Imatis 400 Mg Flm Tablet Kısa Ürün BilgisiAntineoplastik ve İmmünomodülatör Ajanlar KISA ÜRÜN BİLGİLERİ1. BEŞERİ TIBBİ ÜRÜNÜN ADI İMATİS 400 mg film tablet 2. KALİTATİF VE KANTİTATİF BİLEŞİM Etkin madde: Bir film tablet, 400 mg imatinib (mesilat tuzu olarak) içerir. Yardımcı maddeler: Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORM Film tablet. Çok koyu sarı ile kahverengimsi turuncu renkli oval film kaplı tabletler. 4. KLİNİK ÖZELLİKLER 4.1. Terapötik endikasyonlar İMATİS'in endikasyonları: Yeni tanı konmuş Philadelphia kromozomu pozitif kronik faz kronik miyeloid lösemi (KML) hastalarında, Akselere faz Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML) hastalarında, Blastik faz Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML) hastalarında, Diğer tedavilere dirençli Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML) hastalarında, İlk tanısı Philadelphia kromozomu pozitif kronik miyeloid lösemi (KML) olan ancak tedavi ile Philadelphia kromozomu negatif hale gelen kronik/akselere/blastik faz kronik miyeloid lösemi hastalarında, Kronik miyeloid lösemili (KML) olan 3 yaş ve üzerindeki çocuklarda birinci basamak tedavide, FIPILl-PDGFRA füzyon geni laboratuvar incelemeleriyle gösterilen hipereozinofilik sendrom ve sistemik mastositoz hastalarında kullanılabilir. 4.2 Pozoloji ve uygulama şekli Pozoloji/uygulama sıklığı ve süresi: Tedavi, KML, HES ve SM hastalıklarının tedavisinde deneyimi olan bir doktor tarafından başlatılmalıdır. Tedavi, hasta yarar sağladığı sürece devam ettirilmelidir. Kronik Miyeloid Lösemi de (KML) Dozaj Kronik faz KML bulunan hastalar için önerilen İMATİS dozajı 400 mg/gün, hızlanmış faz ya da blast krizi bulunanlar için önerilen dozaj ise 600 mg/gün'dür. İlaca bağlı oluşan ciddi advers etki ve ağır lösemiyle ilişkili nötropeni veya trombositopeni gelişmemiş olması koşuluyla, hastalığın ilerlemesi (herhangi bir zamanda), en az 6 aylık tedaviden sonra tatmin edici bir hematolojik yanıt alınamaması, 12 aylık tedaviye rağmen sitogenetik cevap elde edilmemesi veya daha önce elde edilmiş olan hematolojik ve/veya sitogenetik yanıtın kaybolması gibi durumlarda; kronik fazda hastalık bulunanlarda dozun 400 mg'dan 600 mg'a yükseltilmesi, ya da hızlanmış faz veya blast krizi bulunan hastalarda da dozun 600 mg'dan maksimum 800 mg günlük doza yükseltilmesi düşünülebilir. Hipereozinofilik sendrom (HES) ve sistemik mastositozda (SM) dozaj Yetişkin hipereozinofilik sendrom ve sistemik mastositoz hastalarında önerilen İMATİS dozajı, günde 100 mg'dır. Yanıtsız hallerde 400 mg'a dek çıkılabilir. Bu doz aşılamaz. Advers reaksiyonlar için doz ayarlamalarıHematolojik olmayan advers reaksiyonlarİMATİS kullanıldığında eğer ciddi hematolojik olmayan advers reaksiyon gelişirse, tedavi bu olay ortadan kalkıncaya kadar durdurulmalıdır. Daha sonra, olayın ilk ciddiyetine göre değişecek şekilde tedavi devam ettirilir. Eğer bilirubin, normal sınırın üst limitini (NSÜL) 3 kattan fazla aşacak şekilde yükselirse ya da karaciğer transaminazlarında NSÜL değerinin 5 katından fazla artış olursa, İMATİS, bilirubin düzeyleri <1.5 x NSÜL ve transaminaz düzeyleri <2.5 x NSÜL seviyesine ininceye kadar durdurulmalı ve daha sonra da azaltılmış günlük dozlarla devam ettirilmelidir. Yetişkinlerde doz 400 mg'dan 300 mg'a veya 600 mg'dan 400 mg'a veya 800 mg'dan 600 mg'a, çocuklarda ise 260 mg/m2/gün'den 200 mg/m2/gün'e veya 340 mg/m2/gün'den 260 mg/m2/gün'e düşürülmelidir. Hematolojik advers reaksiyonlar Ağır nötropeni ve trombositopeni geliştiği takdirde dozun azaltılması ya da tedavinin kesilmesi aşağıdaki tabloda belirtildiği şekilde düzenlenmelidir. Tablo 1 Nötropeni ve trombositopeni için doz ayarlamaları
krizi aANC<0.5x109/l ve/veya Trombositler <10 xl09/l 1. Sitopeninin lösemiye bağlı olup olmadığını kontrol edin (kemik iliği aspiratı ya da biyopsisi) 2. Eğer sitopeni lökopeniye bağlı değil ise İMATİS dozunu 400 mg'a düşürünb 3. Eğer sitopeni 2 hafta devam ederse, dozu 300 mg'a düşüründ 4. Eğer sitopeni 4 hafta devam ederse ve hala lösemiyle ilişkili değil ise ANC >1 x 109/l ve trombositler >20 x 109/l oluncaya kadar İMATİS'i durdurun ve daha sonra 300 mg ile tedaviye başlayınd ANC = mutlak nötrofil sayısı aen az 1 aylık tedaviden sonra ortaya çıkan b veya çocuklarda 260 mg/m2 c veya çocuklarda 340 mg/m2 d veya çocuklarda 200 mg/m2 Uygulama şekli: Reçetedeki doz, gastrointestinal etkileri en aza indirmek için yemek sırasında ve büyük bir bardak suyla yutulmalıdır. Günde 400 veya 600 miligramlık dozlar bir defada, 800 miligramlık ise her birinde 400'er miligram olmak üzere sabah ve akşam iki bölümde alınmalıdır. Film-kaplı tabletleri yutamayan hastalarda tablet, bir bardak suda veya elma suyunda dağıtılabilir. İhtiyaç duyulan sayıda tablet, uygun hacimde içeceğin (100 miligramlık tablet için yaklaşık 50, 400 miligramlık tablet için yaklaşık 200 mL) içerisine konarak bir kaşıkla karıştırılır. Meydana gelen süspansiyon, tablet(ler)in tam olarak dağılmasından sonra derhal içilmelidir. Özel popülasyonlara ilişkin ek bilgiler: Karaciğer yetmezliği: İmatinib, temel olarak karaciğer yoluyla metabolize olur. Hafif, orta şiddette veya şiddetli karaciğer fonksiyon bozukluğu olan hastalara, önerilen minimal doz olan günde 400 mg verilmelidir. Bu doz, tolere edilemediği takdirde azaltılabilir (bkz. bölüm 4.4 Özel kullanım uyarıları ve önlemleri, 4.8 İstenmeyen etkiler, 5.1 Farmakodinamik özellikler ve 5.2 Farmakokinetik özellikler). Böbrek yetmezliği: İmatinib ve metabolitleri böbrek yoluyla önemli miktarda atılmazlar. Böbrek yetmezliği olan veya diyaliz uygulanan hastalara böbrek bozukluğu başlangıç dozu olarak, önerilen minimum doz, günlük 400 mg verilebilir. Bununla birlikte, bu hastalarda dikkatli olunması önerilir. Tolere edilememesi halinde doz azaltılabilir ya da etki görülmemesi halinde doz arttırılabilir (bkz 4.4 Özel kullanım uyarıları ve önlemleri). Pediyatrik popülasyon: İMATİS'in KML endikasyonunda 2 yaşın altındaki çocuklarda kullanımıyla ilgili herhangi bir deneyim bulunmamaktadır. İMATİS'in diğer endikasyonlarda 3 yaşından küçük çocuklarda kullanılması konusundaki tecrübelerimiz çok sınırlıdır. Çocuklarda doz uygulaması vücut yüzey alanını (mg/m2) temel almalıdır. Kronik faz ve ilerlemiş faz KML'si olan çocuklar için 340 mg/m2 günlük doz önerilmektedir (toplam doz günde 600 mg'ı geçmemelidir). Tedavi günde bir kere doz uygulaması yoluyla verilebilir ya da alternatif olarak günlük doz iki uygulamaya bölünebilir - sabah bir ve akşam bir (bkz. Bölüm 5.1). Geriyatrik popülasyon: Yaşlılarda spesifik olarak imatinib farmakokinetiği araştırılmamıştır. Katılan hastaların % 20'sinden fazlasının 65 ve daha yukarı yaşlarda olduğu klinik çalışmalarda, yetişkin hastalarda yaşla ilişkili anlamlı farmakokinetik farklılıklar gözlenmemiştir. Yaşlılarda, özel bir doz önerisi gerekli değildir. 4.3. Kontrendikasyonlar Aktif maddeye veya eksipiyanlardan herhangi birine karşı aşırı duyarlılık. 4.4. Özel kullanım uyarıları ve önlemleri İMATİS, başka ilaçlarla eşzamanlı olarak kullanıldığında önemli ilaç etkileşimleri görülme potansiyeli bulunmaktadır. İMATİS, rifampisin ya da diğer güçlü CYP3A4 indükleyicileri, ketokonazol ya da diğer güçlü CYP3A4 inhibitörleri, dar bir terapötik penceresi olan CYP3A4 substratları (örn. siklosporin ya da pimozid) ya da dar bir terapötik penceresi olan CYP2C9 substratları (örn. varfarin ya da diğer kumarin türevleri) ile birlikte alındığında dikkatli olunmalıdır (bkz. Bölüm 4.5). Ateş düşürücü olarak düzenli aralıklarla parasetamol (asetaminofen) alan bir hastanın akut karaciğer yetmezliği nedeniyle öldüğü bildirilmiştir. Etiyolojisi tam olarak bilinmemekle beraber, parasetamol/asetaminofen kullanımında dikkatli olunmalıdır (bkz. