Tafinlar 75 Mg Kapsül Kısa Ürün BilgisiAntineoplastik ve İmmünomodülatör Ajanlar » Antineoplastik İlaçlar (Kanser İlaçları) » Diğer Kanser İlaçları » Protein Kinaz İnhibitörleri KISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADI TAFİNLAR 75 mg kapsül 2. KALİTATİF VE KANTİTATİF BİLEŞİM Etkin madde: Her bir kapsül 75 mg dabrafenibe eşdeğer dabrafenib mesilat içerir. Yardımcı maddeler: Propilen glikol y.m. Yardımcı maddeler için 6.1.'e bakınız. 3. FARMASÖTİK FORM Sert kapsül. Üzeri GS LHF ve 75 mg baskılı opak, koyu pembe renkli kapsüller. 4. KLİNİK ÖZELLİKLER 4.1 Terapötik endikasyonlar TAFİNLAR, daha önce herhangi bir RAF yolağı inhibitörü kullanmamış, ECOG performans skoru 0 veya 1 olan, lokal tedaviler sonrası progresyon göstermiş ve lokal tedavilerin tekrar kullanılamadığı relaps veya metastatik BRAF V600 mutasyonu pozitif olan malign melanom hastalarında tek ajan olarak progresyona kadar kullanımda endikedir. Progresyon sonrası tek ajan veya başka tedavilerle kombinasyon şeklinde kullanılamaz. 4.2 Pozoloji ve uygulama şekli TAFİNLAR tedavisine konusunda uzman ve antikanser tıbbi ürünlerin kullanımında deneyimli bir hekim tarafından başlanmalı ve yakından takip edilmelidir. TAFİNLAR'ı kullanmadan önce hastaların BRAF V600 mutasyonuna sahip oldukları geçerliliği kanıtlanmış bir test kullanılarak doğrulanmalıdır. TAFİNLAR'ın etkililik ve güvenliliği yabanıl tip (wild-type) BRAF melanoması olan hastalarda gösterilmemiştir, bu nedenle TAFİNLAR BRAF yabanıl tip melanoma hastalarında kullanılmamalıdır (bkz. Bölüm 4.4 ve 5.1). Pozoloji: YetişkinlerTAFİNLAR önerilen dozu günde 2 kez 150 mg (iki 75 mg) kapsüldür (toplam günlük doz 300 mg'a karşılık gelir). TAFİNLAR yemekten en az 1 saat önce veya en az 2 saat sonra alınmalı ve dozlar arasında yaklaşık 12 saatlik zaman aralığı olmalıdır. TAFINLAR, hasta uyumunu arttırmak için her gün benzer zamanlarda alınmalıdır. Uygulama sıklığı ve süresi: Tedavi hastalık ilerleyene veya kabul edilemez bir toksisite gelişinceye kadar devam ettirilmelidir (bkz. Tablo 2). Unutulan dozlarEğer bir doz alınmamışsa ve diğer doza 6 saatten daha az bir süre kaldıysa, unutulan doz alınmamalıdır. Doz modifikasyonuDoz modifikasyonu gereksinimlerini etkili bir şekilde yönetmek için 50 mg ve 75 mg'lık iki dabrafenib kapsül formu bulunmaktadır. Advers reaksiyonlar, tedavinin kesilmesi, doz azaltılmasını veya tedavinin bırakılmasını gerektirebilir (bkz. Tablolar 1 ve 2). Kütanöz skuamöz hücre karsinomu (cuSCC) veya yeni primer melanoma advers reaksiyonlarında doz modifikasyonları veya doza ara vermeler önerilmez (bkz. Bölüm 4.4). Eğer hastanın ateşi >38.5°C ise, tedaviye ara verilmelidir. Hastalar enfeksiyon belirtileri ve bulguları açılarından değerlendirilmelidir (bkz. Bölüm 4.4). Önerilen doz seviyesi azaltmaları ve önerilen doz modifikasyonları sırasıyla Tablo 1 ve Tablo 2'de verilmiştir. Günde 2 kez 50 mg'dan daha düşük doza yol açan pozoloji ayarlamaları önerilmemektedir. Tablo 1: Önerilen dabrafenib doz seviyesi azaltmları
Tablo 2: Herhangi bir Advers Olay (AO) derecesine göre dabrafenib doz modifikasyon şeması
Hastanın advers reaksiyonlarının etkili şekilde yönetildiği durumda, doz düşürülmesinde izlenen adımlar doz yükseltilmesinde de göz önünde bulundurulabilir. Doz günde 2 kez 150 mg'ı geçmemelidir. Uygulama şekli: TAFİNLAR kapsüller su ile bütün olarak yutulmalıdır. Kapsüller çiğnenmemeli, ezilmemeli ve dabrafenib'in kimyasal instabilitesi nedeni ile ve yiyecek ve içecek ile karıştırılmamalıdır. Özel popülasyonlara ilişkin ek bilgiler: Böbrek yetmezliği: Hafif veya orta dereceli renal bozukluğu olan hastalarda doz ayarlamasına gerek yoktur. Popülasyon farmakokinetik analizine göre hafif ve orta dereceli renal bozukluk dabrafenibin oral klerensi veya metabolitlerinin konsantrasyonları üzerinde anlamlı etkisi yoktur (bkz. Bölüm 5.2). Ağır renal bozukluğu olan hastalarda klinik veri yoktur ve doz ayarlaması için potansiyel ihtiyaç tespit edilememiştir. TAFİNLAR ağır renal bozukluğu olan hastalarda dikkatli kullanılmalıdır. Karaciğer yetmezliği: Hafif hepatik bozukluğu olan hastalarda doz ayarlamasına gerek yoktur. Popülasyon farmakokinetik analize göre hafif hepatik bozukluğun dabrafenibin oral klerensi veya metabolitleri üzerinde anlamlı etkisi yoktur (bkz. Bölüm 5.2). Orta-ağır hepatik bozukluğu olan hastalarda klinik veriler yoktur ve doz ayarlaması için potansiyel ihtiyaç tespit edilememiştir. Dabrafenib ve metabolitleri için primer eliminasyon yolu hepatik metabolizma ve biliyer sekresyondur ve orta-ağır hepatik bozukluğu olan hastalarda maruziyet artabilir. TAFİNLAR, orta veya ağır hepatik bozukluğu olan hastalarda dikkatli kullanılmalıdır. Pediyatrik popülasyon: TAFİNLAR'ın çocuklarda ve adolesanlarda güvenliliği ve etkililiği gösterilmemiştir (<18 yaş). Klinik veri bulunmamaktadır. Geriyatrik popülasyon: Yaşlı hastalarda doz ve doz aralıkları terapötik ihtiyaçlar ve tolere edilebilirlik ile uyumlu bir biçimde, dikkatlice ayarlanmalıdır. 4.3 Kontrendikasyonlar TAFİNLAR'ın etkin maddesine veya içerisinde bulunan diğer yardımcı maddelerden herhangi birine karşı aşırı duyarlılığı olan hastalarda kontrendikedir. 