Beriate 1000 Iu Iv Enjeksiyon İçin Liyofilize Toz... Kısa Ürün BilgisiKan ve Kan Yapıcı Organlar » Kanama Durdurucu İlaçlar » K Vitamini ve Diğer Hemostatikler » Kan Koagülasyon (pıhtılaşma) Faktörleri » Koagülasyon Faktörü 8 KISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADIBERIATE 1000 IU I.V. enjeksiyon/infüzyon için liyofilize toz içeren flakon2. KALİTATİF VE KANTİTATİF BİLEŞİM Etkin madde:İnsan Koagülasyon Faktör VIII (FVIII: C- Faktör VIII Aktivitesi) 1000 IU (100 IU/mL)BERIATE'ın spesifik aktivitesi yaklaşık olarak 270 IU/mg protein' dir.* *Faktör VIII' in potensi (IU) Avrupa Farmakopesi' nde yer alan kromojenik analiz yöntemi ile hesaplanır. Yardımcı maddeler:Sodyum klorür 45-70 mgSodyum hidroksit (az miktarda pH ayarlamak için) Yardımcı maddeler için bölüm 6.1' e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk veya infüzyonluk çözelti için liyofilize toz Çözelti renksiz, berrak ila hafif opalesan.4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarHemofili A (konjenital Faktör VIII eksikliği) olan hastaların kanamalarının profilaksi sinde ve tedavisinde kullanılır.BERIATE sonradan edinilmiş Faktör VIII eksikliğinin tedavisinde kullanılabilir. BERIATE farmakolojik olarak etkili olacak Von Willebrand Faktör içermez. Bundan dolayı Von Willebrand hastalığında endike değildir.4.2. Pozoloji ve uygulama şekliUygulanacak doz; hastanın klinik durumuna, kanamanın şiddetine, ölçülen FVIII eksikliğinin derecesine göre, bu konuda uzman doktorlar tarafından belirlenmelidir.Pozoloji:Uygulanacak FVIII seviyesi miktarı uluslararası birim (IU) olarak belirlenir ve FVIII ürünleri için güncel WHO standardı ile hesaplanır. Plazmadaki FVIII aktivitesi (FVIII-C) ya yüzde olarak (normal insan plazması) ile ilgili olarak ya da IU (plazmadaki FVIII için belirlenen uluslararası standarda göre) ifade edilir.1 IU FVIII-C normal insan plazmasındaki 1 mL FVIII miktarına eşdeğerdir. Gerekli FVIII miktarı hesaplanırken deneysel bulgular temel alınır. Vücut ağırlığının her 1 kg için 1 IU FVIII plazmadaki FVIII aktivitesini normal aktivitenin % 2 oranında artırır (2 IU/ kg). Gereken dozaj aşağıdaki formüle göre belirlenebilir: Başlangıç dozu: Gereken ünite = Vücut ağırlığı (kg) X İstenilen F VIII artışı (% ya da IU/ dL) X 0,5 Uygulama sıklığı ve süresi:Uygulama sıklığı ve süresi bireyin klinik durumuna göre kişisel olarak belirlenir. Aşağıdaki tabloda belirtilen hemoraji vakalarında FVIII düzeyi, ilgili bölümde belirtilen plazma aktivitesi düzeyinin (normalin %' si olarak ya da IU/ dL) altına düşmemelidir.Uygulanacak doz ve infüzyon sıklığına, ölçülen FVIII seviyesine göre karar verilir. Özellikle büyük cerrahi operasyonlarda koagülasyon analizi ile (plazma FVIII-C) tedavinin yakından takibi gereklidir. FVIII uygulamasına hastanın cevabı, in vivo geri kazanım ve yarılanma ömrü, hastadan hastaya farklılık gösterir. Ciddi Hemofili A hastalığı olanlarda kanamaya karşı uzun süreli profilaksi için 2 ile 3 günlük aralıklarla, vücut ağırlığının her kg için 20- 40 IU Faktör VIII verilmelidir. Eğer gerekiyorsa başka tedavi önlemleri de alınmalıdır. Aşağıdaki tablo; kanama olaylarında ve cerrahi müdahalelerde uygulanabilecek miktarlar konusunda bir kılavuz olarak kullanılabilir.