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri). Hipotiroidism: İmatinib tedavisi sırasında levotiroksin replasmanı yapılan tiroidektomi hastalarında klinik hipotiroidizm olguları bildirilmiştir. Bu tür hastalarda Tiroid Stimulan Hormon (TSH) düzeyleri yakından izlenmelidir. Hepatotoksisite: Karaciğer disfonksiyonu (hafif, orta şiddette ve şiddetli) olan hastalarda, periferik kan sayımları ve karaciğer enzimleri dikkatli bir şekilde izlenmelidir (bkz 4.2 Pozoloji ve uygulama şekli, 4.8 İstenmeyen etkiler, 5.1 Farmakodinamik özellikler, 5.2 Farmakokinetik özellikler). Sıvı retansiyonu: İmatinib alan yeni tanı konulmuş KML hastalarının yaklaşık % 2.5'inde ciddi sıvı retansiyonu (plevra efüzyonu, ödem, pulmoner ödem, asit, yüzeysel ödem) ortaya çıktığı bildirilmiştir. Bu nedenle, hastalarda düzenli aralıklarla kilo kontrolü önerilir. Beklenmedik, ani bir kilo artışı dikkatli araştırılmalı ve gerektiğinde uygun destek tedavisi uygulanmalı ve terapötik önlemler alınmalıdır. Klinik çalışmalarda, yaşlı hastalarda ve daha önceden kardiyak hastalık hikayesi bulunanlarda bu olayların insidanslarının arttığı saptanmıştır. Kalp hastalığı ya da böbrek yetmezliği olan hastalar: Kalp hastalığı, kalp yetmezliği açısından risk faktörleri bulunan veya böbrek yetmezliği hikayesi olan hastalar dikkatlice takip edilmeli, kalp veya böbrek yetmezliğini düşündüren belirti ve semptomları olan her hasta değerlendirilmeli ve tedavi edilmelidir. Miyokardiyum içinde HES hücrelerinin gizli sızdırmasının görüldüğü hipereozinofili sendromu (HES) olan hastalarda izole kardiyojenik şok/sol ventrikül disfonksiyonu, imatinib tedavisine başlanmasıyla beraber oluşan HES hücre degranülasyonu ile ilişkilendirilmiştir. Bu durumun sistemik steroidler kullanılarak, dolaşımı destekleyen önlemler alarak ve imatinib tedavisini geçici olarak durdurarak düzeltilebileceği bildirilmiştir. Miyelodisplastik/miyeloproliferatif hastalıklar (MDS/MPD) ve sistemik mastositoz yüksek eozinofil düzeyleri ile ilişkili olabilir. Bu nedenle, eozinofil düzeylerinin yüksek olduğu MDS/MPD vakalarında, SM vakalarında ve HES vakalarında ekokardiyografik inceleme yapılmalı ve serum troponin düzeyleri ölçülmelidir. Bunlardan birinde anormallik tespit edilirse tedavi başlangıcında imatinible birlikte 1-2 hafta boyunca 1-2 mg/kg dozunda sistemik steroid kullanılması düşünülmelidir. Tümör lizis sendromu: İmatinib ile tedavi edilen hastalarda tümör lizis sendromu (TLS) vakaları bildirilmiştir. TLS meydana gelme olasılığı nedeniyle, İMATİS başlatılmadan önce klinik açıdan anlamlı dehidrasyonun düzeltilmesi ve yüksek ürik asit düzeylerinin tedavisi önerilmektedir (bkz. Bölüm 4.8). Laboratuvar testleri: İMATİS ile tedavi sırasında düzenli olarak tam kan sayımları yapılmalıdır. KML hastalarında imatinib tedavisine, nötropeni ya da trombositopeni eşlik etmiştir. Bununla birlikte, bu sitopenilerin ortaya çıkışı, hastalığın tedavi edildiği evreye bağlıdır ve kronik fazda KML bulunan hastalarla karşılaştırıldığında, hızlanmış fazda KML ya da blast krizinde bulunan hastalarda daha sık olmaktadır. 4.2 Pozoloji ve uygulama şekli bölümünde önerildiği gibi İMATİS tedavisi kesilebilir ya da dozu azaltılabilir. İMATİS alan hastalarda karaciğer fonksiyonu (transaminazlar, bilirubin, alkalin fosfataz) düzenli olarak takip edilmelidir. 4.2 Pozoloji ve uygulama şekli, Hematolojik olmayan advers reaksiyonlar bölümünde önerildiği gibi bu laboratuvar anormallikleri, İMATİS tedavisi kesilerek ve/veya dozu azaltılarak kontrol edilmelidir. İmatinib ve metabolitleri böbrek yoluyla önemli bir miktarda atılmazlar. Kreatinin klerensinin (KrCL) yaşla birlikte azaldığı bilinmektedir ve yaş imatinib kinetiğini anlamlı olarak etkilememektedir. Böbrek fonksiyonları bozuk hastalarda, imatinib plazma maruziyeti böbrek fonksiyonları normal hastalardakinden daha yüksek görünmektedir (muhtemelen, imatinib-bağlayıcı bir protein olan alfa-asit glikoprotein (AGP) plazma düzeylerinin bu hastalarda daha yüksek olması nedeniyle). KrCL ölçümü ile sınıflandırıldığında, hafif (KrCL: 40-59 ml/dakika) ve şiddetli (KrCL: <20 ml/dakika) böbrek bozukluğu olan hastalar arasında maruz kalınan imatinib ile böbrek bozukluğunun derecesi arasında herhangi bir korelasyon yoktur. Ancak, 4.2 Pozoloji ve uygulama şekli bölümünde önerildiği gibi, tolere edilemezse imatinib başlangıç dozu düşürülebilir. Çocuklar ve ergenler: İmatinib kullanan çocuklar ve ergenlik öncesi dönemdeki kişilerde büyüme geriliği raporlanmıştır. Uzun süreli imatinib tedavisinin çocukların gelişimi üzerinde uzun dönem etkileri bilinmemektedir. Bu nedenle imatinib tedavisi gören çocukların yakından takip edilmesi önerilir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri İmatinibin plazma konsantrasyonlarını değiştiren ilaçlar İmatinibin plazma konsantrasyonlarını arttırabilen ilaçlar: Sitokrom P450 izoenzimlerinden CYP3A4 aktivitesini inhibe eden maddeler (örn. indinavir, lopinavir/ritonavir, ritonavir, sakinavir, telaprevir, nelfinavir ve boseprevir gibi proteaz inhibitörleri; ketokonazol, itrakonazol, posakonazol ve varikonazol gibi azol antifungal ajanlar; eritromisin, klaritromisin ve telitromisin gibi belirli makrolidler) metabolizmayı azaltabilir ve imatinib konsantrasyonlarını arttırabilirler. Sağlıklı deneklere tek doz ketokonazol (bir CYP3A4 inhibitörü) ile birlikte uygulandığında, imatinibe maruz kalma durumunda anlamlı bir artış ortaya çıkmıştır (imatinibin ortalama Cmaks ve EAA değerleri sırasıyla % 26 ve % 40 artmıştır). İMATİS, CYP3A4 izoziminin inhibitörleri ile birlikte verilirken dikkatli olunmalıdır. İmatinibin plazma konsantrasyonlarını azaltabilen ilaçlar: CYP3A4 aktivitesini uyaran maddeler (örn. deksametazon, fenitoin, karbamazepin, rifampisin, fenobarbital, fosfenitoin, pirimidon ya da St. John's Worth olarak da bilinen hypericum perforatum) eşzamanlı uygulama İMATİS'e maruz kalmayı azaltabilir. Sağlıklı 14 gönüllünün 8 gün boyunca günde 600 mg rifampisin kullanmasından sonra verilen, 400 miligramlık tek doz imatinib, oral doz klerensini 3.8 kat artırmıştır (%90 güven aralığı 3.5-4.5). Bu artış, ortalama Cmaks, EAA(0-24 saat) ve EAA (0-a,) değerlerinin, daha önce rifampin kullanılmamasına kıyasla sırasıyla %54, %68 ve %74 azalması demektir. Karbamazepin, okskarbazepin, fenitoin, fosfenitoin, fenobarbital ve primidon gibi enzim indükleyici anti-epileptik ilaçlar (AEİ) kullanırken imatinib ile tedavi edilen malign gliyomlu hastalarda da benzer sonuçlar gözlenmiştir. İmatinib için plazma EAA değeri, AEİ'ler kullanmayan hastalarla karşılaştırıldığında %73 oranında azalmıştır. Yayınlanmış iki çalışmada, imatinib ve St. John's Worth içeren bir ürünün birlikte uygulanması imatinib EAA değerinde %30-32'lik bir azalmaya yol açmıştır. Rifampin veya CYP3A4 indüksiyonu yapan diğer ilaçların kullanılmasına ihtiyaç olduğunda, enzim indüksiyon potansiyeli daha az olan, başka ilaçların kullanılması düşünülmelidir. İmatinib ile plazma konsantrasyonu değişebilen ilaçlar: İmatinib, simvastatinin (CYP3A4 substratı) ortalama Cmaks ve EAA değerlerini sırasıyla 2- ve 3.5 kat arttırmaktadır ve bu durum CYP3A4'ün imatinib tarafından inhibe edildiğini göstermektedir. Bu nedenle imatinib, dar bir terapötik pencereye sahip CYP3A4 substratlarıyla (örn. siklosporin, pimozid, takrolimus, sirolimus, ergotamin, diergotamin, fentanil, alfentanil, terfenadin, bortezomib, dosetaksel, kinidin) birlikte uygulandığında dikkatli olunmalıdır. İMATİS, diğer CYP3A4 tarafından metabolize edilen ilaçların da plazma konsantrasyonunu arttırabilir (örn. triazolo-benzodiazepinler, dihidropiridin kalsiyum kanal blokörleri, bazı HMG-KoA redüktaz inhibitörleri, örn. statinler, vs.). İmatinib, aynı zamanda in vitro olarak CYP2C9 ve CYP2C19 aktivitesini de inhibe etmektedir. Varfarin ile eşzamanlı uygulama sırasında protrombin zamanı (PZ) uzaması gözlenmiştir. Kumarinler verildiğinde İMATİS tedavisinin başında ve sonunda ve dozaj değiştirildiğinde kısa vadeli PZ takibi gereklidir. Alternatif olarak, düşük moleküler ağırlıklı heparin düşünülmelidir. İn vitro olarak İMATİS, CYP3A4 aktivitesini etkileyen konsantrasyonların benzeri konsantrasyonlarda sitokrom P450 izoenzimlerinden CYP2D6 aktivitesini de inhibe etmektedir. Günde iki kez 400 mg dozda uygulanan imatinibin CYP2D6-aracılı metoprolol metabolizması üzerinde zayıf bir inhibitör etkisi vardır; metoprolol Cmaks ve EAA değerleri yaklaşık %23 kadar artar. Metoprolol gibi CYP2D6 substratlarının imatinib ile birlikte uygulanması, ilaç-ilaç etkileşimi açısından bir risk faktörü olarak görünmemektedir ve doz ayarlaması gerekli olmayabilir. İmatinib in vitroin vivokoşullarda, 400 mg imatinib ve 1000 mg parasetamol uygulamasının ardından görülmemiştir. Daha yüksek imatinib ve parasetamol dozları çalışılmamıştır. Bu nedenle yüksek dozda İMATİS ve parasetamol eşzamanlı uygulanırken dikkatli olunmalıdır.4.6. Gebelik ve laktasyon Genel tavsiye Gebelik kategorisi D'dir. Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon) Çocuk doğurma potansiyeli bulunan kadınlara tedavi sırasında etkili bir kontrasepsiyon uygulamaları önerilmelidir. Yüksek düzeyde etkili kontrasepsiyon, düzenli ve doğru bir şekilde kullanıldığında düşük bir başarısızlık oranına yol açan (yani, yıl başına %1'den düşük) bir doğum kontrol yöntemidir. Gebelik dönemi Hayvanlar üzerinde yapılan araştırmalar üreme toksisitesinin bulunduğunu göstermiştir (bkz. 5.3 Klinik öncesi güvenlilik verileri). İmatinibin gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. İmatinib alan kadınlarda spontan düşükler ve bebekte konjenital anomalilerle ilgili pazarlama sonrası raporlar mevcuttur. İMATİS, beklenen fayda potansiyel riske ağır basmadığı sürece gebelik sırasında kullanılmamalıdır. Gebelik sırasında kullanılması durumunda, hastaya fötüs üzerindeki potansiyel riskleri hakkında bilgi verilmelidir. Laktasyon dönemi Hem imatinib, hem de aktif metaboliti anne sütüne geçebilir. Süt/plazma oranı imatinib için 0.5, metaboliti için ise 0.9 olarak saptanmıştır; bu da metabolitin süte daha büyük oranda geçtiğini düşündürmektedir. İmatinib ve metabolitinin toplam konsantrasyonu ve bebeklerin maksimum günlük süt alımı düşünüldüğünde, toplam maruziyetin düşük olması beklenir (bir terapötik dozun ~%10'u). Bununla birlikte, bebeğin imatinibe düşük dozlarda maruz kalmasının etkileri bilinmediğinden, İMATİS kullanan anneler bebeklerini emzirmemelidir. Üreme yeteneği/Fertilite İmatinib alan erkek hastalar ve ilacın erkek fertilitesi ve spermatogenezi üzerindeki etkileri ile ilgili insan çalışmaları yapılmamıştır. İMATİS tedavisi gören ve fertilite konusunda endişe duyan erkek hastalar hekimlerine danışmalıdırlar (bkz. Bölüm 5.3). 4.7. Araç ve makina kullanmaya etkisi İmatinib alan hastalarda motorlu araç kazaları bildirilmiştir. Bu raporların büyük kısmında sebebin imatinib olduğundan şüphelenilmemiş olsa da, hastalara imatinib ile tedavi sırasında baş dönmesi, somnolans ya da bulanık görme gibi istenmeyen etkiler yaşayabilecekleri bildirilmelidir. Bu nedenle, araba ya da araç kullanırken dikkatli olunması önerilmelidir. 4.8. İstenmeyen etkiler Güvenlilik profilinin özeti İnsanlardaki klinik kullanımda imatinibin genel güvenlilik profili, 12 yıldan fazla bir dönemi kapsayan imatinib deneyimiyle iyi bir şekilde karakterize edilmiştir. Klinik geliştirme sırasında hastaların büyük kısmı bir zaman noktasında advers olay yaşamıştır. En sık bildirilen ADR'ler (>%10) nötropeni, trombositeponit, anemi, baş ağrısı, dispepsi, ödem, kilo artışı, mide bulantısı, kusma, kas krampları, iskelet-kas ağrısı, ishal, deri döküntüsü, yorgunluk ve abdominal ağrı olmuştur. Olaylar hafif ila orta derecededir ve hastaların yalnızca %2 ila 5'i ilaçla bağlantılı bir olay nedeniyle tedaviyi kalıcı olarak bırakmıştır. Ph+ lösemiler ve solid tümörler arasında güvenlilik profili açısından farklılıklar, Ph+ lösemilerde daha yüksek bir insidans ve şiddette miyelosupresyondur ve bunlar büyük olasılıkla hastalıkla bağlantılı faktörlerden kaynaklanmaktadır. Miyelosüpresyon, GI advers olaylar, ödem ve deri döküntüleri bu hasta popülasyonunda yaygındır. İmatinibe maruziyetten sonra gözlenmiş ve nedensel olarak bağlantılı olabilecek diğer belirgin advers olaylar hepatotoksisite, akut böbrek yetmezliği, hipofosfatemi, şiddetli respiratuar advers reaksiyonlar, tümör lizis sendromu ve çocuklarda büyüme geriliğini kapsamaktadır. Olayların şiddetine bağlı olarak doz ayarlaması gerekli olabilir. Çok az sayıda vakada advers reaksiyonlara bağlı olarak ilacın bırakılması gerekecektir. Advers reaksiyonlar en sık görülen en önce olmak üzere ve şu sınıflandırma uygulanarak sıklıklarına göre sıralanmıştır: Çok yaygın (>1/10); yaygın (> 1/100, <1/10); yaygın olmayan (> 1/1000, <1/100); seyrek (> 1/10,000, <1/1000); çok seyrek (< 1/10,000), izole raporlar dahil. Aşağıda bildirilen advers reaksiyonlar ve sıklıkları, KML için yürütülen çalışmalara dayanmaktadır. KML çalışmalarında gözlenen advers reaksiyonlar Enfeksiyonlar ve enfestasyonlar Yaygın olmayan: Herpes zoster, herpes simplex, nazofarenjit, pnömoni, sinüzit, selülit, üst solunum yolu enfeksiyonu, influenza, idrar yolu enfeksiyonu, gastroenteritis, sepsis. Seyrek: Fungal enfeksiyon Kan ve lenf sistemi hastalıkları Çok yaygın: Nötropeni, trombositopeni, anemi Yaygın: Pansitopeni, febril nötropeni Yaygın olmayan: Trombositemi, lenfopeni, kemik iliği depresyonu, eozinofili, lenfadenopati Seyrek: Hemolitik anemi Metabolizma ve beslenme bozuklukları Yaygın: Anoreksi Yaygın olmayan: Hipokalemi, iştah artışı, hipofosfatemi, iştah azalması, dehidrasyon, gut, hiperürikemi, hiperkalsemi, hiperglisemi, hiponatremi Seyrek: Hiperkalemi, hipomagnezemi Psikiyatrik hastalıklar Yaygın: Uykusuzluk Yaygın olmayan: Depresyon, libido azalması, anksiyete Seyrek: Konfüzyon Sinir sistemi hastalıkları
Çok yaygın: Yaygın: Yaygın olmayan: Seyrek: Baş ağrısı Göz kararması, parestezi, tat duyusu bozuklukları, hipoestezi Migren, somnolans, senkop, periferik nöropati, bellek bozukluğu, siyatik, huzursuz ayak sendromu, tremor, beyin kanaması Kafa-içi basıncının artması, konvülziyon, optik nörit Göz hastalıkları Yaygın: Göz kapağı ödemi, lakrimasyon artışı, konjunktiva kanaması, konjunktivit, göz kuruması, bulanık görme Yaygın olmayan: Göz tahrişi, göz ağrısı, orbita ödemi, sklera kanaması, retina kanaması, blefarit, maküla ödemi Seyrek: Katarakt, glokom, papilödem Kulak ve iç kulak hastalıkları Yaygın olmayan: Vertigo, kulak çınlaması, işitme kaybı Kardiyak hastalıklar Yaygın olmayan: Palpitasyonlar, taşikardi, konjestif kalp yetmezliği, pulmoner ödem Seyrek: Aritmi, atriyal fibrilasyon, kardiyak arest, miyokard enfarktüsü, angina pektoris, perikardiyal efüzyon Vasküler hastalıklar Yaygın: Al basması, kanama Yaygın olmayan: Hipertansiyon, hematom, subdural hematom, periferik soğukluk, hipotansiyon, Raynaud fenomeni Solunum, göğüs bozuklukları ve mediastinal hastalıklar Yaygın: Dispne, burun kanaması, öksürük Yaygın olmayan: Plevra efüzyonu, faringolaringeal ağrı, farenjit Seyrek: Plevra ağrısı, pulmoner fibroz, pulmoner hipertansiyon, pulmoner kanama Bulantı, ishal, kusma, dispepsi, karın ağrısı
Çok yaygın: Yaygın: Yaygın olmayan: Seyrek:
Gastrointestinal hastalıklar Aşırı miktarda barsak gazları, karında gerilme, gastro-özofageal reflü, kabızlık, ağız kuruması, gastrit Stomatit, ağız ülserasyonu, gastrointestinal kanama, geğirme, melena, özofajit, asit, gastrik ülseri, kan kusma, dudak iltihabı, disfaji, pankreatit Kolit, ileus, enflamatuar barsak hastalığı Hepato-biliyer hastalıklar Yaygın: Karaciğer enzimlerinde artış Yaygın olmayan: Hiperbilirübinemi, hepatit, sarılık Seyrek: Karaciğer yetmezliği, hepatik nekroz Deri ve deri altı doku hastalıkları Periorbital ödem, dermatit/egzama/deri döküntüsü.
Çok yaygın: Yaygın: Yaygın olmayan: Seyrek: Kaşıntı, yüz ödemi, deride kuruma, eritem, alopesi, gece terlemeleri, ışığa duyarlılık reaksiyonu Püstüler döküntü, kontüzyon, terlemede artış, ürtiker, ekimoz, çürük eğiliminde artış, hipotrikoz, deride hipopigmentasyon, eksfoliyatif dermatit, tırnak kırılması, folikülit, peteşiler, psoriazis, purpura, deride hiperpigmentasyon, büllöz erupsiyonlar Akut febril nötrofilik dermatoz (Sweet's hastalığı), tırnakta renk kaybı, anjiyonörotik ödem, veziküler döküntü, eritem multiform, lökositoklastik vaskülit, Stevens-Johnson sendromu, akut jeneralize ekzantematöz püstülozis (AGEP). Kas-iskelet bozukluklar, bağ doku ve kemik hastalıkları Kas spazmları ve krampları, miyalji, artralji, kemik ağrısı da dahil olmak üzere kas-iskelet ağrıları.
