Zelboraf 240 Mg Film Kaplı Tablet Kısa Ürün BilgisiAntineoplastik ve İmmünomodülatör Ajanlar » Antineoplastik İlaçlar (Kanser İlaçları) » Diğer Kanser İlaçları » Protein Kinaz İnhibitörleri KISA ÜRÜN BİLGİSİ 1. BEŞERİ TIBBİ ÜRÜNÜN ADIZELBORAF 240 mg film kaplı tablet2. KALİTATİF VE KANTİTATİF BİLEŞİM Etkin madde:Vemurafenib : 240 mgHer tablet, 240 mg vemurafenib (vemurafenib ve hipromelloz asetat süksinat eş çökeltisi şeklinde) içerir. Yardımcı madde(ler):Kroskarmeloz sodyum 29.4 mg (1.278 mmol)Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMFilm kaplı tablet.Bir yüzünde 'VEM' baskısı olan, yaklaşık 19 mm'lik pembemsi beyaz ila turuncu beyaz, oval, bikonveks film kaplı tabletler. 4. KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarZELBORAF; daha önce herhangi bir RAF yolağı inhibitörü kullanmamış, ECOG performans skoru 0 veya 1 olan, lokal tedaviler sonrası progresyon göstermiş ve lokal tedavilerin tekrar kullanılamadığı relaps veya metastatik BRAF V600 mutasyonu pozitif olan malign melanom hastalarında tek ajan olarak progresyona kadar kullanımı endikedir. Progresyon sonrası tek ajan veya başka tedavilerle kombinasyon şeklinde kullanılamaz (bkz. bölüm 5.1).4.2. Pozoloji ve uygulama şekliZELBORAF tedavisi, antikanser tıbbi ürünlerinin kullanımı konusunda deneyimli kalifiye bir hekim tarafından başlatılmalı ve denetlenmelidir.ZELBORAF almadan önce, geçerli bir test yoluyla hastanın tümöründe BRAF V600 mutasyon pozitifliği tespit edilmelidir (bkz. bölüm 4.4 ve 5.1). Pozoloji:ZELBORAF için önerilen doz, günde iki kez 960 mg'dır (240 mg'lık 4 tablet) (günde toplam 1,920 mg'lık doza eşdeğer). ZELBORAF öğünle birlikte veya aç karnına alınabilir fakat sürekli olarak günlük dozların tümünü mide boşken almaktan kaçınılmalıdır (bkz. bölüm 5.2).Uygulama sıklığı ve süresi:Tedavi süresi:ZELBORAF tedavisi, hastalık progresyonuna kadar veya kabul edilemez toksisite oluşana kadar devam etmelidir (bkz. tablo 1). Unutulan dozlar: Unutulan doz, günde iki kez tedavi rejimine devam edilmesi için sonraki dozdan önceki 4 saate kadar alınabilir. Her iki doz aynı zamanda alınmamalıdır. Kusma: ZELBORAF uygulanmasından sonra kusma oluşması durumunda, hasta tıbbi üründen ek doz almamalı ancak tedaviye normal şekilde devam edilmelidir. Pozoloji ayarlamaları Advers ilaç reaksiyonlarının veya QTc uzamasının yönetimi; dozun azaltılmasını, tedaviye geçici olarak ara verilmesini ve/veya tedavinin sonlandırılmasını gerektirebilir (bkz. tablo 1). Günde iki kez 480 mg'dan düşük bir doz veren pozoloji ayarlamaları önerilmemektedir. Kutanöz Skuamöz Hücreli Karsinom (cuSCC) gelişmesi durumunda, ZELBORAF dozunu modifiye etmeksizin tedaviye devam edilmesi önerilmektedir (bkz. bölüm 4.4 ve 4.8). Tablo 1: Herhangi bir advers olayın derecesine dayalı doz modifikasyonu
Daha önce tedavi uygulanmış metastatik melanomlu hastalarda yapılan kontrollü olmayan, açık etiketli bir faz II çalışmada vemurafenib maruziyetine bağlı QT uzaması gözlenmiştir. QTc uzamasının yönetimi spesifik izlem tedbirlerini gerektirebilir (bkz. bölüm 4.4). Tablo 2: QT aralığının uzamasına dayalı doz modifikasyonu
Uygulama şekli:ZELBORAF tabletler suyla birlikte bütün olarak yutulmalıdır. ZELBORAF tabletler çiğnenmemeli veya ezilmemelidir.Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek yetmezliği olan hastalara ilişkin kısıtlı veri mevcuttur. Şiddetli böbrek yetmezliği olan hastalarda yüksek ilaç maruziyeti riski göz ardı edilemez. Şiddetli böbrek yetmezliği olan hastalar yakından izlenmelidir (bkz. bölüm 4.4 ve 5.2).Karaciğer yetmezliği:Karaciğer yetmezliği olan hastalara ilişkin kısıtlı veri mevcuttur. Vemurafenibin karaciğer tarafından uzaklaştırılması nedeniyle, orta ila şiddetli karaciğer yetmezliği olan hastalarda ilaç maruziyeti artabilir ve bu hastalar yakından izlenmelidir (bkz. bölüm 4.4 ve 5.2).Pediyatrik popülasyon:Çocuklarda ve adolesanlarda (<18 yaş) ZELBORAF'ın güvenliliği ve etkililiği belirlenmemiştir. Veri bulunmamaktadır.Geriyatrik popülasyon:65 yaşından büyük hastalarda özel bir doz ayarlaması gerekmemektedir.Beyaz ırktan olmayan hastalar:Beyaz ırktan olmayan hastalarda ZELBORAF'ın güvenliliği ve etkililiği belirlenmemiştir. Veri bulunmamaktadır.4.3. KontrendikasyonlarEtkin maddeye veya