Uygulama şekli:BERIATE intravenöz yolla uygulanır.Ürünü Bölüm 6.6. Beşeri Tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler başlığı altında tarif edildiği şekilde hazırlayınız. Enjeksiyon: Ürün uygulanmadan önce oda sıcaklığına veya vücut sıcaklığına getirilmelidir. Hastanın rahat olacağı hızda damar içine enjekte edilir veya infüzyon şeklinde verilir. İnfüzyonun verilme hızı veya enjeksiyon hızı dakikada 2 mL' yi geçmemelidir. Herhangi bir acil müdahale için hasta sürekli gözlem altında tutulmalıdır. BERIATE uygulanması ile ilgili herhangi bir reaksiyon oluşursa, hastanın gerekli klinik koşullara gelebilmesi için infüzyon oranı azaltılabilir ya da tamamen kesilebilir. Bakınız Bölüm 4.4. 'Özelkullanım uyarıları ve önlemleri''Özel popülasyonlara ilişkin ek bilgiler:Böbrek/ Karaciğer yetmezliği:BERIATE'ın böbrek/karaciğer yetmezliği olan hastalarda kullanımı ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle BERIATE'ın bu hastalarda kullanımında tedbirli olunmalı ve hasta açısından yarar/ zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır.Pediyatrik popülasyon:BERIATE'ın çocuklarda doz ayarlaması ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle BERIATE'ın çocuklarda kullanımında tedbirli olunmalı ve çocuk açısından yarar/ zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır.Geriyatrik popülasyon:BERIATE'ın yaşlı hastalarda kullanımı ile ilgili yapılan bir klinik çalışma yoktur. Bu nedenle BERIATE'ın yaşlı hastalarda kullanımında tedbirli olunmalı ve hasta açısından yarar/ zarar değerlendirmesi yapıldıktan sonra kullanılmalıdır.4.3. KontrendikasyonlarEtkin maddeye veya yardımcı maddelerden herhangi birine aşırı duyarlılığı olan hastalarda kontrendikedir.Virüs GüvenliğiBERIATE insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibi, çeşitli hastalıklara yol açabilen enfeksiyon yapıcı ajanlar içerebilirler. BERIATE' da Varyant Creutzfeldt-Jacob hastalığının bulaşma riski teorik olarak minimumken, klasik Creutzfeldt-Jacob hastalığının bulaşma riski hiçbir kanıtla desteklenmez. Alınan önlemlere rağmen, bu tür ürünler halen potansiyel olarak hastalık bulaştırabilir.Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıp kalmadığının izlenmesi, belirli virüs enfeksiyonlarının halihazırda varlığının test edilmesi ve belirli virüslerin yok edilmesi ve/veya inaktivasyonu ile azaltılmıştır. Bütün bu önlemlere rağmen, bu ürünler hala potansiyel olarak hastalık bulaştırabilirler. Ayrıca, henüz bilinmeyen enfeksiyon yapıcı ajanların bu ürünlerin içersinde bulunma ihtimali mevcuttur.HIV, HBV, HCV gibi zarflı virüsler ve HAV gibi zarflı olmayan virüslerin etkisi için önlemlerin alınmasına