Çok yaygın: Yaygın: Yaygın olmayan: Seyrek: Eklemlerde şişme. Kaslarda ve eklemlerde sertlik Kas zayıflığı, artrit Böbrek ve idrar hastalıkları Yaygın olmayan: Böbrek ağrısı, hematüri, akut böbrek yetmezliği, sık sık idrar yapmak. Üreme sistemi ve meme hastalıkları Yaygın olmayan: Jinekomasti, erektil disfonksiyon, menoraji, düzensiz menstrüasyon, cinsel disfonksiyon, meme başında ağrı, memelerde büyüme, skrotum ödemi Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar Çok yaygın: Sıvı retansiyonu ve ödem, yorgunluk Yaygın: Güçsüzlük, pireksi, anazarka, titreme nöbetleri, kaslarda sertlikler Yaygın olmayan: Göğüs ağrısı, huzursuzluk Laboratuvar bulguları Çok yaygın: Kilo artışı Yaygın: Kilo azalması Yaygın olmayan: Kanda kreatinin düzeyinin yükselmesi, kandaki kreatin fosfokinaz düzeyinin yükselmesi, kandaki laktat dehidrojenaz düzeyinin yükselmesi, kanda alkalin fosfataz düzeyinin yükselmesi Seyrek: Kanda amilaz düzeyinin yükselmesi İmatinib ile yapılan ilave klinik çalışmalardan ve pazarlama sonrası deneyimden aşağıdaki reaksiyon türleri bildirilmiştir. Bunlar arasında, genişletilmiş erişim programlarından ve daha küçük veya devam etmekte olan klinik çalışmalardan bildirilen ciddi istenmeyen olayların yanı sıra kendiliğinden bildirilen vaka raporları da yer almaktadır. Bu reaksiyonların büyüklüğü bilinmeyen bir popülasyondan bildirilmesi nedeniyle, sıklıklarının güvenilir bir biçimde belirlenmesi veya imatinibe maruz kalma ile nedensel bir ilişkinin kesinleştirilmesi her zaman mümkün olmamaktadır. Pazarlama sonrası raporlarda bildirilen istenmeyen reaksiyonlar (Kist ve polipler de dahil olmak üzere) iyi huylu ve kötü huylu neoplazmalar Seyrek: Tümör lizis sendromu Sinir sistemi hastalıkları Yaygın olmayan: Serebral ödem Göz hastalıkları Seyrek: Vitröz kanama Kardiyak hastalıklar Seyrek: Perikardit, kalp tamponadı Vasküler hastalıklar Yaygın olmayan: Tromboz/emboli Çok seyrek: Anafilaktik şok Solunum, göğüs bozuklukları ve mediastinal hastalıklar Yaygın olmayan: Akut respiratuvar yetmezlik, interstisyal akciğer hastalığı Gastrointestinal hastalıklar Yaygın olmayan: İleus/intestinal obstrüksiyon, tümör kanaması/tümör nekrozu, gastrointestinal perforasyon Seyrek: Divertikülit Deri ve deri altı doku hastalıkları Yaygın olmayan: Palmar-plantar eritrodisestezi sendromu (el-ayak sendromu) Seyrek: Liken keratoz, liken planuz Çok seyrek: Toksik epidermal nekroliz Kas-iskelet bozukluklar, bağ doku ve kemik hastalıkları Seyrek: Avasküler nekroz/kalça osteonekrozu, rabdomiyoliz/miyopati Bilinmeyen: Çocuklarda büyüme geriliği Üreme sistemi hastalıkları: Çok seyrek: Hemorajik korpus luteum, hemorajik over kisti Seçilmiş advers ilaç reaksiyonlarıyla ilgili açıklamalar Miyelosüpresyon: Miyelosüpresyon imatinible tedavi edilmiş kanser hastalarında çok yaygındır. Miyelosüpresyon, trombositopeni, nötropeni ve anemi en sık bildirilen 3. ve 4. derece laboratuar anormallikleridir. Genel olarak KML hastalarında imatinible yaşanan miyelosüpresyon genellikle geri dönüşümlüdür ve hastaların büyük kısmında doz kesintisine ya da doz azaltımına neden olmamıştır. Birkaç hastada ilacın bırakılması gerekmiştir. Aynı zamanda pansitopeni, lenfopeni ve kemik iliği depresyonu gibi başka olaylar da bildirilmiştir. Hematolojik supresyon yüksek dozlarda en fazladır ve KML hastalığının evresiyle bağlantılı gibi görünmektedir; 3. ya da 4. derece nötropeni ve trombositopeni, KF KML'de yeni tanısı konmuş hastalarla karşılaştırıldığında (sırasıyla %16.7 ve %8.9) blast ve akselere fazda (sırasıyla %44 ve %63) 4 ila 6 kat daha yüksektir. Bu olaylar dozun azaltılması ya da İMATİS tedavisine ara verilmesi yoluyla tedavi edilebilir; fakat nadiren tedavinin bırakılmasını gerektirir. Hematolojik toksisitelerin insidansı, Ph+ lösemileri olan hastalarla karşılaştırıldığında solid tümörleri olan hastalarda daha düşüktür; 3/4. derece nötropeni ve trombositopeni oranı sırasıyla yaklaşık %10 ve %1'dir. Kanama: SSS ve GI kanamalar, başlangıçta bozulmuş kemik iliği fonksiyonu olan KML hastalarında seyrek değildir. Kanamalar, lösemik hastalardan oluşan, akut hastalık durumuna sahip bir popülasyonda hastalık komplikasyonlarının iyi bilinen bir parçasıdır ve trombositopeniden ve daha seyrek olarak trombosit disfonksiyonundan kaynaklanabilir. Diğer yandan imatinible tedavi sırasında SSS ve GI kanamaları deneyimleyen tüm hastalar trombositopenik değildir. Klinik açıdan anlamlı kanamanın en yaygın belirtisi GI kanamadır; en fazla ilerlemiş KML hastalarında meydana gelmektedir; bu gibi durumlarda kanama, tümör hemorajisi/tümör nekrozundan kaynaklanan tümör kanaması nedeniyle altta yatan hastalığın bir parçası olarak meydana gelebilir. Birinci basamak KML durumunda gözlenmiş GI kanama sıklıkları genellikle en düşük düzeydedir. Ödem ve sıvı tutulumu: Ödem imatinibin en yaygın toksisitesidir ve tüm endikasyonlarda hastaların %50'sinden fazlasında ortaya çıkmaktadır. Ödem dozla bağlantılıdır ve görünüşe göre ortaya çıkışı ve plazma düzeyleri arasında bir korelasyon bulunmaktadır. En yaygın belirtileri periorbital ödemdir ve alt uzuv ödemine göre biraz daha seyrektir. Genellikle spesifik bir tedavi gerekli değildir. Diğer sıvı tutulumu olayları daha seyrek olarak meydana gelir, fakat anatomik yerin konumu nedeniyle ciddi olabilir. En sık görülen sıvı tutulumu olayı plevral efüzyondur ve en fazla ilerlemiş KML'de gözlenmektedir. Kalp yetmezliğinin sıklığı ödem ve sıvı tutulumu olan hastalarda genellikle düşüktür. Diğer gruplara kıyasla ileri evre KML'de daha yüksek olduğu bulunmuştur. Bu durum ileri evre KML hastalarının daha kötü olan tıbbi durumu ile açıklanabilir. Aynı eğilim, ödem ve sıvı tutulumu olan hastalarda böbrek yetmezliği için de gözlenmiştir. Yeni tanısı konmuş KML hastaları üzerinde yapılan bir klinik çalışmada, konjestif kalp yetmezliğine işaret eden olayların sıklığı imatinib grubunda %1.5 ve IFN-alfa grubunda %1.1 olarak belirlenmiştir. Sıklık, transforme KML (akselere faz ya da blast fazı), daha yüksek yaş ya da başlangıç hemoglobin düzeyi 8 g/dL'den düşük olan hastalarda belirgin şekilde daha yüksek olmuştur. Ödem ve sıvı tutulumu olan hastaların çoğu yaşlıdır (>65 yaş). Deri döküntüleri ve şiddetli kutanöz advers reaksiyonlar: Tedavi devam ettirilmesine rağmen azalabilen genel eritematöz, makulopapüler, pruritik deri döküntüsü bildirilmiştir. Bazı hastalarda eşlik eden deri döküntüsü olmaksızın prurit görülebilmektedir ve kimi zaman bir eksfolyatif bileşen bulunmaktadır. Tüm hastalarda olmasa da bazı hastalarda yeniden maruziyet deri döküntüsünün yeniden ortaya çıkmasıyla sonuçlanmıştır. Bu erüpsiyonlar genel olarak antihistaminlere ve topikal steroidlere yanıt vermektedir. Nadiren sistemik steroidler gerekli olmaktadır. Tüm endikasyonlarda imatinible tedavi edilmiş hastaların üçte bire kadar olan kısmında deri döküntüleri gözlenmiştir. Bunlar genellikle pruritiktir ve büyük oranda ön kolda, gövdede ya da yüzde eritematöz, makulopapüler lezyonlar olarak ortaya çıkmaktadır. Deri biyopsileri, karışık bir hücresel infiltratla toksik ilaç reaksiyonunu ortaya çıkarmıştır. Deri döküntülerinin büyük kısmı hafif şiddette ve kendi kendini sınırlayıcı olsa da, daha şiddetli vakalar tedavinin kesilmesini ya da bırakılmasını gerektirebilir. Hepatotoksisite: Nadiren şiddetli olan hepatotoksisite meydana gelebilir ve preklinik ve klinik olarak gözlenmiştir. LFT anormallikleri genellikle transaminazlarda hafif yükselmelerden oluşmaktadır; diğer yandan, hastaların küçük bir kısmında bilirubin düzeyleri de yükselmiştir. Ortaya çıkış süresi genellikle tedavinin ilk iki ayı içindedir; fakat tedavi başlatıldıktan sonraki 6. ila 12 aylar arası gibi geç dönemlerde de görülebilmektedir. Düzeyler, tedaviye 1 ila 4 hafta ara verildikten sonra normal değerlere dönmektedir. Hipofosfatemi: Tüm endikasyonlarda nispeten yaygın bir şekilde düşük serum fosfatı ve hipofosfatemi (3/4. dereceye kadar) gözlenmiştir; diğer yandan, bu bulgunun kökeni ve klinik önemi henüz belirlenmemiştir. İmatinibin insan monositlerinin osteoklastlara farklılaşmasını inhibe ettiği gösterilmiştir. Bu azalmaya, bu hücrelerin resorptif kapasitelerindeki düşüş de eşlik etmiştir. İmatinib varlığında osteoklastlarda doza bağlı RANK-L azalması gözlenmiştir. Osteoklastik aktivitenin sürekli inhibisyonu, artmış PTH düzeyleriyle sonuçlanan düzenleyici yanıtın engellenmesine yardımcı olabilir. Preklinik