogramlar çekilmelidir (bkz. Bölüm 4.2 ve 4.4). Siponimod için spesifik bir antidot yoktur. Diyaliz ya da plazma değişimi siponimodun vücuttan anlamlı bir şekilde uzaklaştırılmasına neden olmaz. 5. FARMAKOLOJIK ÖZELLIKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: İmmünosupresanlar, seçici immünosupresanlar ATC kodu: L04AA42 Etki mekanizmasıSiponimod, bir sfingozin-1-fosfat (S1P) reseptör modülatörüdür. Siponimod, S1P için beş G-protein bağlı reseptörden (GPCR) ikisine (yani S1P1 ve S1P5) seçici olarak bağlanır. Siponimod, lenfositler üzerindeki S1P1 reseptörleri üzerinde fonksiyonel bir antagonist olarakhareket ederek lenf düğümlerinden çıkışı önler. Bu, T hücrelerinin santral sinir sistemine (SSS)yeniden dolaşımını azaltarak merkezi enflamasyonu sınırlandırır. Farmakodinamik etkilerPeriferik kan lenfositlerinde azalmaSiponimod, lenfoid dokularda geri dönüşlü lenfosit sekestrasyonu nedeniyle, ilk dozdan sonraki 6 saat içinde periferik kan lenfosit sayısında doza bağlı bir azalmaya neden olur. Devam eden günlük dozlama ile birlikte lenfosit sayısı azalmaya devam ederek tipik bir CYP2C9*1*1 SPMS hastasında veya *1*2 Japon olmayan SPMS hastasında yaklaşık %0,560(0,271-1,08) hücre/nL lenfosit sayısı şeklindeki dip medyan (%90 GA) değerine ulaşır(başlangıç değerinin %20-30'una karşılık gelir). Günlük dozlama ile düşük lenfosit sayılarıkorunur. SPMS hastalarının büyük çoğunluğunda (%90), tedaviyi bıraktıktan sonraki 10 gün içinde lenfosit sayıları normal aralığa döner. Siponimod tedavisini durdurduktan sonra periferiklenfosit sayısı üzerindeki rezidüel düşürücü etkiler son dozdan sonra 3-4 haftaya kadar devamedebilir. Kalp atım hızı ve ritmiSiponimod, tedavi başlangıcında, kalp atım hızı ve atriyoventriküler iletimde geçici bir azalmaya neden olur (bkz. Bölüm 4.4 ve 4.8); bu etki, mekanik olarak, hücresel hiperpolarizasyona veazaltılmış uyarılabilirliğe yol açan, G-protein kenetli içe rektifiye potasyum (GIRK)kanallarının aktivasyonu ile ilişkilidir. S1P1 reseptörlerindeki fonksiyonel antagonizminedeniyle, siponimodun ilk titrasyonu, idame dozu elde edilene kadar GIRK kanallarını ardışıkolarak duyarsızlaştırır. QTaralısını uzatma potansiyeliSiponimodun terapötik (2 mg) ve supraterapötik (10 mg) dozlarının kardiyak repolarizasyon üzerindeki etkileri ayrıntılı bir QT çalışmasında araştırılmıştır. Sonuçlar siponimod ile QTuzaması ile ilişkili bir aritmojenik potansiyel ortaya koymamıştır. Siponimod, doz sonrası 3 saatsonra plaseboya göre düzeltilmiş başlangıca ayarlı ortalama QTcF'yi (AAQTcF) 5 ms'den fazlaartırmıştır ve maksimum ortalama etki sırasıyla 7,8 ms (2 mg) ve 7,2 ms (10 mg) olmuştur.Bütün zaman noktalarında AAQTcF için tek taraflı %95 güven aralığının üst sınırı 10 ms'ninaltında kalmıştır. Kategorik analiz, tedaviden kaynaklanan 480 ms'nin üzerinde QTc değerleriolmadığını, başlangıca kıyasla 60 ms'den daha fazla QTc artışı olmadığını ve düzeltilmiş veyadüzeltilmemiş QT/QTc değerinin 500 ms'yi aşmadığını ortaya koymuştur. Pulmoner _28 gün boyunca tek veya çoklu dozlarla siponimod tedavisi, zorlu vital kapasitenin %25 ila 75'inin (FEF %25-75) ekspirasyonu sırasında 1 saniyede zorlu ekspiratuvar hacim (FEVı) vezorlu ekspiratuvar