bölüm 6.1'de verilen yardımcı maddelerden herhangi birine karşı aşırı duyarlılığı bulunan kişilerde kontrendikedir.4.4. Özel kullanım uyarıları ve önlemleriZELBORAF kullanmadan önce, geçerli bir test yoluyla hastanın tümöründe BRAF V600 mutasyon pozitifliği tespit edilmelidir. V600E ve V600K dışında seyrek BRAF V600 mutasyonları eksprese eden hastalarda ZELBORAF'ın etkililiği ve güvenliliği kesin olarak belirlenmemiştir (bkz. bölüm 5.1). ZELBORAF, BRAF wild-tip (mutasyonu bulunmayan) malign melanomu olan hastalarda kullanılmamalıdır.Aşırı duyarlılık reaksiyonu ZELBORAF'la ilişkili olarak anafilaksi dahil ciddi aşırı duyarlılık reaksiyonları bildirilmiştir (bkz. bölüm 4.3 ve 4.8). Şiddetli aşırı duyarlılık reaksiyonları; Stevens-Johnson sendromunu, yaygın döküntüyü, eritemi veya hipotansiyonu içerebilir. Şiddetli aşırı duyarlılık reaksiyonları oluşan hastalarda ZELBORAF tedavisi kalıcı olarak sonlandırılmalıdır. Dermatolojik reaksiyonlar ZELBORAF alan hastalarda, pivotal klinik çalışmalarda oluşan nadir Stevens-Johnson sendromu ve toksik epidermal nekroliz vakaları dahil, şiddetli dermatolojik reaksiyonlar bildirilmiştir. ZELBORAF kullanımı ile eozinofili ve sistemik semptomlar ile seyreden ilaç döküntüsü (DRESS Sendromu) riski bildirilmiştir. Şiddetli dermatolojik reaksiyonlar oluşan hastalarda ZELBORAF tedavisi kalıcı olarak sonlandırılmalıdır. QT uzaması Daha önce tedavi uygulanmış metastatik melanomu bulunan hastalarda yapılan kontrollü olmayan, açık etiketli bir faz II çalışmada ZELBORAF maruziyetine bağlı QT uzaması gözlenmiştir (bkz. bölüm 4.8). QT uzaması, Torsades de Pointes dahil ventriküler aritmilerin riskinde artışa neden olabilir. Elektrolit anormallikleri (magnezyum dahil) düzeltilemeyen, uzun QT sendromu olan veya QT aralığını uzattığı bilinen tıbbi ürünleri kullanan hastalarda ZELBORAF tedavisi önerilmemektedir. Tüm hastalarda ZELBORAF tedavisinden önce, tedaviden bir ay sonra ve doz modifikasyonu ardından elektrokardiyogram (EKG) ve elektrolitler (magnezyum dahil) izlenmelidir. Özellikle orta ila şiddetli karaciğer yetmezliği olan hastalarda, tedavinin ilk 3 ayı boyunca her ay ve sonrasında 3 ayda bir ya da klinik olarak endike ise daha sık şekilde izlem yapılması önerilmektedir. QTc >500 milisaniye (msn) olan hastalarda ZELBORAF tedavisine başlanması önerilmemektedir. Tedavi sırasında QTc değeri 500 msn'yi geçerse, ZELBORAF tedavisine geçici olarak ara verilmeli, elektrolit anormallikleri (magnezyum dahil) düzeltilmeli ve QT uzaması ile ilgili kardiyak risk faktörleri (örn. konjestif kalp yetmezliği, bradiaritmiler) kontrol altına alınmalıdır. Tedavi tekrar, QTc değeri 500 msn altında düştükten sonra ve Tablo 1'de açıklandığı gibi daha düşük bir dozda başlatılmalıdır. QTc uzamasının hem >500 msn olması, hem de tedavi öncesi değerlere göre >60 msn değişim göstermesi durumunda ZELBORAF tedavisinin kalıcı olarak sonlandırılması önerilmektedir. Oftalmolojik reaksiyonlar Üveit, iritis ve retinal ven oklüzyonu dahil ciddi oftalmolojik reaksiyonlar bildirilmiştir. Hastalar, oftalmolojik reaksiyonlar açısından rutin şekilde izlenmelidir. Kutanöz Skuamöz Hücreli Karsinom (cuSCC) ZELBORAF ile tedavi uygulanan hastalarda cuSCC vakaları (keratoakantom veya mikst keratoakantom alt tipi olarak sınıflandırılanlar dahil) bildirilmiştir (bkz. bölüm 4.8). Tedaviye başlanmadan önce tüm hastalara dermatolojik değerlendirme yapılması ve tedavi sırasında rutin izlem yapılması önerilmektedir. Şüphelenilen deri lezyonları eksize edilmeli, dermatopatolojik değerlendirmeye gönderilmeli ve yerel tedavi standardına göre tedavi uygulanmalıdır. Reçete yazan uzman, tedavi sırasında ve tedaviden sonra altı aya kadar hastayı her ay cuSCC açısından incelemelidir. cuSCC gelişen hastalarda, doz ayarlaması yapılmaksızın tedaviye devam edilmesi önerilmektedir. ZELBORAF'ın sonlandırılmasını izleyen altı ay boyunca veya başka bir anti-neoplastik tedavi başlatılana kadar izlemeye devam edilmelidir. Hastalara, derilerinde herhangi bir değişiklik olması durumunda hekimlerini bilgilendirmeleri söylenmelidir. Kutanöz Olmayan Skuamöz Hücreli Karsinom (non-cuSCC) Klinik çalışmalarda ZELBORAF kullanan hastalarda non-cuSCC vakaları bildirilmiştir. Hastalara, tedavi başlatılmadan