bulguların klinik önemli henüz açık değildir ve kemik kırıkları gibi iskelete bağlı advers olaylarla ilişkili gösterilmemiştir. Klinik geliştirme programında serum fosfatı tüm çalışmalarda rutin bir şekilde ölçülmemiştir. Her ne kadar başta hipofosfateminin doza bağlı olabileceği düşünülmüş olsa da, yeni tanısı konmuş KML hastalarında güvenlilik sonlanım noktalarının doza bağlılığını araştırmak üzere tasarlanmış Faz III TOPS çalışmasından elde edilen 24 aylık yorumlanabilir bulgular, 400 mg ya da 800 mg alan hastaların %19.1'e karşılık %15.5'inde ve %5.1'e karşılık %0.9'undan 3/4. derece azalmış serum fosfatı ya da serum kalsiyumu gözlendiği gösterilmiştir. Gastrointestinal obstrüksiyon, perforasyon ya da ülserasyon: Şiddetli vakalarda imatinibin neden olduğu lokal tahrişi temsil edebilen GI ülserasyon, tüm endikasyonlarda hastaların küçük bir kısmında gözlenmiştir. Tümör lizis sendromu: Tümör lizis sendromu ve imatinib tedavisi arasında nedensel bir ilişki olası görünmektedir; diğer yandan bazı vakalarda eşzamanlı ilaçlar ve diğer bağımsız riskler karışıklık yaratmaktadır (bkz. Bölüm 4.4). Çocuklarda büyüme geriliği: İMATİS, özellikle pre-pubertal dönemde olanlar olmak üzere çocukların boylarını etkilemektedir. Çocuklarda büyüme geriliği ve İMATİS tedavisi arasındaki nedensel ilişki olasılık dışı bırakılamaz; diğer yandan bazı büyüme geriliği vakaları için sınırlı bilgi bulunmaktadır (bkz. Bölüm 4.4). Şiddetli respiratuar advers ilaç reaksiyonu: İmatinib tedavisiyle kimi zaman ölümcül olan, akut respiratuar yetmezlik, pulmoner hipertansiyon, interstisyal akciğer hastalığı ve pulmoner fibroz gibi şiddetli respiratuar olaylar gözlenmiştir. Vakaların çoğunda, şiddetli respiratuar olaylarla ilişkili olabilecek, daha önce mevcut kardiyak ya da pulmoner durumlar bildirilmiştir. Laboratuvar testi anormallikleri Hematoloji KML'de başta nötropeni ve trombositopeni olmak üzere sitopeniler tüm çalışmaların devamlı bir bulgusu olmuş, >750 mg gibi daha yüksek dozlarda daha sık oldukları düşünülmüştür (faz I çalışma). Bununla birlikte, sitopenilerin ortaya çıkışı, aynı zamanda açıkça hastalığın evresine de bağlı olmuştur. Sitopeniler, yeni tanı konulan KML vakalarında, diğer vakalara kıyasla daha seyrektir. Evre 3 veya 4 nötropenilerin (ANC <1.0xl09/L) ve trombositopenilerin (trombosit sayısı < 50xl09/L) blast krizindeki ve hızlanmış fazdaki sıklığı, yeni tanı konulan kronik faz KML vakalarındakinin 4-6 katıdır. Yeni tanı kronik faz KML vakalarında % 16.7 nötropeni ve % 8.9 trombositopeni görülürken, bu oranlar hızlanmış ve blastik fazda sırasıyla, % 59-64 ve % 44-63 olarak bildirilmiştir. Yeni tanı konulmuş olan kronik faz KML vakalarında evre 4 nötropeni (ANC < 0.5xl09/L) ve trombositopeni (trombosit sayısı <10xl09/L), sırasıyla yalnızca %3.6 ve <%1 oranında görülmüştür. Nötropenik ve trombositopenik periyotların ortalama süresi genellikle sırasıyla 2 ve 3. haftalar arasında ve 3 ve 4. haftalar arasında yer almıştır. Bu olaylar, genellikle İMATİS ile tedavinin dozu azaltılarak ya da tedavi kesilerek kontrol edilebilir, ancak bazı nadir vakalarda kalıcı olarak tedavinin bırakılmasına neden olabilir. Pediyatrik KML hastalarında en sık gözlenen toksisiteler; nötropeni, trombositopeni ve anemi dahil olmak üzere 3 ya da 4. derece sitopeniler olmuştur. Bunlar genellikle ilk birkaç ay içerisinde gerçekleşmektedir. Biyokimya KML hastalarında transaminazlarda (< % 5) ya da bilirubinde (< %1) ciddi artışlar nadir olmuştur (hastaların <% 3'ü) ve genellikle doz azaltılarak ya da kesilerek (bu epizodların ortalama süresi yaklaşık 1 hafta olmuştur) kontrol altına alınmıştır. KML hastaların % 1'inden azında karaciğer laboratuar anormallikleri nedeniyle tedavi sürekli olarak kesilmiştir. Bazıları ölümcül olabilen sitolitik ve kolestatik hepatit ve karaciğer yetmezliği vakaları mevcuttur. 4.9. Doz aşımı Terapötik dozlardan daha yüksek dozlarla deneyim sınırlıdır. İmatinib doz aşımı ile ilgili bireysel vakalar spontan olarak ve literatürde bildirilmiştir. Genellikle, bu vakalarda bildirilen sonuçlar düzelme ya da iyileşme şeklinde olmuştur. Doz aşımı halinde, hasta gözlem altında tutulmalı ve uygun semptomatik tedavi uygulanmalıdır. Farklı doz aralıklarında bildirilen olaylar aşağıda verilmiştir: Erişkinlerde doz aşımı: 1,200 ila 1,600 mg (1 ila 10 gün arasında değişen sürelerle): Bulantı, kusma, diyare, döküntü, eritem, ödem, şişme, yorgunluk, kas spazmları, trombositopeni, pansitopeni, karın ağrısı, baş ağrısı, iştahta azalma. 1,800 ila 3,200 mg (6 gün boyunca günde 3,200 mg'a kadar dozlar): Güçsüzlük, miyalji, CPK düzeyinde yükselme, bilirubin düzeyinde yükselme, gastrointestinal ağrı. 6,400 mg (tek doz): Literatürde yer alan bir vakada, bulantı, kusma, karın ağrısı, pireksi, yüzde şişme, nötrofil sayısında azalma, transaminaz düzeylerinde yükselme görülen bir hasta bildirilmiştir. 8 ila 10 g (tek doz): Kusma ve gastrointestinal ağrı bildirilmiştir. Pediyatrik doz aşımı: 400 mg'lık tek doza maruz kalan 3 yaşındaki bir erkek çocukta kusma, diyare ve anoreksi; 980 mg'lık tek doza maruz kalan 3 yaşındaki diğer bir erkek çocukta ise lökosit sayısında azalma ve diyare görülmüştür. 5. Farmakolojik özellikler 5.1. Farmakodinamik özellikler Farmakoterapötik grup: Protein-tirozin kinaz inhibitörü ATC kodu: LO1XE01 Etki mekanizması: İmatinib küçük bir molekül yapısına sahip bir protein-tirozin kinaz inhibitörüdür; Bcr-Abl tirozin kinaz (TK) aktivitesini ve birçok reseptör TK'yı kuvvetli bir şekilde inhibe etmektedir: Kit, c-Kit proto-onkogen tarafından kodlanan kök hücre faktörü (SCF) reseptörü, diskoidin domen reseptörleri (DDR1 ve DDR2), koloni uyarıcı faktör reseptörü (CSF-1R), trombosit kökenli büyüme faktörü reseptörleri alfa ve beta (PDGFR-alfa ve PDGFR-beta). İmatinib aynı zamanda bu reseptör kinazların aktivasyonunun aracılık ettiği hücresel olayları da inhibe edebilmektedir. İmatinib, in vitro, hücresel ve in vivo düzeylerde kırılma noktalarının yoğunlaştığı bölge-Abelson (Bcr-Abl) tirozin kinazı güçlü bir şekilde inhibe eden bir protein-tirozin kinaz inhibitörüdür. Bileşik, Bcr-Abl pozitif hücre dizilerinde, Philadelphia kromozom pozitif Kronik Miyeloid Lösemi (KML) hastalarının yeni lösemi hücrelerinde selektif olarak proliferasyonu inhibe etmekte ve apopitozisi uyarmaktadır. Ex vivo periferik kan ve kemik iliği örneklerinin kullanıldığı koloni transformasyon tahlillerinde, imatinib KML hastalarındaki Bcr-Abl pozitif kolonilerde selektif inhibisyon göstermektedir. Bileşik in vivoolarak, Bcr-Abl pozitif tümör hücreleri kullanılan hayvan modellerinde tek ajan olarak anti-tümör aktivite gösterir.Kronik Miyeloid Lösemide Klinik Çalışmalar İmatinibin etkinliği, bir bütün olarak elde edilen hematolojik ve sitogenetik yanıt oranlarını ve hastalıksız sağkalım süresini temel alır. Bütün klinik çalışmalarda hastaların %38-40'ının en az 60, %10-12'sinin en az 70 yaşında olduğu bildirilmiştir. Kronik faz, yeni tanı konulmuş:Bu faz III çalışmasında, imatinib monoterapisi, interferon-alfa (IFN) + sitarabin (ARA-C) kombinasyonuyla karşılaştırılmıştır. Kullanılan tedaviye yanıt vermeyen hastaların, kullandıkları tedaviyi bırakarak diğer tedaviyi kullanmalarına izin verilmiştir. İmatinib grubundaki hastalarda günde 400 miligramlık doz kullanılmıştır. IFN grubundaki hastalar, hedef alınan günlük subkütan IFN dozu 5 MIU/m2 + her ayın 10 günü, günde 20 mg/m2 Ara-C kombinasyonu kullanmıştır.16 ülkedeki 177 çalışma merkezinden toplam 1106 (her grupta 553) hasta, randomize edilmiştir. Yaşları 18-70 arasında değişmek üzere medyan 51 olan hastaların %21.9'unun, 60 yaşında ya da daha ileri yaşta olduğu görülmüştür. Verilerin bu analizde kullanılmak üzere derlendiği sıradaki (son hastanın kaydedilmesinden 7 yıl sonra) medyan ilk seçenek tedavi süresi imatinib grubunda 82, IFN (kombinasyon) grubunda 8 aydır. İmatinib ile medyan ikinci seçenek tedavi süresi 64 aydır. İmatinib grubuna ayrılmış olan hastaların % 60'ı, başlangıçta kullandıkları ilaca (imatinib) devam etmektedir. Bu hastalarda ortalama imatinib dozu 403±57 mg'dır. Genel olarak, birinci seçenek olarak imatinib alan hastalarda dağıtılan ortalama günlük doz 406±76 mg'dır. IFN + ARA-C grubundaki hastaların yalnızca %2'si başlangıçta kullandıkları tedaviye devam etmektedir. IFN + ARA-C grubunda başlangıçtaki tedaviye devam etmeyen hastalarda bunun en sık rastlanan nedeni (%14), hastaların verdikleri onayı geri çekmesi; imatinib grubuna geçenlerde ise buna en sık (hastaların %26'sında) yol açan neden, şiddetli intolerans ve hastalığın ilerlemesidir (%14). Primer bitiş noktası, hastalıksız sağkalım süresidir. Sekonder sonlanım noktalarının yanıt verileri de Tablo 2'de gösterilmektedir. Tablo 2 Yeni tanı konulan KML çalışmasındaki yanıt oranları (84 aylık veri)