dikkat edilmelidir. Parvovirus B19 gibi zarflı olmayan virüslere karşı alınan tedbirler sınırlı sayıda olabilir. Parvovirus B19 enfeksiyonu, gebelikte (fetal infeksiyon) ve immün yetmezlik ya da kırmızı kan hücre üretiminde artış olan hastalarda tehlikeli olabilir (hemolitik anemi gibi).Bu nedenle: Düzenli ve tekrarlanan sürelerle BERIATE kullanılması gerekiyorsa, hastalık yapıcı etkenlerin hastalara bulaşmasını önlemek için uygun aşıların (Hepatit A ve Hepatit B aşıları) hastalara yaptırılması önerilebilir. BERIATE'ın her kullanımında doktor tarafından ilacın alınış tarihi, seri numarası ve enjekte edilen ilaç hacmi kayıt altına alınmalıdır. Plazma türevli ürünlerin intravenöz infüzyonunda aşırı duyarlı alerjik tip reaksiyonlar görülebilir. Hastalar aşırı duyarlılık reaksiyonlarının neden olacağı; ürtiker, göğüs sıkışması, göğüste hırıltı, hipotansiyon ve anafilaksi hakkında bilgilendirilmelidir. Bu semptomlar görüldüğü takdirde hasta ilacın kullanımı derhal kesmeli ve doktoruna başvurmalıdır. Hasta şoka girerse; şok tedavisinde geçerli olan tıbbi standart tedaviler uygulanmalıdır. İnhibitör gelişimi Bazı hastalarda Faktör VIII' e karşı inhibitör gelişebilir. Eğer verilen gerekli doz ile beklenen Faktör VIII aktivitesi plazma seviyesine ulaşılamamış ise, ya da kanama kontrol altına alınamamış ise Faktör VIII inhibitörü seviyesi, analiz ile hesaplanır. Hastada inhibitör seviyesi yüksek ise, Faktör VIII tedavisi etkili olmayabilir ve diğer tedavi şekilleri göz önünde bulundurulabilir. Bu inhibitörler genellikle Faktör VIII prokoagülan aktivitesine yönlenmiş IgG immunoglobulinleridir ve modifiye tayin yöntemi kullanılarak her plazma mililitresi için Bethesda Ünitesi (BU)ile miktarlandırılırlar. İnhibitör gelişmesi riski anti-hemofilik Faktör VIII' e maruz kalınma ile ilgilidir, bu risk ürüne maruz kalınan ilk 20 gün için en yüksek düzeydedir. Nadiren, maruz kalınan ilk 100 günden sonra inhibitör gelişimi söz konusudur.Genel olarak insan koagülasyon Faktörü VIII ile tedavi edilen hastalar, uygun klinik takip ve laboratuar testleri kullanılarak inhibitör gelişimine karşı dikkatlice izlenmelidir. Ayrıca Bakınız Bölüm "4.8. İstenmeyen etkiler".Bu tıbbi ürün her 1000 IU de 1 mmol ya da daha fazla sodyum içerebilir. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriİnsan koagülasyon faktör VIII ile diğer tıbbi ürünler arasında bilinen bir etkileşim bulunmamaktadır. Fakat yine de BERIATE diğer tıbbi ürünler ile karıştırılmamalıdır.Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin hiçbir klinik etkileşim çalışması yürütülmemiştir.Pediyatrik popülasyonPediyatrik popülasyona ilişkin hiçbir klinik etkileşim çalışması yürütülmemiştir.4.6. Gebelik ve laktasyon Genel tavsiyeGebelik kategorisi C' dir.