4.4 Özel kullanım uyarıları ve önlemleri TAFİNLAR almadan önce hastaların BRAF V600 mutasyonuna sahip oldukları geçerliliği kanıtlanmış bir test ile doğrulanmalıdır. TAFİNLAR'ın etkililiği ve güvenliliği yabanıl tip BRAF melanoması olan hastalarda gösterilmemiştir, bu nedenle TAFİNLAR BRAF yabanıl tip melanoma hastalarında kullanılmamalıdır (bkz. Bölüm 5.1). Şiddetli enfeksiyöz dışı febril olaylar ve pireksiAteş klinik çalışmalarda bildirilmiştir. Vakaların az bir kısmında enfeksiyon dışı febril olaylar ateş ile tanımlanmış olup normal bazal renal fonksiyonu olan hastalarda buna şiddetli rigor, dehidrasyon, hipotansiyon ve/veya akut renal yetmezlik de eşlik etmektedir. Bu şiddetli enfeksiyöz dışı febril olaylar tipik olarak tedavinin ilk ayı içerisinde başlar. Şiddetli enfeksiyöz dışı febril olaylar klinik çalışmalarda hastaların %1'inde görülür ve doza ara vermeye ve/veya destekleyici bakıma iyi yanıt verir. Hastanın ateşi >38.5°C olduğunda dabrafenib tedavisine ara verilmelidir. Hastalar enfeksiyon bulgu ve belirtileri açısından değerlendirilmelidir. Ateş uygun profilaksi olarak non-steroid antiinflamatuvar tıbbi ürünler veya asetaminofen kullanımı ile düşürüldükten sonra dabrafenib başlanabilir. Eğer ateş diğer şiddetli bulgu veya belirtiler ile birlikteyse TAFİNLAR ateş düştüğünde azaltılmış bir dozda ve klinik olarak uygun şekilde tekrar başlanmalıdır (bkz. Bölüm 4.2). Kütanöz Skuamöz Hücre Karsinomu (cuSCC)cuSCC (keratoakantoma veya mikst keratoakantoma alt tip olarak sınıflanmışları da içeren) vakaları TAFİNLAR tedavisi alan hastalarda bildirilmiştir (bkz. Bölüm 4.8). TAFİNLAR tedavisinden önce, tedavi sırasında, ilk 6 ay, ayda bir ve daha sonra 3 ay aralıklarla deri muayenesi önerilir. Ek bir muayenenin yapılması TAFİNLAR kesildikten 2 ay sonra düşünülmelidir. TAFİNLAR tedavisinin kesilmesinden sonra veya başka bir antineoplastik ajan tedavisine başlayana kadar deri muayenesine 6 ay süreyle devam edilmelidir. cuSCC vakaları dermatolojik eksizyon ile tedavi edilmelidir ve TAFİNLAR tedavisi doz ayarlaması yapılmadan devam edilmelidir. Hastalar yeni lezyon gelişirse hekimlerine bildirmeleri konusunda bilgilendirilmelidir. Yeni primer melanomKlinik çalışmalarda yeni primer melanom bildirilmiştir. Bu vakalar tedavinin ilk 5 ayında tespit edilmiştir ve eksizyon ile tedavi edilmiştir; tedavi modifikasyonuna gereksinim olmamıştır. Deri lezyonlarının izlemi kütanöz skuamöz hücre karsinomu için tarif edildiği şekilde yapılmalıdır. Non-kütanöz malignansiYapılan in vitrodeneyler RAS mutasyon içeren BRAF yabanıl tip hücrelerde BRAF inhibitörlerine maruziyette mitojenle aktive edilen protein kinaz (MAP kinaz) sinyal iletiminde paradoksal aktivasyon oluştuğunu göstermiştir. Bu durum RAS mutasyonları mevcut olduğunda dabrafenib maruziyetiyle kutanöz-olmayan malignitelerin riskinde artışa neden olabilir. Dabrafenib ile gerek başka bir BRAF inhibitörü (kronik miyelomonositik lösemi ve kutanöz-olmayan baş ve boyun SCC'si) gerekse MEK inhibitörü trametinib (kolorektal kanser, pankreatik kanser) ile birlikte uygulandığında RAS'la bağlantılı malignite olguları bildirilmiştir.Tedavinin başlatılmasından önce hastalarda en azından gözle oral mukoza muayenesi ve lenf nodülü palpasyonuyla birlikte bir baş ve boyun muayenesinin yanısıra göğüs/abdomen Bilgisayarlı Tomografi (BT) taraması yapılmalıdır. Tedavi sırasında hastalar klinik olarak gerektiğinde izlenmelidir, buna her 3 ayda bir baş ve boyun mauyenesi ile her 6 ayda bir yapılan göğüs/abdomen BT taraması dahil edilebilir. Tedaviye başlamadan önce veya tedavi bittikten sonra ya da klinik açıdan gerekli olduğu düşünüldüğünde anal muayene ve pelvik muayene (kadınlarda) yapılması da önerilir. Klinik olarak uygun görüldüğünde tam kan sayımı yapılmalıdır. Dabrafenib kullanımı kesildikten sonra kutanöz-olmayan ikincil/tekrarlayan malignite takipleri 6 aya kadar veya başka bir anti-neoplastik terapi başlangıcına kadar devam etmelidir. Anormal bulgular klinik uygulamalara göre tedavi edilmelidir. Böbrek yetmezliğiDabrafenib tedavisi alan hastaların <%1'inde böbrek yetmezliği saptanmıştır. Gözlenen olgular genellikle pireksi ve dehidrasyonla bağlantılı olmuştur ve dozun kesilmesine ve genel destekleyici önlemlere iyi yanıt vermiştir. Granülomatoz nefrit bildirilmiştir (Bkz. Bölüm 4.8). Hastalarda tedavi sırasında rutin olarak serum kreatinin değerleri takip edilmelidir. Kreatininde artış olursa, klinik olarak uygun görüldüğünde dabrafenibin kesilmesi gerekebilir. Dabrafenib böbrek yetmezliği (kreatinin >1.5 x ULN şeklinde tanımlanmıştır) bulunan hastalarda çalışılmamıştır, bu nedenle bu ortamda kullanılmaması gerekir (bkz. Bölüm 5.2). ÜveitÜveit ve irit de dahil olmak üzere oftalmolojik reaksiyonlar bildirilmiştir. Görme ile ilgili bulgu ve belirtiler (ör; görme değişiklikleri, fotofobi ve göz ağrısı) tedavideki hastalarda rutin olarak izlenmelidir. PankreatitDabrafenib tedavisi alan hastaların <%1'inde pankreatit bildirilmiştir. Olaylardan biri dozun verildiği ilk gün görülmüş olup, azaltılmış bir dozda yeniden yükleme yapıldığında rekürens yaşanmıştır. Açıklanamayan abdominal ağrının serum amilaz ve lipaz ölçümü dahil olacak şekilde hemen araştırılması gerekir. Hastalar pankreatit nöbeti sonrasında yeniden dabrafenibe başlarken yakından izlenmelidir. QT uzamasıDabrafenib ile tedavi edilen hastaların %3'ünde en kötü durumda >60 milisaniyelik (ms) bir QTc uzaması gözlenmiştir (Birleştirilmiş güvenlik popülasyonunda bir kişi >500 ms). Düzeltilemeyen elektrolit anormalikleri (magnzeyum dahil), uzun QT sendromu olan veya QT aralığını uzattığı bilinen tıbbi ürünler alan hastalarda dabrafenib tedavisi önerilmez. Dabrafenib tedavisinden önce, tedaviden bir ay sonra ve doz modifikasyondan sonra tüm hastalarda elektrokardiyogram (EKG) ve elektrolit (magnezyum dahil) takibi yapılması zorunludur. Tedavinin ilk 3 ayında ayda bir, sonrasında klinik açıdan uygun olduğunda daha sık olarak