akış (FEF) ile ölçüldüğü üzere, havayolu direncinde klinik olarak anlamlıartışlarla ilişkili değildir. Terapötik olmayan tekli dozlarda (> 10 mg) hafif bir azaltılmış FEVıeğilimi tespit edilmiştir. Birden fazla siponimod dozu, %FEVı ve FEF%25-75'te, doza ve gündüzsaatlerine bağımlı olmayan ve artmış hava yolu direncine dair herhangi bir klinik bulgu ileilişkili olmayan hafif ila orta değişiklikler ile ilişkilendirilmiştir. Klinik etkililik ve güvenlilikSiponimodun etkililiği, SPMS hastalarında günde bir kez 2 mg'lık dozların değerlendirildiği bir faz III çalışmasında araştırılmıştır. SPMS'de A2304 (EXPAND) çalışmasıÇalışma A2304, relapsların yokluğunda veya relapslardan bağımsız olarak önceki 2 yıl içinde belgelenmiş progresyon kanıtı olan, çalışmaya kayıt öncesindeki 3 ay içinde relaps kanıtıbulunmayan ve çalışmaya girdiği tarihte ortanca Genişletilmiş Özürlülük Durum Ölçeği (EDSS)skoru 3,0 ila 6,5 olan SPMS hastalarıyla yürütülen randomize, çift kör, plasebo kontrollü, olayve takip süresi güdümlü, bir faz III bir çalışmasıdır. Başlangıçta ortalama EDSS 6,0'dır. 61 yaşüstü hastalar dahil edilmemiştir. Hastalık aktivitesi ile ilgili olarak, SPMS'de enflamatuvaraktivitenin karakteristik özellikleri relaps veya görüntülemeyle ilişkili olabilir (yani Gd tutan T1lezyonları veya aktif [yeni veya genişleyen] T2 lezyonları). Hastalar, günde bir kez siponimod 2 mg veya plasebo almak üzere 2:1 oranında randomize edilmiştir. Taramada ve her 3 ayda bir ve relaps zamanında klinik değerlendirmelergerçekleştirilmiştir. MRG değerlendirmeleri taramada ve 12 ayda bir yürütülmüştür. Çalışmanın birincil sonlanım noktası, EDSS'de 3 ay boyunca devam eden başlangıçtan en az 1 puanlık artış (başlangıç EDSS'si > 5,5 olan hastalar için 0,5 puanlık artış) olarak tanımlanan 3aylık doğrulanmış özürlülük progresyonunun (CDP) zamanı olmuştur. Başlıca ikincil sonlanımnoktaları, zamanlı 25-adım yürüme testinde (T25W) 3 ayda doğrulanmış başlangıca kıyasla enaz %20 kötüleşme ve T2 lezyon hacminde başlangıca kıyasla değişim olmuştur. Ek ikincilsonlanım noktaları arasında, 6 aylık CDP'ye kadar geçen süre, beyin hacminde yüzde değişimve enflamatuvar hastalık aktivitesi (yıllık nüks oranı, MRG lezyonları) ölçümleri yer almıştır.Sembol Sayı Modalite Testi skorundaki bilişsel işlem hızındaki değişim keşifsel bir sonlanımnokta olarak olmuştur. Çalışma süresi her hasta için değişken olmuştur (ortanca çalışma süresi 21 ay, dağılım: 1 gün ila 37 ay). Çalışma, 1.651 hastanın siponimod 2 mg (N = 1.105) veya plaseboya (N = 546) randomize edilmesini içermiştir; siponimod ile tedavi edilen hastaların %82'si ve plasebo ile tedavi edilenhastaların %78'i çalışmayı tamamlamıştır. Başlangıçta ortanca yaş 49, ortanca hastalık süresi16 yıl ve ortanca EDSS skoru 6,0 olmuştur. Hastaların %64'ünde çalışma girişinden önceki 2yıl içinde relaps görülmemiştir ve %76'sında başlangıç MRG taramasında gadolinyum (Gd)tutan lezyon yoktur. Hastaların %78'i daha önce MS'leri için bir tedavi görmüştür. 3 aylık ve 6 aylık CDP'nin başlama zamanı, siponimod için anlamlı olarak daha geç olmuş, 3 aylık CDP riskinde plasebo ile karşılaştırıldığında %21 azalma (tehlike oranı [HR] 0,79, p =0,0134) ve 6 aylık CDP riskinde plasebo ile karşılaştırıldığında %26'lık azalma (HR 0,74, p =0,0058) izlenmiştir. Şekil 1 EDSS-Kaplan-Meier eğrilerine dayalı 3 aylık ve 6 aylık CDP'li hastalar (tam analiz seti, çalışma A2304)