önce ve tedavi sırasında 3 ayda bir, en azından oral mukozanın görsel muayenesini ve lenf nodu palpasyonunu içeren baş ve boyun muayenesi yapılmalıdır. Hastalara ayrıca, tedaviden önce ve tedavi sırasında 6 ayda bir toraks Bilgisayarlı Tomografi (BT) taraması yapılmalıdır. Tedaviden önce ve sonra veya klinik olarak gerekli görüldüğünde, anal muayenelerin ve pelvik muayenelerin (kadınlar için) yapılması önerilmektedir. ZELBORAF tedavisinin sonlandırılmasının ardından non-cuSCC izlenmesi, 6 aya kadar veya başka bir anti-neoplastik tedavi başlatılana kadar devam etmelidir. Anormal bulgular, klinik uygulamalara göre ele alınmalıdır. Yeni primer melanom Klinik çalışmalarda yeni primer melanomlar bildirilmiştir. Vakalar, eksizyon ile kontrol altına alınmış ve hastalar doz ayarlaması olmaksızın tedaviye devam etmiştir. Deri lezyonlarının izlenmesi, yukarıda kutanöz skuamöz hücreli karsinom için belirtildiği gibi yapılmalıdır. Diğer maligniteler Etki mekanizması baz alındığında ZELBORAF, RAS mutasyonu ile ilişkili kanserlerin progresyonuna neden olabilir (bkz. bölüm 4.8). ZELBORAF RAS mutasyonu ile ilişkili geçmiş veya eş zamanlı kanserleri olan hastalarda dikkatli kullanılmalıdır. Karaciğer hasarı ZELBORAF ile karaciğer laboratuvar anormallikleri oluşabilir (bkz. bölüm 4.8). Tedaviye başlanmadan önce ve tedavi sırasında aylık olarak veya klinik açıdan gerekli olduğunda karaciğer enzimleri (transaminazlar ve alkalen fosfataz) ve bilirubin izlenmelidir. Karaciğer anormallikleri, dozun azaltılmasıyla, tedaviye ara verilmesiyle veya tedavinin sonlandırılmasıyla kontrol altına alınmalıdır (bkz. bölüm 4.2 ve 4.4). Karaciğer yetmezliği Karaciğer yetmezliği olan hastalar için başlangıç dozunda ayarlama yapılmasına gerek yoktur. Hiperbilirubinemi olmaksızın karaciğer metastazları nedeniyle hafif karaciğer yetmezliği olan hastalar, genel önerilere uygun şekilde izlenebilir. Orta ila şiddetli karaciğer yetmezliği olan hastalara ilişkin yalnızca çok kısıtlı veri mevcuttur. Orta ila şiddetli karaciğer yetmezliği olan hastalarda ilaca maruziyet artabilir (bkz. bölüm 5.2). Bu nedenle, uzun süre (birkaç hafta) içinde birikme oluşabildiğinden, özellikle tedavinin ilk birkaç haftasından sonra yakından izlem yapılmalıdır. Buna ek olarak, ilk üç ay boyunca her ay EKG izlemi yapılması önerilmektedir. Böbrek yetmezliği Hafif ila orta düzeyde böbrek yetmezliği olan hastalar için başlangıç dozunda ayarlama yapılması gerekmemektedir. Şiddetli böbrek yetmezliği olan hastalara ilişkin yalnızca çok kısıtlı veri mevcuttur (bkz. bölüm 5.2). ZELBORAF, şiddetli böbrek yetmezliği olan hastalarda dikkatli şekilde kullanılmalı ve hastalar yakından izlenmelidir. Fotosensitivite Klinik çalışmalarda ZELBORAF alan hastalarda hafif ila şiddetli düzeyde fotosensitivite bildirilmiştir (bkz. bölüm 4.8). Tüm hastalara, ZELBORAF kullanırken güneşe maruz kalmaktan kaçınmaları önerilmelidir. Bu tıbbi ürünü kullanırken hastalara, güneş yanığından korunmalarına yardımcı olmak için açık havada koruyucu kıyafet giymeleri ve geniş spektrumlu Ultraviyole A (UVA)/Ultraviyole B (UVB) güneş kremi ve dudak kremi (Koruma Faktörü >30) kullanmaları söylenmelidir. Derece 2 (tolere edilemeyen) veya daha yüksek dereceli fotosensitivitede doz modifikasyonları yapılması önerilmektedir (bkz. bölüm 4.2). ZELBORAF'ın diğer tıbbi ürünler üzerindeki etkileri ZELBORAF, başlıca CYP1A2 tarafından metabolize edilen tıbbi ürünlerin plazma maruziyetini artırabilir ve oral kontraseptifler dahil, başlıca CYP3A4 tarafından metabolize edilen ilaçların plazma maruziyetini azaltabilir. ZELBORAF ile eş zamanlı tedaviden önce, başlıca CYP1A2 veya CYP3A4 yoluyla metabolize edilen tıbbi ürünler için terapötik çerçeve temelinde doz ayarlamaları yapılması dikkate alınmalıdır (bkz. bölüm 4.5 ve 4.6). ZELBORAF'ı varfarin ile eş zamanlı kullanırken dikkatli olunmalı ve ek INR (Uluslararası Normalize Oran) izlemi göz önünde bulundurulmalıdır. Diğer tıbbi ürünlerin ZELBORAF üzerindeki etkisi ZELBORAF farmakokinetiği, P-gp'yi inhibe eden veya etkileyen ilaçlardan (örn. verapamil, klaritromisin, siklosporin, ritonavir, kinidin, dronedaron, amiodaron, itrakonazol, ranolazin) etkilenebilir (bkz. bölüm 4.5). Mümkün olduğunda P-gp, glukuronidasyon, CYP3A4'ün