özellikle orta ve şiddetli karaciğer yetmezliği bulunan hastaların daha fazla takip edilmesi önerilmektedir. QTc >500 ms olan hastalarda dabrafenib tedavisine başlanması önerilmez. Tedavi sırasında QTc 500 ms'yi aştığı takdirde, dabrafenib tedavisine geçici olarak ara verilmeli, elektrolit anormallikleri (magnezyum dahil) düzeltilmeli ve QT uzaması kardiyak risk faktörleri (örn., konjestif kalp yetmezliği, bradiaritmiler) kontrol altına alınmalıdır. QTc 500 ms'in altına indiğinde ve Tablo 2'de açıklandığı gibi düşük bir dozda tedavi yeniden başlatılmalıdır. QTc artışı tedavi öncesi değerlerden hem >500 ms hem de > 60 ms değerlerini karşılarsa dabrafenib tedavisinin kalıcı olarak sonlandırılması önerilir. Diğer maddelerin dabrafenib üzerindeki etkileriDabrafenib bir CYP2C8 ve CYP3A4 substratıdır. Bu enzimlerin güçlü indükleyicileri dabrafenibin etkililiğini azaltabileceğinden bunlardan mümkün olduğunca kaçınılması gerekir (bkz. Bölüm 4.5). Gastrik pH'ı artıran ajanlar dabrafenibin biyoyararlanımını azaltabilir, bundan mümkün olduğunda kaçınmak gerekir (bkz. Bölüm 4.5). Dabrafenibin diğer maddeler üzerindeki etkisiDabrafenib yaygın olarak kullanılan birçok tıbbi ürünün etkinliğinin kaybına yol açabilecek metabolize edici enzimlerin indükleyicisidir (örnekler için Bölüm 4.5'e bakınız). Bu nedenle, dabrafenib tedavisine başlarken bir ilaç kullanımı incelemesi (DUR) yapılması şarttır. Etkinlik izlemesi ve doz ayarı mümkün değilse genellikle dabrafenibin bazı metabolize edici enzim veya taşıyıcıların duyarlı substratları olan tıbbi ürünlerle (bkz. Bölüm 4.5) eşzamanlı kullanımından kaçınılmalıdır. Dabrafenibin varfarinle eşzamanlı kullanımı varfarine maruziyetin azalmasıyla sonuçlanabilir. Dabrafenib varfarinle eşzamanlı kullanıldığında ve dabrafenib tedavisi kesildiğinde dikkat edilmesi, ayrıca Uluslararası Normalleştirilmiş Oran'ın (INR) takip edilmesi önerilir (bkz. Bölüm 4.5). Dabrafenibin digoksin ile eşzamanlı uygulanması digoksin maruziyetinin azalmasına neden olabilir. Digoksin (bir taşıyıcı substratı) dabrafenible eşzamanlı kullanıldığında ve dabrafenib tedavisi kesildiğinde dikkat edilmesi ve ek olarak digoksinin izlenmesi önerilir (bkz. Bölüm 4.5). TAFİNLAR, ihmal edilebilir miktarda propilen glikol ihtiva etmektedir; bu yardımcı maddeye bağlı herhangi bir olumsuz etki beklenmez. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri Diğer tıbbi ürünlerin TAFİNLAR üzerine etkisiKlinik öncesi in vitroçalışmalar, aktif metabolitler hidroksi-dabrafenib ve desmetil-dabrafenib'in CYP3A4 substratları iken, dabrafenibin birincil olarak CYP2C8 ve CYP3A4 ile metabolize edildiğini göstermiştir. Farmakokinetik veriler, ketakonazol ile tekrarlanan doz dabrafenib Cmaks (%26) ve EAA (%57) değerlerinde orta derecede artış ve hidroksi- ve desmetil-dabrafenib EAA artışları (artışlar sırasıyla %48 ve %61) göstermiştir. Karboksi-dabrafenibde EAA'da %33'lük bir azalma bildirilmiştir. Ketokonazolün eş zamanlı verilmesi (CYP3A4 inhibitörü) dabrafenib EAA değerini %57 arttırmıştır. Bu nedenle, CYP2C8 veya CYP3A4'ün kuvvetli inhibitörleri veya indükleyicileri olan tıbbi ürünler TAFİNLAR konsantrasyonlarını sırasıyla arttırır veya azaltır. Mümkün olduğunda, TAFİNLAR verilirken alternatif ajanlar düşünülmelidir. TAFİNLAR ile eş zamanlı olarak kuvvetli inhibitörler (ör; ketokonazol, nefazodon, klaritromisin, ritonavir, sakinavir, telitromisin, itrakonazol, varikonazol, posakonazol, atazanavir) veya indükleyicilerin (ör; rifampin, fenitoin, karbamazepin, fenobarbital, sarı kantaron (St. John's Wort)) kullanılmasına dikkat edilmeli ya da kaçınılmalıdır.Dabrafenib çözünürlüğü pH'ya bağlı olup, daha yüksek pH değerlerinde çözünürlük azalır. Proton pompası inhibitörleri gibi mide asit salınımını inhibe ederek mide pH'sının yükselmesine neden olan tıbbi ürünler dabrafenibin çözünürlüğünü ve biyoyararlanımını azaltabilirler. pH'nın dabrafenibin farmakokinetiğine etkisini değerlendiren bir klinik çalışma yapılmamıştır. pH yükseltici ilaçların kuramsal olarak oral biyoyararlanımı ve maruz kalınan dabrafenib miktarını azaltma riski nedeni ile, eğer mümkünse, mide pH'sını arttıran bu tıbbi ürünleri kullanmaktan dabrafenib tedavisi boyunca kaçınılmalıdır. TAFİNLAR'ın diğer tıbbi ürünler üzerine etkisiTAFİNLAR, insan hepatositlerinde CYP2B6 ve CYP3A4 mRNA seviyelerinde kontrollere kıyasla 32 kata kadar doza bağlı artışlara yol açmıştır ve in vivoolarak CYP3A4'ün orta dereceli indükleyicisi olduğu gösterilmiştir. On iki hastada yapılmış bir klinik çalışmada tek doz midazolamın, CYP3A4 substratı, Cmaks ve EAA değerleri tekrarlayan dozlarda 150 g TAFİNLAR ile birlikte verildiğinde sırasıyla %61 ve %74 azaltmıştır. Bu nedenle TAFİNLAR barsak ve karaciğerde CYP3A4 aracılı metabolizmanın orta dereceli indükleyicisidir. Ayrıca, CYP2B6, CYP2C8, CYP2C9 ve CYP2C19'u ve UGT'leri (glukuronid konjugasyonu yapan enzimleri de) indükleyebilir. TAFİNLAR ve bu enzimlerin indüklemesinden etkilenen tıbbi ürünlerin birlikte verilmesi (ör; hormonal kontraseptifler [bkz. Bölüm 4.6], varfarin veya deksametazon) konsantrasyonların azalmasına ve etkililik kaybına yol açabilir. Bu tıbbi ürünlerin eş zamanlı verilmesi gerekliyse, hastalarda etkililik kaybının izlenmesi veya bu tıbbi ürünlerin değiştirilmesini düşünmelidirler.Taşıyıcı protein p-glikoprotein (Pgp) ile diğer taşıyıcılar ör: MRP-2, BCRP ve OATP1B1/1B3 de indüklenmektedir. Etkileşimin şiddeti değişkenlik gösterse de etkilenen tıbbi ürün sayısının yüksek olduğu düşünülmektedir. Etkinebilecek tıbbi ürünler, belirtilenlerle