**Yetersiz veri, numuneler ile yalnızca iki hasta var Hematolojik yanıt kriterleri (bütün yanıtlar >4 hafta sonra doğrulanmalıdır):Kandaki lökosit sayısı <10 xl09/L, trombosit sayısı <450 xl09/L, miyelosit+metamiyelosit <%5; kanda blast hücresi veya promiyelosit yok, bazofiller <%20, kemik iliği dışında hastalık yokSitogenetik yanıt kriterleri:tam (%0 Ph+ metafazlar), kısmi (%l-35), minor (%36-65) veya minimal (%66-95) Major yanıt (%0-35), hem kısmi hem tam yanıtları içerir [1]Major moleküler yanıt kriterleri:Gerçek-zaman kantitatif revers kriptaz polimeraz zincir reaksiyonuyla ölçülen BCR-ABL transkriptlerinin periferik kanda, başlangıç düzeyine göre en az 3 log azalmasıİmatinib ile birinci seçenek tedavinin kümülatif yanıt oranları Tablo 3'te gösterilmektedir. Tablo 3 Birinci seçenek imatinib tedavisine verilen kümülatif yanıt tahminiTedavide geçen aylar %THY %MSY %TSY
84 ayda tahmin edilen progresyonsuz sağkalım imatinib grubunda %95 güven aralığı ile %81.2 (78, 85) ve kontrol grubunda %60.6 (56.5) olmuştur (p <0.001) (Şekil 1). 84 ayda akselere faza ya da blast krizine ilerleme olmayan hastaların tahmin edilen oranı, IFN grubu ile karşılaştırıldığında imatinib grubunda anlamlı düzeyde yüksek olmuştur (%85.1 (82,89) karşısında %92.5 (90,95), p <0.001) (Şekil 2). Yıllık progresyon hızı tedavide geçirilen süre ile birlikte azalmaktadır. 1 İlerleme kaydedilinceye kadar geçen süre (ITT ilkesi) 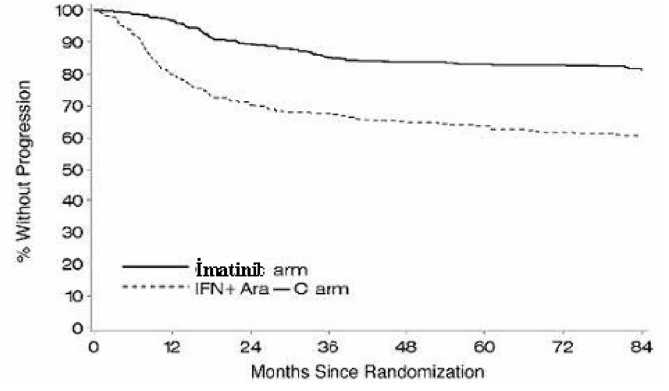 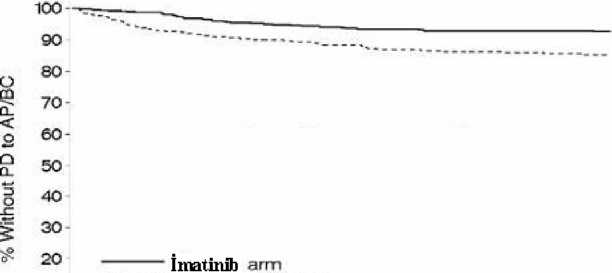
IFN+ Ara C aımO --------------------------------O 12 24 36 48 60 72 84Morıths Since Randomizationİmatinib ve IFN+Ara-C gruplarında, sırasıyla, toplam 71 (%12.8) ve 85 (%15.4) hasta ölmüştür. 84 ayda randomize imatinib ve IFN+Ara-C gruplarında tahmin edilen genel sağkalım, sırasıyla %86.4 (83, 90) ve %83.3 (80, 87) düzeyindedir (p=0.073, log-rank testi). Buna ek olarak, 84 aylık verilere göre imatinib hastalarında yalnızca 31 (%5.6) ölüm (BMT öncesi) KML ile ilişkilendirilmiştir. Buna karşılık IFN+Ara-C hastalarında 40 (%7.2) ölüm KML ile ilişkilendirilmiştir. Yalnızca KML ile ilişkili bu ölümler dikkate alınır ve BMT sonrası ya da diğer nedenlerle meydana gelen bütün ölümler sansürlenirse, tahmin edilen 84 aylık sağkalım oranları %93.6 ve %91.1 olmaktadır (p=0.1, log-rank testi). İmatinib tedavisinin kronik fazdaki, yeni tanı konulmuş KML'deki sağkalım etkisi, aynı rejimde IFN+Ara-C (n=325) kullanılan başka bir Faz III çalışmadan elde edilen birinci verilerle birlikte yukarıda belirtilen imatinib verilerinin retrospektif analizinde ayrıntılı olarak incelenmiştir. Bu yayında, genel sağkalım bakımından imatinibin IFN+Ara-C karşısındaki üstünlüğü kanıtlanmıştır (p<0.001); 42 ay içinde 47 (%8.5) imatinib hastası ve 63 (%19.4) IFN+Ara-C hastası ölmüştür.
Şekil 2 Akselere Faz veya Blast krizinin gelişmesine kadar geçen süre (ITT ilkesi) Sitogenetik yanıtın derecesi imatinib tedavisi uygulanan hastalarda uzun vadeli sonuçlar üzerinde açık bir etkiye sahiptir. 12 ayda TSY (KSY) olan hastalardan 84 ayda AF/BK'ye ilerlemeden kalacağı tahmin edilenlerin oranı %96 (%93) düzeyindedir, 12 ayda MSY olmayan hastaların yalnızca %81'i 84 ayda ileri KML'ye ilerlemeden kalacaktır (genel olarak p<0.001, TSY ve KSY arasında p=0.25). 18 aylık dönüm noktası esas alındığında, tahminler sırasıyla %99, %90 ve %83 olmakta, ayrıca TSY ve KSY arasında istatistiksel olarak anlamlı bir fark (p<0.001) meydana gelmektedir. Moleküler izlem önemli ek prognostik bilgiler sağlamıştır. TSY olan ve 12 ay sonra Bcr-Abl transkriptlerinde en az 3 log azalma olan hastalar için, 60 ayda hastalığın ilerlemeden kalma olasılığı, 12 ayda TSY olmayan hastalarda gözlemlenenden anlamlı düzeyde daha büyüktür (%70, p<0.001). Yalnızca AF/BK'ye ilerleme dikkate alındığında, tahmin edilen olaysızlık oranları, sırasıyla %100, %95 ve %88 olur (genel olarak p<0.001, MMY olan ya da olmayan TSY arasında p=0.007). 18 aylık dönüm noktası kullanıldığında, 60 ayda tahmin edilen AP/BC'sizlik oranları TSY ve MMY olan hastalar için %100, TSY olan ama MMY olmayan hastalar için %98 ve TSY olmayan hastalar için yalnızca %87 olmuştur (genel olarak p<0.001, MMY olan ya da olmayan TSY arasında p=0.105). Bu çalışmada kullanılan dozların günde 400 miligramdan 600 miligrama, daha sonra da 600 miligramdan 800 miligrama yükseltilmesine izin verilmiştir. Dozun günde 800 miligrama artırıldığı 40 hastadaki bazı advers reaksiyonların yüzdesinin, doz artırımından öncesine kıyasla yükseldiği görülmüştür (n=551). Gastrointestinal kanamalar, konjunktivit ve plazmadaki transaminaz ya da bilirübin düzeylerinin yükselmesi; doz artırıldığında daha sık görülen advers reaksiyonlardandır. Diğer advers reaksiyonlar ise doz artırıldıktan sonra, önceye kıyasla daha seyrek veya aynı sıklıkta görülmüştür. Yaşam kalitesi, geçerliliği kanıtlanmış bir enstrüman olan FACT-BRM anketiyle değerlendirildi. Bu anketin bütün bölümlerinde imatinib grubu, IFN + ARA-C grubuna kıyasla istatistik bakımdan anlam taşıyacak şekilde daha yüksek puan aldı. Sonuçlar, hastaların yaşam kalitesinin imatinib tedavisi sırasında iyi durumda kaldığını gösterdi. Kronik faz, interferon tedavisinin başarısız kaldığı hastalar:532 hasta, 400 miligramlık başlangıç dozuyla tedavi edildi. Bu hastalar; hematolojik başarısızlık (%29), sitogenetik başarısızlık (%35) veya interferon intoleransı (%36) olmak üzere başlıca 3 gruptu. Hastalar, bu çalışma öncesinde medyan 14 ay boyunca haftada >25 xl06 IU dozunda interferon kullanmış bulunan, geç kronik faz vakalarıydı ve tanı konulduktan sonra geçen medyan süre 32 aydı. Majör sitogenetik yanıt (tam + kısmi yanıt, kemik iliğinde %0-35 Ph+ metafaz) oranı, bu çalışmadaki başlıca etkinlik parametresi olarak değerlendirildi.Bu çalışmada hastaların % 65'inde (% 53'ü tam olmak üzere) majör sitogenetik yanıt elde edildi (Tablo 4). Hastaların %95'i bu tedaviye tam hematolojik yanıt verdi. Hızlanmış faz:Bu fazdaki 235 KML vakasının ilk 77'sinde tedaviye günde 400 mg ile başlandı; daha sonra çalışma protokolü, daha yüksek imatinib dozlarının kullanılmasına olanak tanıyacak şekilde tadil edildi ve geriye kalan 158 hasta, başlangıçta 600 mg imatinib kullandı.Tam hematolojik yanıt, hiçbir lösemi kanıtının mevcut olmaması (kemik iliğindeki ve kandaki blast hücrelerinin kaybolması, ancak periferik kan tablosunda, tam yanıt için gereken düzelmenin gerçekleşmemesi) veya kronik faz kronik miyeloid lösemiye dönüş olarak tanımlanan tam hematolojik yanıt elde edilme oranı, bu çalışmanın etkinlik konusundaki değerlendirilen primer parametresiydi. Doğrulanmış hematolojik yanıt, hastaların % 71.5'inde elde edildi (Tablo 4). Bu hastalardan % 27.7'sinde ayrıca majör sitogenetik yanıt (% 20.4'ünde tam majör sitogenetik yanıt) alınmış olması önemlidir. 600 mg imatinib kullanan hastalarda bugünkü saptamalara göre tahmini medyan hastalıksız sağkalım ve genel sağkalım oranları, sırasıyla 22.9 ay ve 42.5 ay olarak hesaplandı. Miyeloid blast krizi:Bu çalışma, blast krizi gelişmiş olan 260 hasta üzerinde yapıldı. Bu hastaların 95'i (%37'si), hızlanmış faz veya yine blast krizi nedeniyle daha önce de kemoterapi görmüştü (önceden tedavi edilmiş olan hastalar), 165 (%63) hastada ise daha önce kemoterapi uygulanmamıştı (önceden tedavi edilmemiş olan hastalar). Başlangıç dozu, ilk 37 hastada 400 miligramdı; daha sonra yapılan protokol tadilatı, daha yüksek dozların kullanılmasına olanak verdiğinden, diğer 223 hasta, başlangıçta 600 mg imatinib kullandı.Primer etkinlik parametresi, hızlanmış faz çalışmasında olduğu gibi yine tam hematolojik yanıt, lösemi kanıtının mevcut olmaması veya kronik faza dönüş olarak tanımlanan, hematolojik yanıt oranıydı. Hastaların %31'inde hematolojik yanıt elde edildi. 600 mg imatinib kullanan hastalardaki hematolojik yanıt oranı, 400 mg imatinib kullanmış olanlara kıyasla daha yüksekti (%16'ya karşılık %33, p=0.0220). Daha önceden tedavi edilmemiş ve tedavi edilmiş hastaların mevcut tahmini ortalama sağkalımı sırasıyla 7.7 ve 4.7 aydır.