güçlü indükleyicileriyle (örn. rifampisin, rifabutin, karbamazepin, fenitoin veya St. John's Wort [hiperisin]) eş zamanlı uygulamadan kaçınılmalıdır (bkz. bölüm 4.5). ZELBORAF'ın etkililiğini korumak için daha düşük indükleyici potansiyele sahip alternatif tedavi dikkate alınmalıdır. İpilimumab ile eşzamanlı kullanımı ZELBORAF (960 mg BID veya 720 mg BID) ve ipilimumabın (3 mg/kg) eşzamanlı kullanıldığı bir faz I çalışmasında transaminazlarda ve bilirubinde asemptomatik derece 3 artışlar (ALT/AST normal üst limitin 5 katı, total bilirubin normal üst limitin 3 katı) raporlanmıştır. Bu ön verilere dayanarak ZELBORAF ve ipilimumabın eşzamanlı kullanımı önerilmemektedir. ZELBORAF, 1920 mg'lık günlük dozunda (240 mg'lık 8 tablet), 10.226 mmol (veya 235.2 mg) sodyum içermektedir. Bu durum, kontrollü sodyum diyetinde olan hastalar için göz önünde bulundurulmalıdır. 4.5. Diğer tıbbi ürünlerle etkileşim ve diğer etkileşim şekilleriVemurafenibin CYP substratları üzerindeki etkileri15 gün boyunca vemurafenib uygulamasından sonra tek doz kafein eş zamanlı uygulandığında CYP1A2 inhibisyonu gözlenmiştir. Bu durum, vemurafenib tedavisinden sonra kafein plazma maruziyetinde ortalama 2.5 kat artışa (maksimum 10 kata kadar) neden olmuştur. Vemurafenib, başlıca CYP1A2 tarafından metabolize edilen maddelerin plazma maruziyetini artırabilir ve doz ayarlamaları dikkate alınmalıdır. 15 gün boyunca vemurafenib uygulamasından sonra tek doz midazolam eş zamanlı uygulandığında CYP3A4 indüksiyonu gözlenmiştir. Bu durum, vemurafenib tedavisinden sonra midazolam plazma maruziyetinde ortalama %32 azalmaya (maksimum %80'e kadar) neden olmuştur. Vemurafenib, başlıca CYP3A4 tarafından metabolize edilen maddelerin plazma maruziyetini azaltabilir. Buna dayalı olarak, vemurafenib ile eş zamanlı kullanılan CYP3A4 tarafından metabolize edilen kontraseptif ilaçların etkililiği azalabilir. Dar terapötik çerçeveye sahip CYP3A4 substratları için doz ayarlamaları dikkate alınmalıdır (bkz. bölüm 4.4 ve 4.6). 10 |iM vemurafenib konsantrasyonunda, in vitrovemurafenib tarafından CYP2B6'nın hafif indüksiyonu kaydedilmiştir. Hastalarda kararlı durumda (yaklaşık 50 |ig/mL) gözlenen 100 |iM'lik plazma düzeyinde vemurafenibin eş zamanlı uygulanan bupropiyon gibi CYP2B6 substratlarının plazma konsantrasyonlarını azaltıp azaltmadığı bilinmemektedir.15 gün boyunca vemurafenib uygulamasından sonra tek doz varfarin eş zamanlı uygulandığında, bazı hastalarda varfarin maruziyetinde artış gözlenmiştir (ortalama %20) (bkz. bölüm 4.4). Melanomlu hastalarda vemurafenib varfarin (CYP2C9) ile eş zamanlı uygulanırken dikkatli olunmalıdır. Vemurafenib CYP2C8'i in-vitro olarak inhibe eder. Bu bulgunun in-vivo olarak ilişkisi bilinmemektedir ancak eşzamanlı uygulanan CYP2C8 substratları üzerindeki klinik etki riski göz ardı edilemez. Vemurafenibin uzun yarılanma ömrü nedeniyle, eş zamanlı uygulanan tıbbi ürün üzerinde vemurafenibin tam inhibitör etkisi, vemurafenib tedavisinde 8 günden önce gözlenmeyebilir. Vemurafenib tedavisi sonlandırıldıktan sonra, izleyen tedaviyle etkileşimin önlenmesi için 8 günlük bir arınma dönemi gerekli olabilir. Vemurafenibin madde taşıma sistemleri üzerindeki etkileri In vitroçalışmalar, vemurafenibin P-gp ve BCRP efluks taşıyıcılarının bir inhibitörü olduğunu göstermiştir. Bu bulgunun klinik önemi bilinmemektedir. Vemurafenibin, P-gp tarafından taşınan (aliskiren, kolşisin, digoksin, everolimus, feksofenadin gibi) veya BCRP tarafından taşınan (metotreksat, mitoksantron, rosuvastatin gibi) diğer ilaçların maruziyetini artırabileceği göz ardı edilemez.Kanser ilaçlarının çoğu P-gp ve/veya BCRP substratıdırlar ve bu nedenle ZELBORAF ile etkileşim riski söz konusu olabilir. Vemurafenibin diğer taşıyıcılar üzerindeki olası etkisi günümüzde bilinmemektedir. Eş zamanlı uygulanan ilaçların vemurafenib üzerindeki etkileri In vitroçalışmalar, CYP3A4 metabolizmasının ve glukuronidasyonun vemurafenib metabolizmasından sorumlu olduğunu göstermektedir. Safradan atılımın bir diğer önemli eliminasyon yolu olduğu düşünülmektedir. CYP3A4'ün ve/veya taşıyıcı protein aktivitesinin güçlü indükleyicilerinin veya inhibitörlerinin vemurafenib maruziyeti üzerindeki etkisini gösteren klinik veriler