sınırlı olmamakla birlikte, aşağıda listelenmiştir: Analjezikler (örneğin fentanil, metadon) Antibiyotikler (örneğin Klaritromisin, doksisiklin) Kanser ilaçları (örneğin Kabazitaksel) Antikoagulanlar (örneğin asenokumarol, varfarin (bkz.bölüm 4.4)) Antiepileptikler (örneğin karbamazepin, fenitoin, primidon, valproik asit) Antipsikotikler (örneğin haloperidol) Kalsiyum kanal blokerleri (örneğin diltiazem, felodipin, nikardipin, nifedipin, verapamil) Kardiyak glikozidler (örneğin digoksin, bkz.Bölüm 4.4) Kortikosteroidler (örneğin deksametason, metilprednisolon) HIV antiviralleri (örneğin amprenavir, atazanavir, darunavir, delavirdin, efavirenz, fosamprenavir, indinavir, lopinavir, nelfinavir, saquinavir, tipranavir) Hormonal kontraseptifler (bkz. Bölüm 4.6) Hipnotikler (örneğin diazepam, midazolam, zolpidem) İmmunosupresanlar (örneğin Siklosporin, takrolimus, sirolimus) CYP3A4 ile metabolize olan statinler (örneğin atorvastatin, simvastatin) Enzim indüksiyonunun başlangıcı dabrafenibin 3 günlük tekrarlayan dozlarından sonraya tekabül eder. Dabrafenibin kullanımının bırakılmasından sonra indüksiyonun ortadan kalkışı dereceli olarak gerçekleşir. Duyarlı CYP3A4, CYP2B6, CYP2C8, CYP2C9 ve CYP2C19, UDP glukoronosil transferaz (UGT) ve taşıyıcı substratlarının konsantrasyonları artabilir. Bu nedenle hastalar toksisite açısından monitörize edilmelidirler. Bu ajanların dozajlarının yeniden ayarlanması gerekebilir. İn vitroolarak dabrafenib CYP3A4'ün mekanizma bazlı bir inhibitörüdür. Bu nedenle, CYP3A4'ün geçici inhibisyonu tedavinin ilk birkaç gününde gözlemlenebilir.TAFİNLAR'ın substrat transport sistemleri üzerine etkisiDabrafenib insan organik anyon taşıyıcı polipeptit (OATP) 1B1 (OATP1B1) ve OATP1B3'ün in vitroinhibitörüdür ve klinik önemi göz ardı edilemez. Bu nedenle, dabrafenib ve statinler gibi OATB1B1 veya OATP1B3 substratlarıyla birlikte uygulandığında dikkat edilmesi önerilir.Dabrafenib ve dabrafenibin metabolitleri olan hidroksi-dabrafenib, karboksi-dabrafenib ve desmetil-dabrafenib in vitroortamda insan organik anyon taşıyıcı (OAT) 1 ve OAT3'ün inhibitörleri olduğundan, klinik maruziyete dayanarak ilaç etkileşimi riski minimumdur. Dabrafenibin ve desmetil-dabrafenibin ayrıca insan meme kanseri direnç proteininin (BCRP) orta dereceli inhibitörler olduğu gösterilmiştir; ancak, klinik maruziyete dayanarak ilaç etkileşim riski minimum düzeydedir.Yiyeceklerin TAFİNLAR üzerine etkisiHastalar TAFİNLAR'ı, yiyeceklerin dabrafenibin emilimi üzerine etkileri nedeniyle yemeklerden en az bir saat önce veya iki saat sonra almalıdırlar (bkz. Bölüm 5.2). 4.6 Gebelik ve laktasyon Genel tavsiye Gebelik kategorisi: D Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon) Çocuk doğurma potansiyeline sahip kadınlar tedavi sırasına ve tedaviden sonra 4 hafta boyunca etkin kontrasepsiyon yöntemlerini kullanmalıdırlar. TAFİNLAR hormonal kontraseptiflerin etkinliğini azaltabilir ve kontrasepsiyon için alternatif bir yöntem kullanılması gerekir (bkz. Bölüm 4.5). Gebelik dönemi TAFİNLAR'ın gebe kadınlarda kullanımı ile ilişkili hiçbir veri yoktur. Hayvan çalışmaları reprodüktif toksisite göstermiştir (bkz. Bölüm 5.3). TAFINLAR gebelik sırasında ve kontrasepsiyon kullanmayan çocuk doğurma potansiyeli olan kadınlarda önerilmez. Eğer gebelik sırasında TAFİNLAR kullanılırsa veya hasta TAFİNLAR alırken hamile kalırsa, ilacın fetüse olası tehlikesi konusunda hasta bilgilendirilmelidir. Laktasyon dönemi TAFİNLAR'ın insan sütüne geçip geçmediği bilinmemektedir. Birçok tıbbi ürün insan sütüne geçtiği için emen çocuk için riski düşünülmelidir. Emzirme veya TAFİNLAR'ın kesilmesi konusunda çocuğun emzirilmesinin yararı ile TAFİNLAR tedavisinin anneye yararı değerlendirilerek bir karar verilmelidir. Üreme yeteneği/Fertilite İnsanlara ilişkin veri bulunmamaktadır. Hayvanlarda erkek üreme organlarında advers etkiler görülmüştür (bkz. Bölüm 5.3). Erkek hastalar geri dönüşümsüz spermatogenez bozukluğu riski konusunda bilgilendirilmelidir. 4.7 Araç ve makine kullanımı üzerindeki etkiler TAFİNLAR'ın araç ve makine kullanımına etkileri hakkında çalışma yapılmamıştır. TAFİNLAR'ın farmakolojisinde bu tür aktivitelerdeki etkisi minimal düzeydedir. Hastanın değerlendirme, motor veya kognitif yeteneklerini gerektiren işlerdeki yeteneği düşünülürken hastanın klinik durumu ve TAFİNLAR'ın advers olay profili göz önünde bulundurulmalıdır. Hastalar bu tür aktiviteleri etkileyebilecek halsizlik ve göz problemleri açısından bilgilendirilmelidir. 4.8 İstenmeyen etkiler Güvenlilik profili özeti Güvenlilik profili beş klinik monoterapi çalışmasındaki ve 578 melanoma hastasının verilerine dayanmaktadır. Dabrafenib ile en sık bildirilmiş olan advers reaksiyonlar (ADR) (> %15) hiperkeratoz, baş ağrısı, pireksi, artralji, halsizlik, bulantı, papillom, alopesi, döküntü ve kusmadır. Melanoma hastalarında bildirilmiş olan ADR'ler aşağıda MedDRA vücut sistemi organ sınıflaması ve sıklıklarıyla listelenmiştir. Sıklık tanımlaması: çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). (Kistler ve polipler dahil olmak üzere) İyi huylu ve kötü huylu neoplazmalar Çok yaygın: Papilloma Yaygın: Kütanöz skuamöz hücreli karsinomb, seboreik keratoz, akrokordon (deri etiketleri), bazal hücre karsinomu Yaygın olmayan: Yeni primer melanoma Bağışıklık sistemi hastalıkları Yaygın olmayan: Hipersensitivite, panikülit (özellikle deri altında yer alan yağlı ara doku katmanında ortaya çıkan iltihap) Metabolizma ve beslenme hastalıkları Çok yaygın: İştah azalması Yaygın: Hipofosfatemi, hiperglisemi