Çocuk doğurma potansiyeli bulunan kadınlar/ Doğum kontrolüBERIATE'ın çocuk doğurma potansiyeli bulunan kadınlarda kullanıldığında üreme kapasitesini etkileyip etkilemediği bilinmemektedir. Bu nedenle çocuk doğurma potansiyeli bulunan kadınlarda planlanmış bir gebelikten önce uygun bir alternatif tedaviye geçilmelidir.Gebelik dönemiBERIATE'ın gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir ve insanlara yönelik potansiyel risk bilinmemektedir. Bu nedenle BERIATE gerekli olmadıkça gebelik döneminde kullanılmamalıdır. Buna rağmen, BERIATE tedavisinin durdurulup durdurulmayacağına/ tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verilirken, BERIATE'nin anne açısından faydası dikkate alınmalıdır.Laktasyon dönemiBERIATE'ın anne sütüyle atılıp atılmadığı bilinmemektedir. Buna rağmen, emzirmenin durdurulup durdurulmayacağına ya da BERIATE tedavisinin durdurulup durdurulmayacağına/ tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verilirken, emzirmenin çocuk açısından faydası ve BERIATE tedavisinin emziren anne açısından faydası dikkate alınmalıdır.Üreme yeteneği/FertiliteBERIATE'ın fertilite üzerinde doğrudan veya dolaylı olarak zararlı etkilerinin olduğu hakkında yeterli veri bulunmamaktadır.4.7. Araç ve makine kullanımı üzerindeki etkilerAraç ve makine kullanmaya etkisi yoktur.4.8. İstenmeyen etkilerAşağıdaki sıklık grupları kullanılmıştır:Çok yaygın (> 1/10); yaygın (>1/100 ila <1/10);yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000);çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) Bağışıklık sistemi hastalıklarıÇok seyrek: Alerjik (hipersensitivite) reaksiyonlar; anjioödem, enjeksiyon veya infüzyon bölgesinde yanma, batma, üşüme hissi, al basması, jeneralize ürtiker, baş ağrısı, ürtiker, hipotansiyon, letarji, mide bulantısı, huzursuzluk, taşikardi, göğüste daralma, titreme, kusma, hırıltılı soluma. Bazen bu yan etkiler şok içeren ciddi anafilaksiye neden olabilir. Bilinmiyor: Hemofili A hastalarında faktör VIII' e karşı nötralize antikorlar (inhibitörler) gelişebilir. Faktör VIII'e karşı inhibitör gelişimi yetersiz klinik cevap oluşturur. Bu durumda kontrol edilemeyen kanamalara neden olur. Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar:Çok Seyrek: Uygulama yerinde yanma, batma Ateş 4.9. Doz aşımı ve tedavisiİnsan pıhtılaşma Faktör VIII' e ait doz aşımı belirtileri bildirilmemiştir.5. FARMAKOLOJİK ÖZELLİKLER5.1. Farmakodinamik özelliklerFarmakoterapötik grubu: Antihemorajikler: Koagülasyon Faktör VIIIATC kodu: B02BD02 Etki MekanizmasıBERIATE, insan vücudunda bulunan endojen FVIII ile aynı etkileri gösterir. Hemofilik hastalara infüzyon yapıldığı zaman FVIII hastanın dolaşımındaki VWF' e bağlanır. Aktive edilmiş Faktör VIII, aktive edilmiş Faktör IX kofaktörü gibi davranarak, Faktör X' un aktive edilmiş Faktör X' a dönüşmesini hızlandırır. Aktive olan Faktör X, protrombini trombin haline çevirir. Daha sonra trombin fibrinojeni fibrine dönüştürür ve pıhtılaşma gerçekleştirilir. Hemofili A, cinsiyet ile bağlantılı, FVIII seviyesinin kanda düşmesi sonucu oluşan kan koagülasyon bozukluğunun görüldüğü kalıtsal bir hastalıktır. Eklem yerlerinde, kaslarda ya da iç organlarda spontan olarak ya da bir kaza veya cerrahi bir müdahale sonucunda durdurulamayan aşırı kanama oluşur. Yerine koyma tedavisi ile Faktör VIII seviyesi yükseltilir. Tedavi faktör eksikliğinde geçici düzelme sağlar ve kanama durdurulur. 