Plasebo karşısında 6 aylık CDP'ye kadar geçen sürefi
Q ü «10-
Çalışmadan elde edilen sonuçlar, 3 aylık ve 6 aylık CDP'ye kadar geçen sürede, cinsiyet, yaş, çalışma öncesi relaps aktivitesi, başlangıç MRG hastalık aktivitesi, hastalık süresi ve başlangıçtaözürlülük düzeylerine dayalı tanımlı alt gruplarda siponimod ile değişken fakat istikrarlı bir riskazalması göstermiştir. Aktif hastalığı olan hasta alt grubunda (n = 779) (çalışmadan önceki 2 yıl içinde relapsı olan hastalar ve/veya başlangıçta Gd tutan T1 lezyonlarının varlığı olarak tanımlanır) tedavibaşlangıcındaki özellikler genel popülasyona benzer olmuştur. Ortanca yaş 47, ortanca hastalıksüresi 15 yıl ve başlangıçtaki ortanca EDSS skoru 6,0'dır. Siponimod ile tedavi edilen aktif hastalıklı hastalarda 3 aylık ve 6 aylık CDP'nin başlama zamanı, plaseboya kıyasla sırasıyla %31 (tehlike oranı [HR] 0,69; %95 GA: 0,53, 0,91) ve %37 (HR 0,63;%95 Cl: 0,47, 0,86) oranlarında daha geç olmuştur. ARR (doğrulanmış relapslar) plasebo ilekarşılaştırıldığında %46 azalmıştır (ARR oranı 0,54; %95 GA: 0,39, 0,77). 24 ay boyunca Gdtutan T1 ağırlıklı lezyonların kümülatif sayısında bağıl oran azalması, plaseboya kıyasla %85olmuştur (vuku oranı 0,155; %95 GA: 0,104, 0,231). T2 lezyon hacmi değişimi ve beyin hacmideğişim yüzdesindeki (12 ve 24 aylık ortalamalar) plaseboya kıyasla farklar sırasıyla -1163 mm3(%95 GA: -1484, -843 mm3) ve %0,141 (%95 GA: 0,020, %0,261) bulunmuştur.
Riskli hasta sayısıO Siponimod O Pİ2ceİKiSiponirrtDdPlueba5162652541154472255911715120 4â 1514964 245112376164439221516»5 Siponimod (N=516|PlacebojN=265]Hastalık aktivitesi belirtileri ve semptomları olmayan (çalışmadan önceki 2 yıl içinde relapsı olmayan hastalar ve/veya başlangıçta Gd tutan T1 lezyonları olmayan hastalar olarak tanımlanır)hasta alt grubunda (n = 827), 3 aylık ve 6 aylık CDP üzerindeki etkiler küçük olmuştur (riskazalmaları sırasıyla %7 ve %13). 5.2 Farmakokinetik özelliklerGenel özelliklerEmilim:Siponimodun çoklu oral uygulamasından sonra maksimum plazma konsantrasyonlarına (Cmaks) ulaşma süresi yaklaşık 4 saattir (aralık: 2 ila 12 saat). Siponimod emilimi geniştir (> %70, idrarlaatılan radyoaktivite miktarına ve dışkıdaki sonsuzluğa ekstrapole edilen metabolit miktarına bağlıolarak). Siponimodun mutlak oral biyoyararlanımı yaklaşık %84'tür. 10 gün boyunca günde birkez verilen 2 mg siponimod için, 10. günde ortalama 30,4 ng/ml Cmaks ve ortalama 558 h*ng/ml 20 EAAtau değerleri gözlenmiştir. Siponimodun günde bir kez çoklu uygulamalarından sonra kararlı duruma yaklaşık 6 gün sonra ulaşılmıştır. Tmaks'ta tek bir dozdan sonra 8 saatlik bir gecikmeye rağmen, gıda alımının siponimodun sistemik maruziyeti (Cmaks ve EAA) üzerinde hiçbir etkisi olmamıştır, bu nedenle siponimod yemeklerdenbağımsız olarak alınabilir (bkz. Bölüm 4.2). Dağılım:Siponimod vücut dokularına ortalama 124 litre dağılım hacmi ile dağıtılır. Plazmada bulunan siponimod fraksiyonu insanlarda %68'dir. Siponimod kan-beyin bariyerini