hematolojik yanıt kriterleri (bütün yanıtlar >4 hafta sonra doğrulanmış olmalıdır):THY: çalışma 0110 [kandaki WBC <10 x109/L, trombosit sayısı <450x109/l, miyelosit + metamiyelosit <5% , kanda blast veya promiyelosit yok, bazofiller <%20, kemik iliği dışında hastalık yok] ve çalışma 0102 ve 0109 [ANC>1.5 xl09/L, trombosit sayısı >100 x 109/l, kanda blast hücresi yok, BM blast hücresi oranı<%5ve BM dışında hastalık yok]NEL: THY ile aynı kriterler; yalnızca ANC >1 x109/L ve trombosit sayısı >20 xl09/L (çalışma 0102 ve 0109'da) RTC: BM ve PB blast hücresi oranı <%15; BM ve PB blast hücresi + promiyelosit oranı <%30, PB bazofil oranı <%20, dalak ve karaciğer hariç BM dışında hastalık yok (çalışma 0102 ve 0109'da) ANC = mutlak nötrofil sayısı, BM = kemik iliği, PB = periferik kan, WBC = lökosit sayısı 2Major yanıt = tam (%0 Ph+ metafaz) + kısmi (% l-35) yanıtPediatrik hastalar.Açık-etiketli, çok merkezli, tek kollu bir faz II çalışmaya, tanısı yeni konmuş ve tedavi edilmemiş kronik fazda KML'si olan toplam 51 pediyatrik hasta katılmıştır ve hastalara 340 mg/m2/gün dozla imatinib tedavisi uygulanmıştır. İmatinib tedavisiyle yeni tanı konmuş pediyatrik KML hastalarında hızlı bir yanıt sağlanmış, 8 haftalık tedaviden sonra THY oranı %78 olmuş ve 3-10 aylık tedaviden sonra tam sitogenetik yanıtın (TSY) %65 oranında (yetişkinlerde gözlenene yakın) gerçekleştiği de gözlenmiştirKronik faz KML'si (n=15), blast krizi aşamasında KML'si ya da Ph+ ALL'si (n=16) olan, daha önce ağır tedavi uygulanmış (%45'ine daha önce BMT ve %68'ine daha önce çoklu ajan kemoterapisi) toplam 31 pediatrik hasta bir doz yükseltme faz 1 çalışmaya kaydedilmiştir. Hastalar 260 mg/m2/gün ve 570 mg/m2/gün aralığında dozlarla tedavi edildi. Sitogenetik verileri mevcut olan 13 KML vakasından 7'sinde (%54) tam, 4'ünde (%31) kısmi olmak üzere %85'inde MSY elde edildi. SM ile İlgili Klinik Çalışmalar Abl, Kit ya da PDGFR protein tirozin kinazlarla ilişkili yaşamı tehdit edici hastalıkları olan farklı hasta popülasyonlarında imatinibin test edildiği açık-etiketli, çok merkezli bir faz II klinik çalışma (çalışma B2225) yürütülmüştür. Bu çalışmada tedavi edilen ve 45'inde hematolojik hastalıklar, 140'ında da çeşitli solid tümörler bulunan 185 hastadan 5'inde SM saptanmıştır. SM hastaları günlük 100 mg ila 400 mg imatinib ile tedavi edilmiştir. Yayınlanmış 10 vaka raporu ve vaka serisinde, yaşları 26 ila 85 arasında değişen 25 SM hastası daha bildirilmiştir. Bu hastalara da günlük 100 mg ila 400 mg dozda imatinib uygulanmıştır. SM için tedavi edilen toplam popülasyonun (30 hasta) 10'unda (%33) tam hematolojik yanıt, 9'unda (%30) kısmi hematolojik yanıt elde edilmiştir (toplam yanıt oranı %63). Sitogenetik anormallikler yayınlanmış raporlarda ve çalışma B2225'te tedavi edilen 30 hastanın 21'inde değerlendirilmiştir. Bu 21 hastanın sekizinde FIP1L1-PDGFR-alfa füzyon kinaz saptanmıştır. Çalışma B2225'te tedavi edilen hastalarda medyan tedavi süresi 13 ay olmuş (aralık: 1.4-22.3 ay), yayınlanmış literatürde yanıt veren hastalarda ise aralık 1 ay ila 30 ayın üzerinde bir süre arasında değişmiştir. Sonuçlar Tablo 5'te verilmiştir. Tablo 5 SM'de Elde Edilen Yanıt
HES ile İlgili Klinik Çalışmalar Abl, Kit ya da PDGFR protein tirozin kinazlarla ilişkili yaşamı tehdit edici hastalıkları olan farklı hasta popülasyonlarında imatinibin test edildiği açık-etiketli, çok merkezli bir faz II klinik çalışma (çalışma B2225) yürütülmüştür. Bu çalışmada, toplam 185 hastadan (45'i hematolojik hastalık, 140'ında çeşitli solid tümörler saptanmıştır) HES'i olan 14 hasta günde 100 mg ila 1000 mg dozda imatinib ile tedavi edilmiştir. Yayınlanmış 35 vaka raporu ve vaka serisinde, HES'i olan ve yaşları 11 ila 78 arasında değişen 162 hasta daha bildirilmiştir. Bu hastalara, günde 75 mg ila 800 mg dozda imatinib verilmiştir. HES için tedavi edilen toplam popülasyonun (176 hasta) 107'sinde (%61) tam hematolojik yanıt, 16'sında ise (%9) kısmi hematolojik yanıt elde edilmiştir (toplam yanıt oranı %70). Yayınlanmış raporlarda ve çalışma B2225'te, tedavi edilen 176 hastadan 117'sinde sitogenetik anormallikler değerlendirilmiştir. Bu 117 hastanın 61'i, FIP1L1-PDGFR-alfa füzyon kinaz pozitif bulunmuştur. Tüm bu FIPlLl-PDGFR-alfa füzyon kinaz pozitif hastalarda, tam hematolojik yanıt elde edilmiştir. 115 hastada FIP1L1-PDGFR-alfa füzyon ya negatif bulunmuştur, ya da bilinmemektedir. Bunların 62'sinde (%54) ya tam (n=46) ya da kısmi (n=16) hematolojik yanıt elde edilmiştir. Sonuçlar Tablo 6'da verilmiştir. Tablo 6 HES'de Elde Edilen Yanıt Sitogenetik anormallik Hasta sayısı Tam hematolojik yanıt Kısmi hematolojik yanıtPozitif FIPlLl-PDGFR-alfa füzyon kinaz 61 61 0Negatif FIP1L1-PDGFR- alfa füzyon kinaz 56 12 9 Bilinmeyen sitogenetik anormallik 59 34 7 Genel toplamlar 176 107 (%61) 16 (%9) Ayrıca, vaka raporlarında araştırmacılar tarafından, semptomatoloji ve diğer organ fonksiyon bozukluğu anormalliklerinde iyileşmeler bildirilmiştir. Kalp, sinir, cilt/ciltaltı dokusu, solunum/toraks/mediasten, kas-iskelet/bağ dokusu/vasküler ve gastrointestinal organ sistemlerinde iyileşmeler bildirilmiştir. Karaciğer yetersizliği olan hastalarda yapılan klinik çalışmalar Çeşitli derecelerde (hafif, orta şiddette veya şiddetli; karaciğer fonksiyon bozukluğunun sınıflandırılması için bkz Tablo 7) karaciğer yetersizliği olan hastalarda yapılan bir çalışmada imatinibe ortalama maruz kalım (doza göre normalize edilmiş EAA değeri), karaciğer fonksiyonu normal olan hastalara kıyasla artmamıştır. Bu çalışma sırasında hafif karaciğer bozukluğu olan hastalarda günde 500 mg, diğer hastalarda günde 300 mg imatinib, güvenle kullanılmıştır. Orta-ileri derecede şiddetli karaciğer yetersizliği olan hastalarda yalnızca 300 miligramlık doz kullanılmıştır ama farmakokinetik analiz bunun, 400 miligramla güvenle yükseltilebileceğini göstermiştir (bkz 4.2 Pozoloji ve uygulama şekil; 4.4 Özel kullanım uyarıları ve önlemleri; 4.8 İstenmeyen Etkiler ve 5.2 Farmakokinetik Özellikler bölümleri) Tablo 7 Karaciğer yetersizliğinin sınıflandırılması
SGOT, serum glutamik oksaloasetik transferaz Böbrek yetmezliği olan hastalarda yürütülen klinik çalışmalar Değişen derecelerde (hafif, orta ve şiddetli - böbrek fonksiyonu sınıflandırması için bkz. aşağıda Tablo 8) böbrek yetmezliği olan hastalarla yürütülen bir çalışmada, maruz kalınan ortalama imatinib (doz normalize EAA), böbrek fonksiyonları normal olan hastalarla karşılaştırıldığında 1.5-2 kat artmıştır, bu da, imatinibin güçlü bir biçimde bağlandığı bir protein olan AGP'nin plazma düzeyinde benzer bir artışa karşılık gelir. Maruz kalınan imatinib ile böbrek bozukluğunun şiddeti arasında hiçbir korelasyon gözlemlenmemiştir. Bu çalışmada, hafif böbrek yetmezliği olan hastalarda günlük 800 mg ve orta düzeyde böbrek yetmezliği olan hastalarda günlük 600 mg güvenle kullanılmıştır. Sınırlı sayıda hasta kaydedildiği için orta düzeyde böbrek yetmezliği olan hastalarda günlük 800 mg dozu test edilmemiştir. Aynı şekilde, şiddetli böbrek yetmezliği olan yalnızca 2 hasta düşük (100 mg) doza kaydedilmiş ve daha yüksek dozların hiçbiri test edilmemiştir. Çalışmaya hiçbir hemodiyaliz hastası kaydedilmemiştir. Literatür verileri, son evre böbrek hastalığı olan ve hemodiyaliz uygulanan bir kişide günlük 400 mg dozun çok iyi tolere edildiğini göstermiştir. Diyaliz, imatinibin plazma kinetiklerini engellememiştir. Böbrekler yoluyla atılım imatinib için minör bir eliminasyon yolu olduğundan, şiddetli böbrek yetmezliği olan ve diyaliz uygulanan hastalara 400 mg'lık başlangıç dozu ile tedavi uygulanabilmektedir. Ancak, bu hastalarda dikkatli olunması önerilmektedir. Tolere edilememesi halinde doz azaltılabilir ya da etki görülmemesi halinde doz arttırılabilir (bkz. 4.2 Pozoloji ve uygulama şekli; 4.4 Özel kullanım uyarıları ve önlemleri ve 5.2 Farmakokinetik özellikler bölümleri). Tablo 8 Böbrek fonksiyonu sınıflandırması