Sinir sistemi hastalıkları Çok yaygın: Baş ağrısı Göz hastalıkları Yaygın olmayan: Üveit Solunum, göğüs bozuklukları ve mediastinal hastalıklar Çok yaygın: Öksürük Gastrointestinal hastalıklar Çok yaygın: Bulantı, kusma, ishal Yaygın: Kabızlık Yaygın olmayan: Pankreatit Deri ve deri altı doku hastalıkları Çok yaygın: Hiperkeratoz, alopesi, döküntü, palmar-plantar eritrodisestezi sendromu Yaygın: Deride kuruluk, pruritus, aktinik keratoz, deri lezyonu, eritem Kas-iskelet bozuklukları, bağ doku ve kemik hastalıkları Çok yaygın: Artralji, miyalji, ekstremite ağrısı Böbrek ve idrar hastalıkları Yaygın olmayan: Renal yetmezlik, akut renal yetmezlik, nefrit Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar Çok yaygın: Pireksi, halsizlik, titreme, asteni Yaygın: Inflüenza benzeri hastalık Araştırmalar Yaygın: Sol ventrikül ejeksiyon fraksiyonu (LVEF) azalması Yaygın olmayan: QT uzaması Birleşik olarak verilmiş terimlera. Papilloma, deri papillomu b. Kütanöz skuamöz hücreli karsinom: SCC, deri SCC'si, in situ SCC (Bowen hastalığı) ve keratoakantoma Kütanöz skuamöz hücreli karsinom Seçili advers reaksiyonlarla ilgili açıklamalarPireksi Klinik çalışmalarda ateş bildirilmiştir. Klinik çalışmalara katılan hastaların % 1'inde başlangıç böbrek fonksiyonu normal olan olgularda şiddetli titreme, dehidrasyon, hipotansiyon ve/veya akut böbrek yetmezliği eşliğinde enfeksiyöz-olmayan ciddi ateş olayları saptanmıştır. Infeksiyöz-olmayan bu ciddi ateş olaylarının başlangıcı tipik olarak tedavinin ilk ayının içindedir. Infeksiyöz olmayan ciddi ateş olayı yaşayan hastalar doza ara verilmesine ve/veya doz azaltımına ve destekleyici tedaviye iyi yanıt vermiştir (Bkz. Bölüm 4.2 ve 4.4). Kutanöz skuamoz hücreli karsinom Dabrafenib tedavisi alan hastaların %9'unda kutanöz skuamoz hücreli karsinomlara (keratoakantom veya karma keratoakantom alt tipi) rastlanmıştır. Olayların yaklaşık %70'i ortanca başlangıç süresi 8 hafta olacak şekilde tedavinin ilk 12 haftasında ortaya çıkmıştır. Kutanöz skuamoz hücreli karsinom gelişen hastaların %96'sı tedaviye doz modifikasyonu olmadan devam etmiştir. Yeni primer melanom Dabrafenib ile yapılan klinik çalışmalarda yeni primer melanomlar bildirilmiştir. Olgular eksizyon yoluyla tedavi edilmiş, doz modifikasyonu gerekmemiştir (Bkz. Bölüm 4.4). Non-kütanöz malignansi BRAF inhibitörlerine maruz kalan BRAF yabanıl tip hücrelerde MAP-kinaz sinyal iletiminin aktivasyonu RAS mutasyonları gibi kutanöz-olmayan malignite riskinin artmasına yol açabilir (Bkz. Bölüm 4.4). Dabrafenible RAS kökenli malignite olgularına rastlanmıştır. Hastaların klinik olarak uygun bir şekilde izlenmesi gerekir. QT uzaması Birleştirilmiş güvenlik popülasyonunda bir olguda QTcB >500 ms idi, hastaların sadece %3'ü >60 ms değerinde en kötü senaryo QTc uzaması yaşamıştır. LVEF azalması Hastaların %1'inde LVEF'de azalma bildirilmiştir, olguların çoğu asemptomatik ve tersine çevrilebilir durumda olmuştur. LVEF değeri kurumsal normalin alt sınırından daha düşük olan hastalar dabrafenible yapılan klinik çalışmalara dahil edilmemiştir. Artralji Dabrafenible yapılan klinik çalışmalarda artralji çok yaygın olarak (%25) bildirilse de, bunlar çoğunlukla Derece 1 ve 2 şiddette olmuştur, Derece 3 yaygın olmamıştır (< %1), Derece 4 hiç bildirilmemiştir. Hipofosfatemi Dabrafenible yapılan klinik çalışmalarda hipofosfatemi yaygın olarak (%7) bildirilmiştir. Bunların yaklaşık yarısının (%4 ) Derece 3 şiddetinde olduğuna dikkat etmek gerekir. Pankreatit Dabrafenib tedavisi alan hastalarda pankreatit bildirilmiştir. Açıklanamayan abdominal ağrının serum amilaz ve lipaz ölçümü de yapılacak şekilde hemen araştırılması gerekir. Pankreatit nöbeti sonrasında yeniden dabrafenibe başlarken hastaların yakından izlenmesi gerekir (Bkz. Bölüm 4.4). Böbrek yetmezliği Pireksiye bağlı prerenal azotemi veya granülomatöz nefrit nedeniyle böbrek yetmezliği yaygın olmamıştır; ancak dabrafenib böbrek yetmezliği (kreatinin >1.5 x ULN olarak tanımlanmıştır) bulunan hastalarda çalışılmamıştır. Bu ortamda dikkatli bir şekilde kullanılmalıdır (Bkz. Bölüm 4.4). Fotosensitivite Fotosensitivite advers olayı, başka bir BRAF inhibitörü ile yapılmış klinik çalışmalarda bir advers reaksiyon olarak tespit edilmiştir, dabrafenib tedavisi alan hastaların %2'sinde (9/578) ve DTIC tedavisi alanların %5'inde (3/59) gözlemlenmiştir. Şüpheli istenmeyen etkilerin bildirilmesiTıbbi ürünün ruhsatlandırılmasından sonra şüpheli istenmeyen etkilerin bildirilmesi önemlidir. Tıbbi ürünün risk/yarar dengesinin sürekli olarak izlenebilmesine olanak sağlar. Sağlık çalışanlarının tüm şüpheli istenmeyen etkileri Sağlık Bakanlığı Türkiye İlaç ve Tıbbi Cihaz Kurumu, Risk Yönetimi Dairesi, Türkiye Farmakovijilans Merkezi (TÜFAM)'ni e-posta: [email protected] tr [email protected]; tel: 444 5 475 kullanarak bildirmeleri gerekmektedir.Özel popülasyonlara ilişkin ek bilgiler: Geriyatrik popülasyon: TAFİNLAR klinik çalışmalarındaki toplam hasta sayısı (N = 578), 65 yaş ve üzeri için %22 ve 75 yaş ve üzeri için %6'dır. Daha genç hastalarla (< 65) karşılaştırıldığında, >65 yaşındaki çoğu hasta çalışma ilacında doz azaltımı (%22 vs. %12) veya ilaca ara vermeye (%39 vs. %27) yol açan advers olaylar yaşanmıştır. Ek olarak, yaşlı hastalar genç hastalarla karşılaştırıldığında daha ciddi advers olaylar yaşamıştır (%41 vs. %22). Etkililikte bu hastalar ile genç hastalar arasında genel farklılıklar gözlenmemiştir. 4.9 Doz aşımı ve tedavisi TAFİNLAR doz aşımı konusunda şu anda çok kısıtlı deneyim bulunmaktadır. TAFİNLAR doz aşımı için spesifik bir tedavi yoktur. Eğer doz aşımı görülürse, hasta gerekli uygun izlem ile desteklenerek tedavi edilmelidir. 