bulunmamaktadır. Vemurafenib, CYP3A4'ün, glukuronidasyonun ve/veya taşıyıcı proteinlerin güçlü inhibitörleriyle (örn. ritonavir, sakuinavir, telitromisin, ketokonazol, itrakonazol, vorikonazol, posakonazol, nefazodon, atazanavir) kombinasyon halinde kullanılırken dikkatli olunmalıdır.P-gp, glukuronidasyon ve/veya CYP3A4'ün güçlü indükleyicileriyle (örn. rifampisin, rifabutin, karbamazepin, fenitoin veya St. John's Wort [hypercium perforatum]) eş zamanlı uygulama, vemurafenibin optimumdan düşük maruziyetine neden olabilir ve bu durumdan kaçınılmalıdır.In vitroçalışmalar, vemurafenibin efluks taşıyıcıları olan P-gp'nin ve BCRP'nin bir substratı olduğunu göstermiştir. P-gp ve BCRP indükleyicilerinin ve inhibitörlerinin vemurafenib maruziyeti üzerindeki etkileri bilinmemektedir. Vemurafenib farmakokinetiğinin, P-gp'yi (örn. verapamil, siklosporin, ritonavir, kinidin, itrakonazol) veya BCRP'yi (siklosporin, gefitinib gibi) etkileyen ilaçlardan etkilenebileceği göz ardı edilemez.Vemurafenibin diğer taşıyıcı proteinlerin de substratı olup olmadığı günümüzde bilinmemektedir. Özel popülasyonlara ilişkin ek bilgilerHiçbir etkileşim çalışması yapılmamıştır.Pediyatrik popülasyon:Hiçbir etkileşim çalışması yapılmamıştır.4.6. Gebelik ve laktasyon Genel tavsiyeGebelik kategorisi DÇocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon)Çocuk doğurma potansiyeli bulunan kadınlar tedavi boyunca ve tedaviden sonra en az 6 ay boyunca etkili kontrasepsiyon kullanmalıdır.Vemurafenib, hormonal kontraseptiflerin etkililiğini azaltabilir (bkz. bölüm 4.5). Gebelik dönemiVemurafenibin gebe kadınlarda kullanımıyla ilgili veri bulunmamaktadır.Vemurafenib, sıçan veya tavşan embriyolarında/fetüslerinde teratojenisite kanıtı göstermemiştir (bkz. bölüm 5.3). Hayvanlarda yapılan çalışmalarda, vemurafenibin plasentayı geçtiği belirlenmiştir. ZELBORAF, anneye sağladığı olası yararlar fetüs üzerindeki olası riskten fazla olmadıkça gebe kadınlara uygulanmamalıdır. Laktasyon dönemiVemurafenibin anne sütüne geçip geçmediği bilinmemektedir. Yenidoğanlar/bebekler üzerindeki risk göz ardı edilemez. Emzirmenin sonlandırılması veya ZELBORAF tedavisinin sonlandırılması ile ilgili karar verilirken, çocuğun emzirilmesiyle sağlanan yarar ile tedavinin kadına sağladığı yarar dikkate alınmalıdır.Üreme yeteneği/FertiliteFertilite üzerindeki etkiyi değerlendirmek üzere hayvanlarda vemurafenib ile ilgili spesifik çalışma yapılmamıştır. Bununla birlikte, sıçanlarda ve köpeklerde yapılan tekrarlı doz toksisitesi çalışmalarında, üreme organları üzerinde histopatolojik bulgu saptanmamıştır (bkz. bölüm 5.3).4.7. Araç ve makine kullanımı üzerindeki etkilerZELBORAF'ın araç ve makine kullanımı üzerindeki etkileri incelenmemiştir. Hastalar, araç kullanmaya engel olacak şekilde yorgunluk veya göz sorunları oluşabileceğini unutmamalıdır.4.8. İstenmeyen etkilerGüvenlilik profilinin özetiZELBORAF ile bildirilen en yaygın advers ilaç reaksiyonları (AİR) (>%30); artralji, yorgunluk, döküntü, fotosensitivite reaksiyonu, bulantı, alopesi ve kaşıntıyı içerir. CuSCC çok yaygın şekilde bildirilmiştir ve çoğunlukla lokal eksizyon yoluyla tedavi edilmiştir. Advers reaksiyonların özeti Melanomlu hastalarda bildirilen AİR'ler, MedDRA vücut sistemi organ sınıfına, sıklığa ve şiddet derecesine göre aşağıda listelenmiştir. Sıklığın sınıflandırılmasında aşağıdaki kural kullanılmıştır: Çok yaygın (>1/10), yaygın (>1/100 ila <1/10), yaygın olmayan (>1/1,000 ila <1/100), seyrek (>1/10,000 ila <1/1,000), çok seyrek (<1/10,000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Bu bölümde AİR'ler; BRAF V600 mutasyonu pozitif bulunup cerrahi olarak çıkarılamayan veya metastatik melanomu bulunan yetişkin hastalarda yapılan bir faz III randomize açık etiketli çalışmadan ve yanısıra, daha önce uygulanan en az bir sistemik tedavinin başarısız olduğu BRAF V600 mutasyonu pozitif bulunan metastatik melanomlu hastalarda yapılan bir faz II tek kollu çalışmadan 468 hastaya ait bulgulara dayanmaktadır (bkz. bölüm 5.1). Ek olarak tüm klinik çalışmalardan elde edilen güvenlilik raporlarından alınan AİR'ler de raporlanmıştır. Belirtilen tüm terimlerde, faz II ve faz III klinik çalışmalarda gözlenen en yüksek oran temel alınmıştır. Her sıklık grubunda AİR'ler, azalan