5.2. Farmakokinetik özellikler İmatinibin farmakokinetiği 25 - 1000 mg'lık bir doz aralığında değerlendirilmiştir. Plazma farmakokinetik profilleri 1. günde ve plazmada kararlı düzeylerin elde edildiği 7. ya da 28. günde analiz edilmiştir. Emilim:Kapsül formülünün ortalama mutlak biyoyararlanımı % 98'dir. Bir oral dozu takiben plazma imatinib eğri altında kalan alan (EAA) değerlerinde, yüksek oranda bir hastalar arası değişkenlik (% 40-60) görülmüştür. Yüksek yağ içeren bir gıda ile birlikte verildiğinde, imatinibin emilim oranı minimal düzeyde azalmış (Cmaks'da % 11 azalma ve tmaks'da 1.5 saatlik uzama), açlık koşullarına göre EAA değerinde küçük bir azalma (% 7.4) olmuştur. Dağılım:Klinik açıdan uygun konsantrasyonlarda kullanılan imatinibin plazma proteinlerine bağlanması yaklaşık % 95 olmuş, in vitro deneyler temelinde, daha çok albümin ve alfa-asit-glikoproteine, az miktarda da lipoproteine bağlanmıştır. Biyotransformasyon:İnsanlarda, dolaşımdaki temel metaboliti ana ilaç ile in vitro benzer etkinlikte olduğu gösterilmiş N-demetillenmiş piperazin (CGP71588) türevidir. Bu metabolitin plazma EAA değerinin imatinibin EAA değerinin sadece % 16'sı olduğu bulunmuştur. N-demetile metabolitin plazma proteinlerine bağlanması, asıl bileşikteki gibidir. Eliminasyon:İmatinibin 14C-işaretli tek oral dozundan sonra, dozun yaklaşık % 81'i 7 gün içinde feçesle (dozun % 68'i) ve idrarla (dozun % 13'ü) itrah edilmiştir. Değişmemiş durumdaki imatinib, dozun % 25 ' ini (% 5 idrar, % 20 feçes) oluşturmuştur, geriye kalan kısım metabolitlerdir. Doğrusallık / doğrusal olmayan durum :Sağlıklı gönüllülerde oral uygulamanın ardından, imatinib t1/2 değeri yaklaşık 18 saat olması günde tek doz şeklindeki pozolojinin uygun olduğu izlenimini vermektedir. Oral olarak 25-1000 mg imatinib uygulandıktan sonra artan dozla birlikte ortalama EAA artışı doğrusal bir seyir izlemiştir. Tekrarlanan dozlarda imatinib kinetiğinde değişiklik olmamış ve günde bir kez uygulandığında birikim, kararlı ilaç konsantrasyonunun 1.5-2.5 katı olmuştur. Farmakokinetik / farmakodinamik ilişkiler: Popülasyon farmakokinetikleri Popülasyon farmakokinetiği analizlerine göre yaşın dağılım hacmi üzerinde küçük bir etkisi olmuştur (> 65 yaşındaki hastalarda % 12 artış). Bu değişimin klinik açıdan anlamlı olmadığı düşünülmüştür. Vücut ağırlığının imatinib klerensi üzerindeki etkisine bakıldığında, 50 kg ağırlığındaki bir kişide klerensin 8.5 l/s, olması beklenirken, 100 kg ağırlığındaki bir kişideki klerens 11.8 l/s'e yükselmektedir. Bu değişiklikler vücut ağırlığına göre bir doz ayarlaması yapılması için yeterli olarak kabul edilmemiştir. Cinsiyetin imatinib kinetiği üzerinde etkisi olmamıştır. Yeni tanı konulmuş KML vakalarındaki Faz III çalışmada diğer popülasyon farmakokinetiği analizleri, kovariyans faktörlerinin ve birlikte kullanılan diğer ilaçların gerek klerens, gerekse hacim üzerindeki etkilerinin küçük olduğunu, doz ayarlamasına ihtiyaç bırakmadığını göstermiştir. Çocuklarda farmakokinetik Bir Faz I ve Faz II çalışmasında oral imatinib, pediatrik hastalarda da, erişkin hastalardaki gibi hızla emilmiştir. Çocuklarda 260 ve 340 mg/m2 imatinible elde edilen EAA değerleri, erişkinlerde sırasıyla 400 ve 600 mg imatinible elde edilenler gibidir. 340 mg/m2 imatinibin birinci ve sekizinci günlerdeki EAA(0-24 saat) değerleri bu ilacın, tekrarlanan dozlardan sonra 1.7 kat biriktiğini göstermiştir. Organ fonksiyonu bozukluğu İmatinib ve metabolitleri böbrek yoluyla anlamlı miktarda atılmazlar. Böbrek fonksiyonlarında hafif ve orta şiddette bozukluk olan hastalar, böbrek fonksiyonları normal hastalardan daha yüksek plazma değerlerine sahip görünmektedir. Artış yaklaşık olarak 1.5-2 kattır ve imatinibin güçlü bir biçimde bağlandığı plazma AGP değerinde 1.5 katlık bir artışa karşılık gelir. Böbrek bozukluğu olan hastalarda imatinibin serbest ilaç klerensi muhtemelen böbrek fonksiyonları normal hastalardakinin bir benzeridir çünkü böbrekler yoluyla atılım imatinib için minör bir eliminasyon yolunu oluşturmaktadır (bkz. 4.2 Pozoloji ve uygulama şekli, 4.4 Özel kullanım uyarıları ve önlemleri ve 5.1 Farmakodinamik özellikler). Farmakokinetik analiz sonuçlarının kişiden kişiye değişikliklerin söz konusu olduğunu göstermesine rağmen, değişik derecelerde karaciğer yetersizliği olan hastalardaki imatinibe ortalama maruz kalım, karaciğer fonksiyonları normal olan hastalara kıyasla yükselmemiştir (bkz 4.2 Pozoloji ve uygulama şekli, 4.4 Özel kullanım uyarıları ve önlemleri, 4.8 İstenmeyen Etkiler, 5.1 Farmakodinamik Özellikler ve 5.2 Farmakokinetik Özellikler bölümleri) 5.3. Klinik öncesi güvenlilik verileri İmatinibin güvenlilik farmakolojisi, tekrarlanan doz toksisitesi, genotoksisite ve üreme toksisitesi çalışmalarında değerlendirilmiştir. Kemik iliği, periferik kan, lenfoid doku, gonadlar ve gastrointestinal kanal, imatinibin farmakolojik etkisi altında kalan hedef-organlardandır. Diğer hedef organlar arasında karaciğer ve böbrek yer almaktadır. İmatinib, sıçanlarda embriyotoksik ve teratojen etki göstermiştir. Preklinik fertilite ve erken embriyonik gelişim çalışmasında fertilite etkilenmemiştir; diğer yandan, yüksek doz uygulanmış erkek sıçanlarda daha düşük testis ve epididimal ağırlıklar ve azalmış hareketli sperm sayısı gözlenmiştir. Sıçanlarda preklinik pre ve postnatal çalışmada, ilk nesil yavrularda da fertilite imatinibden etkilenmemiştir. Sıçanlarda juvenil gelişim toksikolojisi çalışmasında yeni hedef organ belirlenmemiştir. (doğum sonrası 10 ila 70. gün). Juvenil toksikoloji çalışmasında, ortalama pediyatrik maruziyet olarak önerilen en yüksek doz olan 340 mg/m2 düzeyinin yaklaşık 0.3 ila 2 katı düzeylerde, büyüme üzerinde geçici etkiler ve vaginal açılma ve prepusyal ayrılmada gecikme gözlenmiştir. Ayrıca, ortalama pediyatrik maruziyet olarak önerilen en yüksek doz olan 340 mg/m2 düzeyinin yaklaşık 2 katı düzeylerde, juvenil hayvanlarda (yaklaşık olarak sütten kesilme döneminde) mortalite gözlemlenmiştir. 2 yıllık sıçan karsinojenisite çalışmasında 15, 30 ve 60 mg/kg/gün olarak imatinib uygulanması, erkeklerde 60 mg/kg/gün dozunda ve dişilerde >30 mg/kg/gün dozunda yaşam süresi üzerinde istatistiksel açıdan anlamlı azalmaya neden olmuştur. Ölenlerde yapılan histopatolojik inceleme, ölümün temel nedeni ya da öldürülme nedeni olarak kardiyomiyopati (her iki cinsiyet), kronik ilerleyici nefropati (dişiler) ve prepusyal bez papillomunu ortaya koymuştur. Neoplastik değişiklikler açısından hedef organlar böbrekler, mesane, üretra, prepusyal ve klitoral bez, ince bağırsak, paratiroid bezleri, adrenal bezler ve glandüler-olmayan mide olmuştur. Neoplastik lezyonlar bulunan çeşitli hedef organlardaki etki görülmeyen düzeyler (NOEL) şu şekilde saptanmıştır: böbrekler, mesane, üretra, ince bağırsak, paratiroid bezleri, adrenal bezler ve glandüler-olmayan mide için 30 mg/kg/gün ve prepusyal ve klitoral bez için 15 mg/kg/gün. Prepusyal/klitoral bezde papilloma/karsinoma 30 ve 60 mg/kg/gün olarak saptanmıştır ve bu değer, insandaki günlük maruziyetin (EAA değerine dayanarak 400 mg/gün ya da 800 mg/gün) yaklaşık 0.5 ila 4 ya da 0.3 ila 2.4 katına, ve çocuklardaki günlük maruziyetin (EAA değerine dayanarak 340 mg/m2) 0.4 ila 3.0 katına karşılık gelmektedir. 60 mg/kg/gün ile renal adenoma/karsinoma, mesane ve üretra papülomu, ince bağırsak adenokarsinomları, paratiroid bezi adenomları, adrenal bezlerde benign ve malign medüller tümörler ve glandüler-olmayan mide papillomaları/karsinomaları görülmüştür. Sıçan karsinojenisite çalışmalarından elde edilen bu bulguların insanlar için anlamı bilinmemektedir. Klinik çalışmalardan elde edilen güvenlilik verilerinin ve spontan advers olay bildirimlerinin bir analizi, genel popülasyonla karşılaştırıldığında imatinib ile tedavi edilen hastalarda genel malignite insidansında artışla ilgili bir kanıt ortaya koymamıştır. İlk klinik çalışmalarda saptanmayan non-neoplastik lezyonlar kardiyovasküler sistem, pankreas, endokrin organlar ve dişlerle ilgili olmuştur. En önemli değişiklikler bazı hayvanlarda kalp yetmezliği belirtilerine yol açan kardiyak hipertrofi ve dilatasyonu içermiştir. 6. FARMASÖTİK ÖZELLİKLER 6.1. Yardımcı maddelerin listesi Tablet çekirdeği:Mikrokristalin selüloz Hidroksi propil metil selüloz Krospovidon Kolloidal silikon dioksit Magnezyum stearat Film kaplama [Opadry II Orange (85F230022)]Polivinil alkol Polietilen glikol (Macrogol)/PEG 3350 Sarı demir oksit Talk Titanyum dioksit Kırmızı demir oksit 6.2 Geçimsizlikler Geçerli değildir. 6.3 Raf ömrü 24 aydır. 6.4 Saklamaya yönelik özel tedbirler 30 °C'nin altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriği Şeffaf PVC/Aklar- Alu folyo blister ambalaj Bir kutu içinde 30 tablet ve kullanma talimatı ile birlikte sunulmaktadır. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklar Kontrolü Yönetmeliği'ne uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİ DEVA Holding A.Ş. Halkalı Merkez Mah. Basın Ekspres Cad. No:1 34303 Küçükçekmece/İSTANBUL 8. RUHSAT NUMARASI 241/33 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİ İlk ruhsat tarihi: 12.03.2012 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ 27 /27 |
İlaç BilgileriImatis 400 Mg Flm TabletEtken Maddesi: Imatinib Mezilat Atc Kodu: LO1XE01 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.