5. FARMAKOLOJİK ÖZELLİKLER 5.1 Farmakodinamik özellikler Farmakoterapötik grup: Antineoplastik ajanlar, protein kinaz inhibitörü, ATC kodu: L01XE23 Etki mekanizmasıDabrafenib kuvvetli, seçici bir ATP-RAF kinaz yarışmalı inhibitörüdür ve BRAF V600E, BRAF V600K ve BRAF V600D enzimleri için sırasıyla IC50 değerleri 0,65; 0,5 ve 1,84 nM'dür. BRAF'deki onkogenik mutasyonlar RAS/RAF/MEK/ERK yolaklarının yapısal aktivasyonuna ve tümör hücre büyümesinin uyarılmasına yol açar. BRAF mutasyonları melanomada yaklaşık %50 olmak üzere spesifik kanserlerde de yüksek frekansta tespit edilmiştir. En sık görülen BRAF mutasyonu V600E ve daha sonra en sık görülen V600K, bütün kanser hastalarında görülen BRAF mutasyonlarının %99'unu oluşturur. Bir dizi az görülen ikame vardır; bunların arasında V600D, V600G, V600M ve V600R bulunur. Biyokimyasal testlerden elde edilen preklinik veriler, dabrafenibin kodon 600 mutasyonlarını aktive ederek BRAF kinazları inhibe ettiğini göstermiştir (Tablo 3). Tablo 3: Dabrafenibin RAF kinazlara karşı kinaz inhibe edici etkinliği
in vitroin vivoolarak inhibe eder.İn vitro ve hayvan modellerinde BRAF V600 mutant melanoma hücre dizilerinde dabrafenibin farmakodinamik bir biyolojik aşağı akım belirtecinin (fosforile ERK) baskılanması gösterilmiştir. BRAF V600 mutasyon pozitif melanoması olan hastalarda dabrafenib verilmesi başlangıca göre tümör fosforile ERK'nin inhibisyonuna yol açmıştır. BRAF mutasyon durumunun tespitiDabrafenib alınmasından önce geçerliliği kanıtlanmış bir test ile hastaların BRAF V600 mutasyon pozitif tümör durumu doğrulanmalıdır. Faz II ve III klinik çalışmalarda uygunluk taraması için, en son tümör örneğinde BRAF V600 mutasyon değerlendirme testi kullanılarak merkezi BRAF V600 mutasyonunun değerlendirmesi gereklidir. Primer tümör veya metastatik bir alandan tümör Response Genetics Inc. (RGI)tarafından sadece araştırma amaçlı geliştirilmiş (IUO) bir test ile değerlendirilmiştir. RGI IUO, formalin ile fiske edilmiş parafine batırılmış (FFPE) tümör dokudan elde edilmiş DNA'ya uygulanmış allele özgü polimeraz zincir reaksiyonu (PCR) testidir. Test özellikle V600E ve V600K mutasyonları arasında ayırım yapmak üzere özel olarak geliştirilmiştir. Sadece BRAF V600E veya V600K mutasyonu pozitif tümörleri olan hastalar bu çalışmaya katılmak üzere uygun bulunmuştur.Sonrasında tüm hasta örnekleri CE işareti taşıyan geçerliliği oluşturulmuş bioMerieux (bMx) THxID BRAF testi kullanılarak yeniden test edilmiştir. bMx TH xTDBRAF testi, FFPE tümör dokusundan alınan DNA üzerinde uygulanan alele-özgün bir PCR'dır. Test BRAF V600E ve V600K mutasyonlarını yüksek bir duyarlılıkla tespit edecek şekilde tasarlanmıştır (FFPE dokusundan alınan DNA kullanılarak yabanıl tip sıralamasından oluşan bir tabanda %5 V600E ve V600K sekansına inerek). Geriye dönük iki-yönlü Sanger sıralama analizleriyle yapılan klinik-dışı ve klinik çalışmalar testin daha az yaygın BRAF V600D mutasyonunu ve V600E/K601E mutasyonunu da daha düşük bir duyarlılıkla tespit ettiğini göstermiştir. Klinik-dışı ve klinik çalışmalarda THxID BRAF testiyle mutasyon pozitif çıkan, ardından referans yöntem kullanılarak sıralaması yapılan örneklerde testin özgünlüğünün %94 olduğu saptanmıştır.Klinik çalışmalarDabrafenibin etkililik ve güvenliği dabrafenibi daha önce tedavi görmemiş BRAF V600E mutasyonu pozitif ileri evre (rezekte edilemeyen Evre III) veya metastatik (Evre IV) melanom hastalarında dakarbazin (DTIC) ile karşılaştıran randomize, açık etiketli bir Faz III çalışmasında [BREAK 3] değerlendirilmiştir. V600E dışındaki BRAF mutasyonlarıyla oluşan melanom hastaları çalışmaya dahil edilmemiştir. Bu çalışmanın primer amacı dabrafenibin araştırmacının değerlendirmesine göre progresyonsuz sağkalım (PFS) açısından etkililiğini DTIC ile karşılaştırmalı olarak değerlendirmektir. DTIC grubundaki hastaların ilk progresyonun bağımsız radyografik teyidi sonrasında dabrafenibe çapraz geçiş yapmasına izin verilmiştir. Başlangıç özellikleri tedavi grupları arasında dengelenmiştir. Hastaların %60'ı erkek, %99.6'sı Kafkas kökenli; ortalama yaş 52'dir, hastaların %21'i >65 yaşında, %98.4'ünde ECOG durumu 0 veya 1 puan olup ve hastaların %97'sinde metastatik hastalık vardır. 19 Aralık 2011 sonlanım tarihli verilerle yapılan önceden belirlenmiş analiz, PFS'den oluşan primer sonlanım noktasında anlamlı iyileşme (HR = 0.30; %95 GA 0.18, 0.51; p <0.0001) olduğunu göstermiştir. Primer analizden ve 6 aylık ek takip zamanı bulunan post-hoc analizden elde edilen etkililik sonuçları Tablo 4'te özetlenmektedir. 18 Aralık 2012 sonlanım tarihine dayalı başka bir post-hoc analizinden elde edilen genel sağkalım verileri Şekil 1'de gösterilmektedir. Tablo 4: Daha önce tedavi edilmemiş hastalarda etkililik (BREAK-3 Çalışması, 25 Haziran 2012)
25 Haziran 2012 sonlanım tarihi itibariyle DTCI grubuna randomize edilen 63 hastanın 35'i (%55.6) dabrafenibe geçiş yapmıştır ve dabrafenibe randomize edilen hastaların %63'ü ve DTIC grubuna randomize edilen hastaların % 79'unda progresyon görülmüştür veya yaşamını yitirmiştir. Çapraz geçiş sonrasında medyan ilerlemesiz sağkalım 4.4 aydır. Tablo 5: Birincil analiz ve post hoc analizlerden elde edilen sağkalım verileri
18 Aralık 2012 sonlanım tarihine dayalı başka bir post-hoc analizinden elde edilen genel sağkalım verileri 12 ayda DTCI ve dabrafenib tedavilerinde sırasıyla %63 ve %70 genel sağkalım oranı olduğunu ortaya koymuştur.