şiddete göre sunulmaktadır ve toksisite değerlendirmesine yönelik NCI-CTCAE v4.0 (ortak toksisite kriterleri) kullanılarak bildirilmiştir. ZELBORAF ile tedavi edilen hastalarda ortaya çıkan ve Faz II veya Faz III çalışmalardan ve tüm klinik çalışmalardaki güvenlilik raporlarından bildirilen AİR'lerEnfeksiyonlar ve enfestasyonlarYaygın : Folikülit(Kist ve polipler de dahil olmak üzere) iyi huylu ve kötü huylu neoplazmalarÇok yaygın : Deride SCC (c), seboreik keratoz, deri papillomuYaygın : Bazal hücreli karsinom, yeni primer melanom+ Yaygın olmayan : Kutanöz olmayan skuamöz hücreli karsinom (Non-cuSCC*+) Metabolizma ve beslenme hastalıklarıÇok yaygın : İştah azalmasıSinir sistemi hastalıklarıBaş ağrısı, disguzi (tat alma bozukluğu)
Çok yaygın Yaygın Yaygın olmayan 7. sinir paralizi, baş dönmesi Periferik nöropati Göz hastalıklarıYaygın : ÜveitYaygın olmayan : Retinal ven oklüzyonu Vasküler hastalıklarYaygın olmayan : VaskülitSolunum, göğüs bozuklukları ve mediastinal hastalıklarÇok yaygın : ÖksürükGastrointestinal hastalıklarÇok yaygın : İshal, kusma, bulantı, kabızlıkDeri ve deri altı doku hastalıklarıÇok yaygın : Fotosensitivite reaksiyonu, aktinik keratoz, döküntü, makülo-papülerdöküntü, papüler döküntü, kaşıntı, hiperkeratoz, eritem, alopesi, cilt kuruluğu, güneş yanığı Yaygın : Palmar-plantar eritrodizestezi sendromu, eritema nodosum, keratoz pilaris Yaygın olmayan : Toksik epidermal nekroliz (d), Stevens-Johnson sendromu (e) Kas-iskelet bozuklukları, bağ doku ve kemik hastalıklarıÇok yaygın : Artralji, miyalji, ekstremite ağrısı, kas-iskelet ağrısı, sırt ağrısıGenel bozukluklar ve uygulama bölgesine ilişkin hastalıklarıÇok yaygın : Yorgunluk, ateş, periferik ödem, asteniAraştırmalarÇok yaygın : GGT yükselmesi (b)Yaygın : ALT yükselmesi (b), alkalen fosfataz yükselmesi (b), bilirubin yükselmesi (b), kilo kaybı, QT uzaması Yaygın olmayan : AST yükselmesi (b) * Klinik çalışmalardaki güvenlilik raporlarından alınan vakalar +Tıbbi ürün ve advers olay arasındaki nedensellik ilişkisi en azından olasılık dahilindedir Belli advers reaksiyonlara ilişkin açıklama Karaciğer enzimlerinde yükselme (b) Faz III klinik çalışmada bildirilen karaciğer enzimi anormallikleri, bazal durumdan derece 3 veya 4 enzim anormalliklerine değişen hastaların oranları şeklinde aşağıda verilmektedir: Çok yaygın: GGT Yaygın: ALT, alkalen fosfataz, bilirubin Yaygın olmayan: AST Derece 4'e ALT, alkalen fosfataz, bilirubin yükselmeleri olmamıştır. Kutanöz skuamöz hücreli karsinom (c) (cuSCC) Vemurafenib ile tedavi edilen hastalarda cuSCC vakaları bildirilmiştir. Çalışmalarda, ZELBORAF uygulanan hastalarda cuSCC insidansı yaklaşık %20 olmuştur. Bağımsız bir merkezi dermatopatoloji laboratuarı tarafından değerlendirilen eksize lezyonların büyük bölümü SCC-keratoakantom alt tipi olarak veya mikst keratoakantom özelliklerine sahip olarak sınıflandırılmıştır (%52). Diğer olarak sınıflandırılan çoğu lezyon (%43) benign deri lezyonları olmuştur (örn. verruca vulgaris, aktinik keratoz, benign keratoz, kist/benign kist). CuSCC, ilk kez görülene kadar geçen medyan süre 7 ila 8 hafta olmak üzere, genellikle tedavinin erken aşamasında oluşmuştur. CuSCC oluşan hastaların yaklaşık %33'ünde cuSCC >1 kez meydana gelmiş ve arada geçen medyan süre 6 hafta olmuştur. CuSCC vakaları tipik olarak basit eksizyonla kontrol altına alınmış ve hastalar genellikle doz modifikasyonu olmaksızın tedaviye devam etmiştir (bkz. bölüm 4.2 ve 4.4). Kutanöz olmayan skuamöz hücreli karsinom (non-cuSCC) Klinik çalışmalarda vemurafenib ile tedavi edilen hastalarda non-cuSCC vakaları bildirilmiştir. Kutanöz olmayan skuamöz hücreli karsinomun izlemi, bölüm 4.4'te belirtildiği gibi yapılmalıdır. Yeni primer melanom Klinik çalışmalarda yeni primer melanomlar bildirilmiştir. Bu vakalar, eksizyon ile kontrol altına alınmış ve hastalar doz ayarlaması olmaksızın tedaviye devam etmiştir. Deri lezyonlarının izlemi, bölüm 4.4'te belirtildiği gibi yapılmalıdır. Aşırı duyarlılık reaksiyonları (d) ZELBORAF'la ilişkili olarak anafilaksi dahil ciddi aşırı duyarlılık reaksiyonları bildirilmiştir. Şiddetli aşırı duyarlılık reaksiyonları; Stevens-Johnson sendromunu, jeneralize döküntüyü, eritemi veya hipotansiyonu içerebilir. Şiddetli aşırı