5.2. Farmakokinetik özellikler Genel özelliklerEmilim:Uygulama yeri açısından (intravenöz) ilaç direkt kana karışır. Dağılım:Plazma konsantrasyon-zaman eğrisinin altında kalan alan 0.4 saat x kg/mL (standart sapma 0.2). Ortalama tutulma zamanı (MRT) 17 saattir (standart sapma 5.5 saat) Biyotransformasyon:Plazma Faktör VIII aktivitesi mono veya biekspotensiyel bozulmayla azalır. Eliminasyon:Eliminasyon yarılanma ömrü ortalama 12 saat olmakla birlikte, 5-22 saat arasında değişir. Ortalama değer yaklaşık 12 saattir. Ortalama klerans 3 mL/saat/ kg'dır (standart sapma 1.5 mL/saat/kg) Doğrusallık/ Doğrusal olmayan durum:Bilgi bulunmamaktadır. 5.3. Klinik öncesi güvenlilik verileriGenel ToksisiteTekrarlanan doz ile toksisite çalışmaları heterelog proteinlere karşı geliştirilen antikorlar nedeniyle yapılamamıştır. İnsanlar için vücut ağırlığındaki kilogram başına tavsiye edilen dozun birkaç katı laboratuar hayvanlarında denenmiş ve bir toksisite oluşmamıştır. Mutajenite Klinik deneyler insan plazma pıhtılaştırma Faktörü VIII için hiçbir tümörojenik veya mutajenik etki bildirmediğinden, özellikle heterolog türler üzerine deneysel çalışmalar yapılması zorunlu değildir. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiSakarozAminoasetik asit (glisin) Kalsiyum klorür Sodyum klorür Sodyum hidroksit Enjeksiyonluk su 6.2. GeçimsizliklerBu tıbbi ürün, çözücüsü ve seyreltilmiş olarak diğer tıbbi ürünlerle karıştırılmamalıdır.6.3. Raf ömrü24 aydır.6.4. Saklamaya yönelik özel tedbirlerBERIATE'ı +2 °C ila +8°C' de (buzdolabında) saklayınız. Dondurmayınız.Donmuş ürünü çözüp kullanmayınız. Isı ve ışıktan koruyunuz. Işıktan korumak için orijinal ambalajında saklayınız. Flakonu direkt ısıdan koruyunuz. Flakon vücut sıcaklığından (37°C) daha fazla ısıtılmamalıdır. Sulandırılan ürün dondurulmamalıdır. Ürün açıldıktan sonra kimyasal ve fiziksel stabilitesini 25 °C' de 8 saat korur. Ürünün mikrobiyolojik kontaminasyona karşı korunması için hemen kullanılmalıdır. Eğer hemen kullanılmayacaksa ürün oda sıcaklığında 8 saatten fazla tutulmamalıdır. 6.5. Ambalajın niteliği ve içeriğiTip I camdan yapılmış renksiz enjeksiyonluk flakonlar vakum altında kauçuk tıpa, alüminyum kapak ve plastik disk ile kapatılmıştır.BERIATE 1000 IU Liyofilize toz içeren 1 adet flakon 10 mL enjeksiyonluk su içeren 1 adet flakon 1 adet transfer seti 1 adet disposable filtre iğnesi 6.6. Beşeri Tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerGenel talimatlar:Çözelti berrak veya hafif opak olmalıdır. Hazırlanan ürünler filtre/çekme işleminden sonra (aşağıya bakınız) ve uygulama öncesinde herhangi bir partikül varlığı veya renk değişimi açısından görsel olarak incelenmelidir. Bulanık veya kalıntılar (birikinti/partikül) içeren çözeltileri kullanmayınız. Hazırlama: Çözücüyü oda sıcaklığına getiriniz. Toz ve çözücü flakonlarının kapaklarının çıkarıldığından ve stoperlerin aseptik bir solüsyonla silinerek Mix2Vial paketinin açılmasından önce kendiliğinden kuruduğundan emin olunuz. 