Şekil 1 İlerlemesiz Sağkalım - daha önce tedavi edilmemiş hastalar (ITT popülasyon) 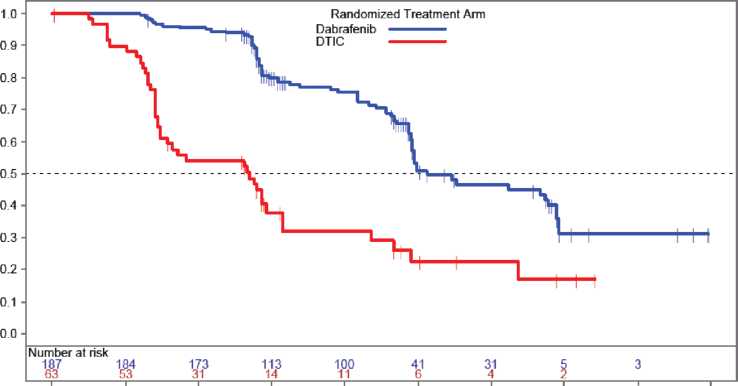
Randomizasyondan İtibaren Geçen Süre (Ay)scS O a>M cS m. S*5« us Beyin metastazı olan hastalar (Faz II çalışma sonuçları (BREAK-MB)BREAK-MB, histolojik olarak doğrulanmış (Evre IV) BRAF mutant pozitif (V600E veya V600K), beyin metastazı gelişmiş melanoma hastalarının dabrafenibe intrakraniyal yanıtını değerlendirmek üzere tasarlanmış çok merkezli, açık etiketli, iki kohortlu, Faz II çalışmasıdır. Hastalar Kohort A (beyin metastazı için daha önce lokal tedavi almamış olan hastalar) veya Kohort B (beyin metastazı için daha önce lokal tedavi almış olan hastalar) olarak ayrılmıştır. Bu çalışmanın birincil sonlanım noktası, BRAF V600E mutasyonu pozitif beyin metastazı yapmış metastatik melanoması olan hastalarda araştırıcı tarafından değerlendirilmiş olan genel intrakraniyal yanıt oranı (OrRR)'dır. Araştırıcı değerlendirmesine göre doğrulanmış OIRR ve diğer etkililik sonuçları Tablo 6'da verilmiştir. Tablo 6: Beyin metastazı olan hastalarda etkililik verileri (BREAK-MB Çalışması)
a. Doğrulanmış yanıt. b. Bu çalışma 0IRR<%10 (tarihsel sonuçlara dayalı) null hipotezini OIRR >%30 alternatif hipotezi lehine desteklemek veya red etmek için BRAF V600E mutasyonu pozitif hastalarda tasarlanmıştır Daha önce tedavi almamış veya en az bir sistemik tedavisi başarısız olmuş hastalar (Faz II çalışma sonuçları [BREAK-2])BRF113710 (BREAK-2), doğrulanmış BRAF V600E veya BRAF V600K mutasyonu pozitif melanoması olan histolojik olarak doğrulanmış metastatik melanomalı (Evre IV) 92 hasta üzerinde yapılmış çok merkezli, global, tek kollu bir Faz II çalışmadır. Hastalar tedavi almamış (n= 15) veya metastatik ortamda daha önce tedavi almış (n= 77) (ör; kemoterapi, immünoterapi, daha önceki hedefli terapi vb.) hastalardır. Araştırıcı, BRAF V600E metastatik melanomalı (n=76) birincil etkililik hasta popülasyonunda doğrulanmış yanıt oranını %59 (%95 GA: 48,2; 70,3) olarak değerlendirmiştir (tam yanıt %7'dir). Medyan PFS 6,3 ay (%95 GA: 4,6; 7,7) ve medyan yanıt süresi 5,2 ay (%95 GA: 3.9, hesaplanamaz)'dur. Daha önceki sistemik terapi yanıtı anlamlı derecede etkilememiştir. Araştırıcı, BRAF V600K mutasyonu pozitif metastatik melanoma hastalarının ikincil etkililik popülasyonunda (n=16) doğrulanmış yanıt oranını %13 (%95 GA: 0,0; 28,7) olarak değerlendirmiştir ve medyan yanıt süresi 5,3 ay olarak bulunmuştur (%95 GA: 3,7; 6,8). V600K hasta popülasyonunda tam yanıt yoktur. 5.2 Farmakokinetik özellikler Genel Özellikler Dabrafenibin farmakokinetiği tek dozdan sonra ve yaklaşık 12 saat aralıklı olarak günde 2 kez 150 mg tekrarlanan dozdan sonra BRAF V600 mutasyonu pozitif metastatik melanoma hastalarında tespit edilmiştir. Emilim:Dabrafenib oral olarak emilir. Dozdan 2 saat sonra pik plazma konsantrasyonuna ulaşır. Oral dabrafenibin ortalama mutlak biyoyararlanımı %95'dir (%90 GA: %81, %110). Dabrafenib maruziyeti (Cmaks ve EAA) doz ile orantılı olarak 12 ve 300 mg tek doz olarak verilmesinden sonra artmıştır, fakat bu artış günde 2 kez tekrarlanan doz alımından sonraki artıştan daha azdır. Tekrarlanan dozlar ile maruziyette bir azalma gözlemlenmiştir; bu da kendi metabolizmasının indüklenmesine bağlı olabilir. Ortalama birikim EAA Gün 18/Gün 1 oranları 0,73'dür. Günde 2 kez 150 mg verilmesinden sonra, geometrik ortalama Cmaks, EAA(0-t) ve predoz konsantrasyonu (Ct) sırasıyla 1478 ng/ml, 4341 ng*sa/ml ve 26 ng/m'dir. Dabrafenibin yiyecekler ile birlikte verilmesi biyoyararlanımı (Cmaks ve EAA sırasıyla %51 ve %31'e düşer) azaltmıştır ve açlık durumu ile karşılaştırıldığında dabrafenib kapsüllerinin absorpsiyonunu geciktirmiştir. Dağılım:Dabrafenib birincil olarak insan plazma proteinine bağlanır (%99,7 bağlı). İntravenöz mikrodoz verilmesinden sonra dağılımın sabit durum hacmi 46 L'dir. Dabrafenib in vitroolarak, insan p-glikoproteini (Pgp) ve fareye ait BCRP substratıdır. Ancak bu taşıyıcıların dabrafenibin oral biyoyararlanımı ve atılımı üzerindeki etkileri asgaridir ve Pgp ya da BCRP inhibitörleri ile klinik olarak anlamlı ilaç etkileşimi gösterme riski düşüktür.Ne dabrafenib ne de 3 ana metabolitinin Pgp inhibitörü oldukları in vitroolarak gösterilememiştir.Biyotransformasyon:Dabrafenib metabolizması birincil olarak CYP2C8 ve CYP3A4 aracılığıyla hidroksi-dabrafenib oluşturmaktadır; bu ise daha sonra CYP3A4 aracılığıyla karboksi dabrafenib oluşturmak üzere oksitlenir. Karboksi-dabrafenib enzimatik olmayan bir işlem ile desmetil dabrafenib oluşturmak üzere dekarboksillenir. Karboksi-dabrafenib safra ve idrarda atılır. Desmetil-dabrafenib bağırsaklarda oluşur ve tekrar emilir. Desmetil-dabrafenib CYP3A4 aracılığıyla oksidatif metabolitlerine metabolize olur. Hidroksi-dabrafenib terminal yarılanma ömrü ana ürünün yarılanma ömrü olan 10 saate paralel iken karboksi- ve desmetil- metabolitleri daha uzun yarılanma ömrüne sahiptir (21-22 saat). Tekrarlanan doz alımlarından sonra hidroksi, karboksi ve desmetil defabrenibin ortalama metabolit/ ebeveyn EAA oranları sırasıyla 0,9;11 ve 0,7'dir. Maruziyet, rölatif güç ve farmakokinetik özelliklere göre hidroksi ve desmetil dabrafenibin her ikisi de dabrafenibin klinik aktivitesine katkıda bulunmakta iken karboksi-dabrafenibin aktivitesi anlamlı değildir. Eliminasyon:IV mikrodozdan sonra terminal yarılanma ömrü 2,6 saattir. Dabrafenib terminal yarılanma ömrü oral verilmesinden sonra uzamış terminal faza göre 8 saattir. IV plazma klerensi 12 L/saattir. Fekal yoldan eliminasyon oral doz kullanımından sonraki majör eliminasyon yoludur; %71 radyoaktif doz iken idrar aracılığıyla eliminasyon radyoaktivitenin %23'üdür. Doğrusallık/doğrusal olmayan durum:Veri bulunmamaktadır. Hastalardaki karakteristik özellikler Karaciğer yetmezliği:Dabrafenibin farmakokinetiği hafif dereceli karaciğer yetmezliği (Ulusal Kanser Enstitüsü [NCI] sınıflaması temel alınmıştır) olan 65 hastanın alındığı klinik çalışmalardaki popülasyon analizine dayanılarak karakterize edilmiştir. Dabrafenibin oral klerensi bu hastalar ve normal hepatik fonksiyonu olan hastalar arasında anlamlı derecede