kolaylıkla geçer.Siponimodun proteinlere bağlanması sağlıklı kişilerde ve karaciğer veya böbrek yetmezliği olanhastalarda >%99,9'dur. Biyotransformasyon:Siponimod, esas olarak sitokrom P450 2C9 (CYP2C9) (%79,3) ve daha az bir ölçüde sitokrom P450 3A4 (CYP3A4) (%18,5) tarafından geniş ölçüde metabolize edilir. Ana metabolitler M3 ve M17'nin farmakolojik aktivitesinin, insanlarda siponimodun klinik etkisine ve güvenliliğine katkıda bulunması beklenmemektedir. In vitro araştırmalar, siponimod ve onun ana sistemik metabolitleri M3 ve M17'nin, araştırılan tüm CYP enzimleri ve taşıyıcıları için günde bir kez 2 mg'lık terapötik dozda klinik olarakanlamlı herhangi bir ilaç-ilaç etkileşim potansiyeli göstermediğini ve klinik araştırmagerektirmediğini göstermiştir. CYP2C9 polimorfiktir ve genotip, iki oksidatif metabolizma yolunun genel eliminasyona fraksiyonel katkılarını etkiler. PBPK modellemesi, CYP2C9 genotipine bağımlı diferansiyel birinhibisyon ve CYP3A4 yolaklarının indüksiyonunu göstermektedir. İlgili genotiplerde CYP2C9metabolik aktivitesi daha az olduğundan, CYP3A4 ile etkileşen maddelerin siponimod maruziyetiüzerinde daha büyük bir etkisinin olması beklenmektedir (bkz. Bölüm 4.5). Eliminasyon:MS hastalarında 3,11 l/saatlik görünür bir sistemik klirens (CL/F) hesaplanmıştır. Siponimodun görünen eliminasyon yarılanma ömrü yaklaşık 30 saattir. Siponimod, esas olarak metabolizma ve daha sonra safra/feçal atılım yoluyla sistemik dolaşımdan elimine edilir. İdrarda değişmemiş siponimod saptanmamıştır. Doğrusallık / doğrusal olmayan durum:Siponimod konsantrasyonu, 0,3 mg ila 20 mg/gün arasındaki çoklu siponimod dozlarından sonra dozla orantılı bir şekilde artar. 6 gün süreyle günde bir kez doz uygulamasından sonra kararlı durum plazma konsantrasyonlarına ulaşılır ve kararlı durum düzeyleri başlangıç dozundan yaklaşık 2 ila 3 kat daha yüksektir. 6 günsonra 2 mg siponimod klinik terapötik dozuna ulaşmak için bir titrasyon rejimi kullanılır vekararlı durum plazma konsantrasyonlarına ulaşmak için 4 gün daha dozlama gerekir. Belirli gruplardaki veya özel popülasyonlardaki hastalardaki karakteristik özelliklerCYP2C9 genotipiCYP2C9 genotipi siponimod CL/F'yi etkiler. İki popülasyon farmakokinetik analizi CYP2C9*1*1 ve*1*2 olgularının büyük ölçüde metabolize edici,*2*2 ve*1*3 olgularının orta 21 düzeyde metabolize edici ve *2*3 ve*3*3 olgularının zayıf metabolize edici olarak davrandığını göstermiştir. CYP2C9*1*1 olgularıyla karşılaştırıldığında, CYP2C9*2*2,*1*3,*2*3 ve*3*3genotipleri olan bireylerde CL/F değerleri sırasıyla %20, %35-38, %45-48 ve %74 dahadüşüktür. Dolayısıyla siponimod maruziyeti, CYP2C9*2*2,*1*3,*2*3 ve*3*3 olgulardasırasıyla %25, %61, %91 ve %284 daha yüksektir. (bkz. Tablo 4) (bkz. bölüm 4.2 ve 4.4). CYP2C9 için daha az sıklıkta meydana gelen başka polimorfizmler de vardır. Siponimodun farmakokinetiği bu tür olgularda değerlendirilmemiştir. *5, *6, *8 ve *11 gibi bazıpolimorfizmler, enzim fonksiyonunun azalması veya kaybı ile ilişkilidir. CYP2C9 *5, *6, *8 ve*11 allellerinin Afrika kökenli popülasyonlarda yaklaşık %10, Latinler/Hispaniklerde %2 veBeyazlar ve Asyalılarda <%0.4 birleşik sıklığa sahip olduğu tahmin edilmektedir.