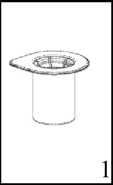 l.Kapağı soyarak Mix2Vial paketini açınız. Mix2Vial' i blister paketinden çıkarmayınız. 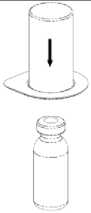 2
22. Çözücü flakonu düz ve temiz bir yüzey üzerine yerleştiriniz ve flakonu sıkıca tutunuz. Blister paketi ile birlikte aldığınız Mix2Vial setinin mavi adaptör ucunun tepe noktasını çözücü flakonun stoperinin üzerine doğru itiniz. 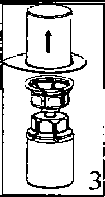 3. Kenarından tutarak ve dikey şekilde yukarıya doğru çekerek, blister paketini Mix2Vial setinden dikkatlice çıkarınız. Sadece blister paketini çekerek çıkardığınızdan ve Mix2Vial setini çıkarmadığınızdan emin olun. 4. Toz flakonunu düz ve sağlam bir zemin üzerine yerleştiriniz. Çözücü flakonunu takılı olan Mix2Vial setiyle birlikte ters çevirin ve saydam adaptör ucunun tepe noktasını, toz flakonun stoperinden aşağıya doğru bastırınız. Çözücü otomatik olarak toz flakonunun içine akacaktır. 5. Bir elinizle Mix2Vial setinin ürün tarafını, diğer elinizle de çözücü tarafını tutunuz ve ürünün çözülmesi sırasında aşırı köpük oluşmasını önlemek için seti dikkatli bir şekilde iki parçaya ayırınız. Çözücü flakonunu mavi Mix2Vial adaptörü takılı şekilde atınız. 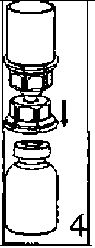 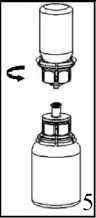 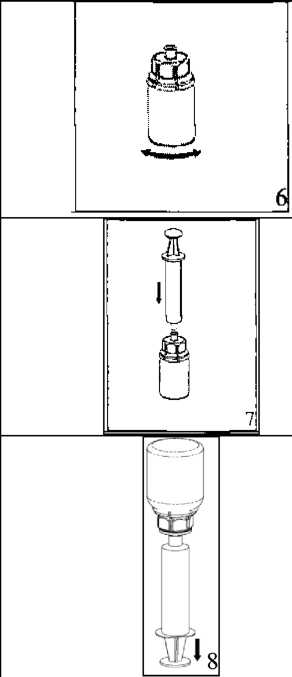 6. Tozun tamamen çözündüğünden emin oluncaya kadar ürün flakonunu saydam adaptör takılı şekilde hafifçe sağa sola çeviriniz. Çalkalamayınız. 7. Boş, steril bir şırıngaya hava çekiniz. Ürün flakonu yukarıya doğru iken, şırıngayı Mix2Vial'ın Luer Lock bağlantı parçasına bağlayınız. Ürün flakonuna hava enjekte ediniz. 8. Şırınganın pistonunu basılı tutarak sistemi baş aşağı çeviriniz ve konsantreyi, pistonu yavaşça geriye çekerek şırıngaya alınız. 9. Çözelti şırıngaya aktarıldıktan sonra, şırınganın gövdesini sıkıca tutunuz (şırınganın sapını aşağıya bakacak şekilde tutarak) ve saydam Mix2Vial adaptörünü şırıngadan ayırınız. ¥
9 Solüsyonu hemen yavaş intravenöz enjeksiyonla veya infüzyonla uygulayınız Ürünle dolu şırıngaya kan girmemesine son derece dikkat edilmelidir. Kullanılmamış olan ürünler ya da artık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmelik lerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİFarma-Tek İlaç San. ve Tic. Ltd. Şti.Şerifali Mah. Bayraktar Bulvarı. Beyan Sok. No:12 34775 Ümraniye/İstanbul 8. RUHSAT NUMARASI179. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİ |
İlaç BilgileriBeriate 1000 Iu Iv Enjeksiyon İçin Liyofilize Toz...Etken Maddesi: Insan Koagülasyon Faktör Viii Atc Kodu: B02BD02 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.