farklı değildi (%4'lük fark). Ek olarak, hafif dereceli karaciğer yetmezliğinin dabrafenib metabolit plazma konsantrasyonlarında anlamlı etkisi olmamıştır. Orta-ağır karaciğer yetmezliği olan hastalara dabrafenib verilirken dikkatli olunmalıdır (bkz. Bölüm 4.2). Böbrek yetmezliği:Dabrafenibin farmakokinetiği, popülasyon analizi yapılmış klinik çalışmalara alınmış olan 233 hafif renal bozukluğu olan hastada (GFR 60-89 ml/dk/1.73 m2) karakterize edilmiştir. Hafif veya orta dereceli renal bozuklukta dabrafenibin oral klerensi düşük olmuştur (her iki kategori için <%6) ve klinik olarak anlamlı değildir. Ek olarak, hafif ve orta şiddetteki renal bozukluğun hidroksi, karboksi ve desmetil dabrafenib plazma konsantrasyonlarında önemli bir etkisi olmamıştır. Ağır renal bozukluğu olan hastalarda veri bulunmamaktadır (bkz. Bölüm 4.2). Dabrafenibin farmakokinetik özellikleri klinik çalışmalara kaydolan hafif şiddette böbrek yetmezliği (GFR 60-89 ml/dk/1.73 m2) bulunan 233 hasta ve orta şiddette böbrek yetmezliği bulunan (GFR 30-59 ml/dk/1.73 m2) hastalarda popülasyon analizi kullanılarak çıkarılmıştır. Hafif veya orta şiddette böbrek yetmezliğinin dabrafenibin oral klerensi üzerindeki etkisi fazla olmamıştır (her iki kategoride de <%6) ve klinik açıdan önemli olmamıştır. Buna ek olarak, hafif ve orta şiddette böbrek yetmezliğinin hidroksi-, karboksi- ve desmetil-dabrafenib plazma konsantrasyonlarında önemli etkisi olmamıştır. Şiddetli böbrek yetmezliği bulunan olgularda veri mevcut değildir (Bkz. Pozoloji ve Uygulama). Geriyatrik popülasyon:Popülasyon farmakokinetik analizine göre, yaşın dabrafenib farmakokinetiği üzerinde önemli bir etkisi yoktur. Karboksi ve desmetil dabrafenib plazma konsantrasyonları için yaşın 75'ten büyük olması önemli bir belirteç olmuştur; >75 yaş hastalarda <75 yaş hastalara kıyasla %40 daha fazla maruziyet saptanmıştır. Vücut ağırlığı ve cinsiyet:Popülasyon farmakokinetik analizine göre, cinsiyet ve ağırlığın dabrafenibin oral klerensini etkilediği, ayrıca ağırlık oral hacmin dağılımı ve dağılma klerensini de etkilemiştir. Bu farmakokinetik farklılıklar klinik olarak anlamlı kabul edilmemiştir. Irk:Dabrafenib farmakokinetiğinin ırk üzerindeki potansiyel etkisini değerlendirmek için yeterli veri yoktur. Pediyatrik popülasyon:Pediyatrik hastalarda dabrafenib farmakokinetiğini araştıran çalışmalar yapılmamıştır. 5.3 Klinik öncesi güvenlilik verileri Dabrafenib ile karsinojenite çalışmaları yapılmamıştır. Dabrafenib, bakteri ve kültüre edilmiş memeli hücrelerindeki in vitroin vivokemirgen mikronükleus değerlendirme testinde mutajenik veya klastojenik değildir.Birleşik dişi fertilitesinde, sıçanlarda erken embriyonik ve embriyofetal gelişim çalışmalarında overiyen korpora lutea sayıları hamile dişilerde 300 mg/kg/gün seviyesine düşmüştür (EAA'ya göre insan klinik maruziyetinin yaklaşık 3 katı), fakat östrojen döngüsü, çiftleşme veya fertilitede etkileri yoktu. Embriyonal letalite ve ventriküler septal defektler de dahil olmak üzere gelişim toksisitesi 300 mg/kg/gün dozunda ve gecikmiş iskelet gelişimi ve > 20 mg/kg/gün seviyesinde azalmış fetal ağırlık (EAA'ya dayalı insan klinik maruziyetinin > 0,5 katı) görülmüştür. Dabrafenib ile erkek fertilite çalışmaları yapılmamıştır. Fakat tekrarlanan doz çalışmalarında sıçanlarda ve köpeklerde (EAA'ya dayalı insan klinik maruziyetinin >0,2 katı) testiküler dejenerasyon/deplesyon görülmüştür. Sıçan ve köpeklerdeki testiküler değişiklikler 4 haftalık iyileşme döneminden sonra da devam etmiştir (bkz. Bölüm 4.6). Köpeklerde (EAA'ya göre klinik maruziyette >2 kat) koronar arter dejenerasyon/nekroz ve/veya hemoraji ve atriyal fibrovasküler proliferasyon dahil olmak üzere kardiyovasküler etkiler görülmüştür. Sıçanlarda artmış hepatik arteriyel dejenerasyon ve inflamasyon ile birlikte spontan kardiyomiyosit dejenerasyonu (spontan kardiyomiyopati) görülmüştür (klinik maruziyetin >0.5 katı). Akciğerlerin bronkoalveolar inflamasyonu birçok köpekte >20 mg/kg/gün (EAA değerine göre insan klinik maruziyetinin >9 katı) dozunda görülmüştür ve yüzeysel ve/veya zorlu solunum ile birliktedir. Dabrafenib verilen köpek ve sıçanlarda geri dönüşümlü hematolojik etkiler gözlemlenmiştir. Köpek ve sıçanlarda, 13 haftaya kadar olan çalışmalarda retikülosit sayısı ve/veya kırmızı küre kütlesi gözlenmiştir (sırasıyla klinik maruziyetin >10 ve 1,4 katı). Farelerde jüvenil toksisite çalışmaları, büyüme üzerinde etki (uzun kemik boyunun daha kısa olması), renal toksisite (tübüler çökeltiler, artmış kortikal kist ve tübüler bazofili sıklığı, üre ve kreatinin konsantrasyonlarında geri dönüşümlü artışlar), testiküler toksisite (dejenerasyon ve tübüler genişleme) ve vajinal açıklığın erken oluşması (over ağırlıklarında ya da dişi üreme organlarında morfolojik değişikliklerin eşlik etmediği) gözlenmiştir. Dabrafenibin, fare fibroblastı yapılan in vitro3T3 Nötral Kırmızı Tutulumu (NRU) deneyinde fototoksik olduğu görülmüştür.6. FARMASÖTİK ÖZELLİKLER 6.1 Yardımcı maddelerin listesi Mikrokristalin selüloz Magnezyum stearat, bitkisel kaynaklı Kolloidal silikon dioksit Kırmızı demir oksit (E172) Titanyum dioksit (E171) Hipromelloz (E464) Siyah demir oksit (E172) Şelak n-Butil alkol İzopropil alkol Propilen glikol Amonyum hidroksit 6.2 Geçimsizlikler Geçerli değil. 6.3 Raf ömrü 24 ay. 6.4 Saklamaya yönelik özel tedbirler 30°C'nin altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriği Vidalı plastik kapaklı, opak, beyaz, yüksek yoğunluklu polietilen (HDPE) şişe ve silika jel kurutucu madde. Ambalaj boyutları: 28 veya 120 sert kapsül. Tüm ambalaj boyutları pazarda bulunmayabilir. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİ GlaxoSmithKline ilaçları San. ve Tic. A.Ş. Büyükdere Cad. No:173, 1. Levent Plaza B Blok, 34394 1. Levent/Istanbul Tel:0 212 339 44 00 Faks: 0 212 339 45 00 8. RUHSAT NUMARASI 2014/339 9. İLK RUHSAT/RUHSAT YENİLENME TA RİHİilk ruhsat tarihi: 22.04.2014 Ruhsat yenileme tarihi: 10. KÜB'ün YENİLENME TARİHİ 20 |
İlaç BilgileriTafinlar 75 Mg KapsülEtken Maddesi: Dabrafenib Mesilat Atc Kodu: L01XE23 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.