Yaşlı hastalarPopülasyon farmakokinetiğinin sonuçları, yaşlı hastalarda (65 yaş ve üstü) doz ayarlamasının gerekli olmadığını göstermektedir. Klinik çalışmalara 61 yaşın üzerindeki hasta dahiledilmemiştir. Siponimod yaşlılarda dikkatli kullanılmalıdır (bkz. Bölüm 4.2). CinsiyetPopülasyon farmakokinetiğinin sonuçları, cinsiyete dayalı doz ayarlamasının gerekli olmadığını göstermektedir. Irk/etnik kökenTek doz farmakokinetik parametreler, sağlıklı Japon ve Beyaz bireyler arasında farklılık göstermemiştir ki bu da siponimodun farmakokinetiği üzerinde etnik duyarlılığın olmadığınıgösterir. Böbrek yetmezliğiHafif, orta veya şiddetli böbrek yetmezliği olan hastalarda siponimod doz ayarlamasına gerek yoktur. Ortalama siponimod yarılanma ömrü ve Cmaks (toplam ve plazma proteinlerinebağlanmamış), ciddi böbrek yetmezliği olan hastalar ile sağlıklı kişiler arasında benzer olmuştur. Toplam ve bağlanmamış EAA'lar sağlıklı olgulara kıyasla sadece biraz artmıştır (%23-33). Son dönem böbrek yetmezliği veya hemodiyalizin siponimodun farmakokinetiği üzerindeki etkileriaraştırılmamıştır. Siponimodun yüksek plazma protein bağlanmasına (>%99,9) bağlı olarak,hemodiyalizin toplam ve bağlanmamış siponimod konsantrasyonunu değiştirmesi beklenmez vebu hususlara dayanarak herhangi bir doz ayarlaması öngörülmez. Karaciğer yetmezliğiSiponimod, ciddi karaciğer yetmezliği olan hastalarda kullanılmamalıdır (bkz. Bölüm 4.3). Hafif veya orta şiddette karaciğer yetmezliği olan hastalarda siponimod için doz ayarlamasına gerekyoktur. Bağlanmamış siponimod farmakokinetiği (EAA), incelenen 0,25 mg tek doz için sağlıklıolgulara kıyasla, orta ve şiddetli karaciğer yetmezliği olanlarda sırasıyla %15 ve %50 dahayüksektir. Siponimodun ortalama yarı ömrü karaciğer yetmezliğinde değişmemiştir. 5.3 Klinik öncesi güvenlilik verileriFarelerde, sıçanlarda ve maymunlarda tekrarlı doz toksisite çalışmalarında, siponimod, lenfoid sistemi (lenfopeni, lenfoid atrofi ve azaltılmış antikor tepkisi) belirgin şekilde etkilemiş olup bu,S1P1 reseptörlerindeki birincil farmakolojik aktivitesi ile tutarlıdır (bkz. Bölüm 5.1). Hayvan türlerindeki doz sınırlayıcı toksisiteler, farelerde nefrotoksisite, sıçanlarda vücut ağırlığı gelişimi ve maymunlarda advers SSS ve gastrointestinal etkiler olmuştur. Kemirgenlerdekitoksisitenin ana hedef organları akciğer, karaciğer, tiroid, böbrek ve rahim/vajinadır.Maymunlarda ayrıca kas ve cilt üzerindeki etkiler gözlenmiştir. Bu toksisiteler, 2 mg/gün idamedozunda EAA bazında insan maruziyetinden 30 kat daha yüksek sistemik siponimoddüzeylerinde gelişmiştir. Siponimod herhangi bir fototoksik veya bağımlılık potansiyeli göstermemiştir ve in vitro ve in vivo koşullarda genotoksik etki sergilememiştir. KarsinojeniteKarsinojenite araştırmalarında, siponimod, farelerde lenfoma, hemanjiyom ve hemanjiyosarkomu indüklerken erkek sıçanlarda foliküler adenom ve tiroid bezinin karsinomusaptanmıştır. Bu tümör bulguları ya