duyarlılık reaksiyonları oluşan hastalarda ZELBORAF tedavisi kalıcı olarak sonlandırılmalıdır (bkz. bölüm 4.4). Dermatolojik reaksiyonlar (e) ZELBORAF alan hastalarda, pivotal klinik çalışmalarda oluşan nadir Stevens-Johnson sendromu ve toksik epidermal nekroliz vakaları dahil, şiddetli dermatolojik reaksiyonlar bildirilmiştir. Şiddetli dermatolojik reaksiyonlar oluşan hastalarda ZELBORAF tedavisi kalıcı olarak sonlandırılmalıdır. QT uzaması Günde iki kez 960 mg ZELBORAF uygulanan 132 hastada yapılan açık etiketli, kontrollü olmayan bir faz II QT alt çalışmasından (NP22657) elde edilen merkezileştirilmiş EKG verilerinin analizi, vemurafenib maruziyetine bağlı QTc uzaması olduğunu göstermiştir. Ortalama QTc etkisi, tedavinin ilk ayından sonra 12-15 msn arasında sabit kalmıştır ve en büyük ortalama QTc uzaması (15.1 msn; üst %95 GA: 17.7 msn) ilk 6 ay içinde gözlenmiştir (n=90 hasta). İki hastada (%1.5) tedaviyle >500 msn değerlerinde mutlak QTc oluşmuştur (CTC Derece 3) ve yalnızca bir hastada (%0.8) başlangıca göre >60 msn'lik QTc değişimi görülmüştür (bkz. bölüm 4.4). Pazarlama sonrası bildirilen yan etkiler (Kist ve polipler de dahil olmak üzere) iyi huylu ve kötü huylu neoplazmalarBilinmiyor : Kronik miyelomonositik lösemi (KMML) progresyonu (bkz bölüm 4.4)**: Önceden var olan NRAS mutasyonlu kronik miyelomonositik lösemi Deri ve deri altı doku hastalıklarıBilinmiyor : Eozinofili ve sistemik semptomlar ile seyreden ilaç döküntüsü (DRESS Sendromu) (bkz bölüm 4.4).Özel popülasyonlara ilişkin ek bilgilerGeriyatrik popülasyonFaz III çalışmada, ZELBORAF tedavisi uygulanan cerrahi olarak çıkarılamayan veya metastatik melanomlu 336 hastadan 94'ü (%28) >65 yaşındadır. Yaşlı hastalarda (>65 yaş) cuSCC, iştah azalması ve kardiyak hastalıklar dahil advers reaksiyonların oluşma olasılığı daha yüksektir.CinsiyetZELBORAF ile yapılan klinik çalışmalar sırasında, erkeklere göre kadınlarda daha sık bildirilen derece 3 advers reaksiyonlar döküntü, artralji ve fotosensitivite olmuştur.4.9. Doz aşımı ve tedavisiVemurafenib doz aşımı için spesifik antidot bulunmamaktadır. Advers reaksiyonlar gelişen hastalar, uygun semptomatik tedavi almalıdır. Klinik çalışmalarda vemurafenib ile doz aşımı vakaları gözlenmemiştir. Şüpheli doz aşımı durumunda, vemurafenib tedavisi durdurulmalı ve destek tedavisi başlatılmalıdır.5.1. Farmakodinamik özelliklerFarmakoterapötik grup: Antineoplastik ajanlar, protein kinaz inhibitörü ATC kodu: L01XE15Etki mekanizması ve farmakodinamik etkiler Vemurafenib düşük molekül ağırlıklı, oral olarak kullanılan bir BRAF serin-treonin kinaz inhibitörüdür. Amino asit pozisyonu 600'de valini substitüte eden BRAF genindeki mutasyonlar, yapısal olarak sürekli aktif BRAF proteinlerine yol açar ve bu proteinler, normalde proliferasyon için gerekli olan büyüme faktörlerinin yokluğunda hücre proliferasyonuna neden olabilir. Biyokimyasal tayinlerden elde edilen klinik öncesi veriler, vemurafenibin aktive edici kodon 600 mutasyonları olan BRAF kinazları güçlü şekilde inhibe edebildiğini göstermiştir (Tablo 3). Tablo 3: Vemurafenibin farklı BRAF kinazlara karşı kinaz inhibitör aktivitesi
Bu inhibitör etki, V600-mutant BRAF eksprese eden mevcut melanom hücre serilerinde yapılan ERK fosforilasyon ve hücresel anti-proliferasyon tayinlerinde doğrulanmıştır. Hücresel anti-proliferasyon tayinlerinde, V600 mutasyonlu hücre serilerinde (V600E, V600R, V600D ve V600K mutasyonlu hücre sıraları) karşı IC50 0.016 ila 1.131 ^M arasında olurken, BRAF wild-tip hücre sıralarına karşı inhibitör konsantrasyon sırasıyla 12.06 ve 14.32 |iM olmuştur. BRAF mutasyon durumunun saptanması Vemurafenib almadan önce, geçerli bir test yoluyla hastaların BRAF V600 mutasyon-pozitif tümör durumu doğrulanmalıdır. Faz II ve faz III klinik çalışmalarda, uygun hastalar gerçek zamanlı polimeraz zincir reaksiyonu (cobas 4800 BRAF V600 Mutasyon Testi) tayini kullanılarak belirlenmiştir. Bu test CE işareti taşımaktadır ve formalinle fikse edilen, parafine gömülü (FFPE) tümör dokusundan izole edilen DNA'nın BRAF mutasyon durumunu değerlendirmek için kullanılır. Baskın BRAF V600E mutasyonunu yüksek duyarlılıkla saptamak üzere tasarlanmıştır (FFPE ile elde edilen DNA'dan wild-tip dizilimi fonunda, %5'e kadar