fareye özgü olarak kabul edilmiştir ya da özellikle hassasolan sıçan türlerinde metabolik karaciğer adaptasyonlarına atfedilebilirdir ve insanlar açısındanilgisi şüphelidir. Fertilite ve üreme toksisitesiSiponimod, günlük 2 mg dozda insan sistemik maruziyetine (EAA) dayalı olarak yaklaşık 19 kat güvenlilik marjına karşılık gelen, test edilen en yüksek doza kadar sıçanlarda erkek ve dişidoğurganlığını etkilememiştir. Siponimoddan etkilenen reseptörün (sfenosin-1-fosfat reseptörü) embriyogenez sırasında vasküler oluşumda rol oynadığı bilinmektedir. Sıçan ve tavşanlarda yapılan embriyofetal gelişim çalışmalarında siponimod, maternal toksisite olmaksızın embriyotoksik etkilere neden olmuştur. Her iki türde de doğum öncesi mortaliteartmıştır. Sıçanlarda dış, iskelet ve iç organ malformasyonları olan (örn. yarık damak ve şekilsizklaviküller, kardiyomegali ve ödem) daha fazla sayıda fetüs kaydedilirken, tavşan fetüslerindeağırlıklı olarak iskelet ve iç organ varyasyonları gözlenmiştir. Sıçanlarda yapılan prenatal ve postnatal gelişim çalışmasında, daha yüksek ölü yavru (ölü doğan veya doğum sonrası 4. günden önce ölü bulunan) ve malforme yavru (ürogenital malformasyonları ve/veya artmış anogenital mesafeli olan erkek yavrular; her iki cinsiyetten ödemli, şişmiş yumuşak kafataslı veya bükülmüş arka ayaklı yavrular) sayısı gözlenmiştir.Embriyofetal (sıçanlar ve tavşanlar) ve pre/postnatal (sıçanlar) gelişim için ilgili NOAEL'lerdemaruziyet düzeyleri (EAA), günlük 2 mg'lık bir dozda insan sistemik maruziyetinin (EAA)altında olmuştur ve dolayısıyla herhangi bir güvenlik payı bulunmamaktadır. 6. FARMASÖTİK BİLGİLER6.1 Yardımcı maddelerin listesiLaktoz monohidrat (sığır kaynaklı) Mikrokristalin selüloz Krospovidon (Tip A) Gliserol dibehenat Kolloidal susuz silika Polivinil alkol (kısmen hidrolize) Titanyum dioksit (E171) Kırmızı demir oksit (E172) Siyah demir oksit (E172) Talk Soya lesitini (E322) Ksantan sakızı 6.2 GeçimsizliklerGeçerli değildir. 6.3 Raf ömrüÜrün açılmamış olarak buzdolabında (2-8°C'de) saklanmak koşulu ile 24 ay raf ömrüne sahiptir. Ürün açıldıktan sonra 25°C'nin altındaki oda sıcaklığında 3 ay saklanabilir. 6.4 Saklamaya yönelik özel tedbirler2 °C - 8 °C arasında buzdolabında muhafaza ediniz. Ürün açıldıktan sonra 25°C'nin altındaki oda sıcaklığında 3 ay saklanabilir. 6.5 Ambalajın niteliği ve içeriğiCüzdan içerisinde 12 film kaplı tablet içeren PA/alu/PVC/alu blister ambalajlarda titrasyon paketleri. 84 ve 120 film kaplı tablet içeren PA/alu/PVC/alu blister ambalajlarda paketler. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİFarmanova Sağlık Hizmetleri Limited Şirketi Kavacık/Beykoz/İstanbul 8. RUHSAT NUMARASI2022/437 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 12.08.2022 Ruhsat yenileme tarihi: - 10. KÜB'ÜN YENİLEME TARİHİ |
İlaç BilgileriAdmiraz 0,25 Mg Film Kaplı TabletEtken Maddesi: Siponimod Fumarik Asit Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.