V600E dizilimi). Retrospektif dizilim analizlerinin yapıldığı klinik dışı ve klinik çalışmalar, bu testin ayrıca daha az yaygın olan BRAF V600D mutasyonlarını ve V600K mutasyonlarını daha düşük bir duyarlılıkla saptadığını göstermiştir. Klinik dışı ve klinik çalışmalardan sağlanan, gerçek zamanlı polimeraz zincir reaksiyonu tayini testiyle mutasyon-pozitif bulunan ve ek olarak sekans yoluyla analiz edilen numuneler arasında (n=920), Sanger ve 454 sekans yoluyla hiçbir numune wild-tip olarak belirlenmemiştir. Klinik etkililik ve güvenlilik Vemurafenibin etkililiği, bir faz III klinik çalışmadaki (NO25026) 336 hastada ve bir faz II klinik çalışmadaki (NP22657) 132 hastada değerlendirilmiştir. Tüm hastaların, gerçek zamanlı polimeraz zincir reaksiyonu tayini testine göre BRAF V600 mutasyonları bulunan ileri evre melanom olması gerekmiştir. Daha önce tedavi uygulanmamış hastalarda yapılan Faz III çalışmanın (NO25026) bulguları Açık etiketli, çok merkezli, uluslararası, randomize bir faz III çalışma, daha önce tedavi uygulanmamış BRAF V600E mutasyon-pozitif olup cerrahi olarak çıkarılamayan veya metastatik melanomlu hastalarda vemurafenibin kullanımını desteklemektedir. Hastalar, vemurafenib (günde iki kez 960 mg) veya dakarbazin (3 haftada bir 1. gün 1000 mg/m ) ile tedavi almak üzere randomize edilmiştir. Toplam 675 hasta vemurafenibe (n=337) veya dakarbazine (n=338) randomize edilmiştir. Hastaların çoğu erkek (%56) ve beyaz ırktandır (%99), medyan yaş 54'tür (%24'ü >65 yaşındadır), tüm hastaların ECOG performans durumu 0 veya 1'dir ve hastaların büyük bölümünde M1c hastalık mevcuttur (%65). Çalışmanın ortak birincil etkililik sonlanım noktaları genel sağkalım (OS) ve progresyonsuz sağkalım (PFS) olmuştur. Veri kesim tarihi 30 Aralık 2010 olmak üzere önceden belirlenen ara analizde, ortak birincil sonlanım noktaları OS'de (p<0.0001) ve PFS'de (p<0.0001) (katmanlandırılmamış log-rank testi) anlamlı iyileşmeler gözlenmiştir. Veri Güvenlilik İzleme Kurulu'nun (DSMB) önerisiyle, bu bulgular Ocak 2011'de açıklanmış ve çalışma, dakarbazin hastalarının vemurafenibe geçmesine izin verilecek şekilde düzenlenmiştir. Daha sonra, tablo 4'te açıklandığı gibi post-hoc sağkalım analizleri yapılmıştır. Tablo 4: Çalışma veri kesim tarihine göre, önceden tedavi uygulanmamış BRAF V600 mutasyon-pozitif melanomlu hastalarda genel sağkalım (N=338 dakarbazin, N=337 vemurafenib)
Şekil 1: Genel sağkalııııa ilişkin Kaplan-Meier eğrileri - önceden tedavi uygulanmamış hastalar (veri kesim tarihi 20 Aralık 2012)risk altındakiler&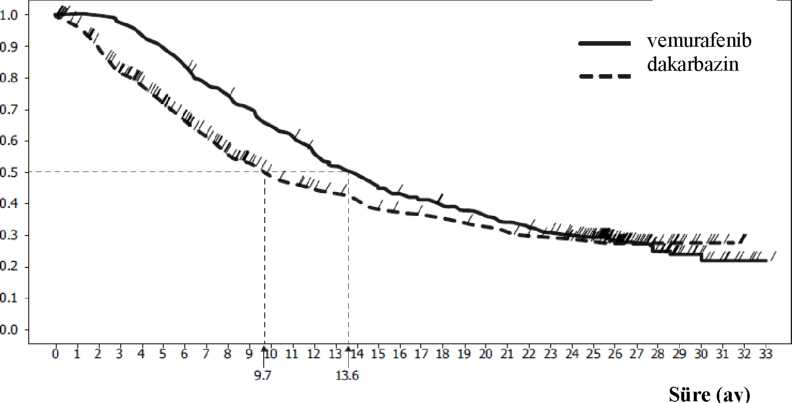 i 33Ö 306 276 243 217193172154 126110 97 91 82 79 76 68 65 63 60 58 55 51 48 46 41 36 28 20 17 11 8 4 0 0 b 337 336335 326 314300 281 260 248 232 214 203183 171 161 148140135 129 123117110104 98 91 81 56 43 30 17 13 8 4 1
i 33Ö 306 276 243 217193172154 126110 97 91 82 79 76 68 65 63 60 58 55 51 48 46 41 36 28 20 17 11 8 4 0 0 b 337 336335 326 314300 281 260 248 232 214 203183 171 161 148140135 129 123117110104 98 91 81 56 43 30 17 13 8 4 1dakarbazin vemurafenib Tablo 5, prognostik faktörler olarak yayımlanan önceden belirlenmiş tüm sınıflandırma değişkenleri için tedavi etkisini göstermektedir. Tablo 5: LDH'ye, tümör evresine ve ECOG durumuna göre, önceden tedavi uygulanmamış BRAF V600 mutasyonu pozitif olan melanomlu hastalarda genel sağkalım (post-hoc analizi, veri kesim tarihi 20 Aralık 2012, cross-over sırasında
Tablo 6, önceden tedavi uygulanmamış BRAF V600 mutasyon-pozitif melanomlu hastalarda genel yanıt oranını ve progresyonsuz sağkalımı göstermektedir. Tablo 6: Önceden tedavi uygulanmamış BRAF V600 mutasyon-pozitif melanomluhastalarda genel yanıt oranı ve progresyonsuz sağkalım__
(i) 30 Aralık 2010 itibarıyla PFS için toplam 549 hasta ve genel yanıt oranı için 439 hasta değerlendirilebilir olmuştur. (j) 1 Şubat 2012 itibarıyla 675 hasta PFS'in post-hoc analiz güncellemesi için değerlendirilebilir olmuştur. NO25026'da, tümörleri retrospektif sekans yoluyla analiz edilen 673 hastadan toplam 57 hastanın BRAF V600K mutasyon-pozitif melanomu olduğu bildirilmiştir. Hasta sayısının az olması nedeniyle kısıtlanmasına karşın, V600K-pozitif tümörleri olan bu hastalarda yapılan etkililik analizleri OS, PFS ve doğrulanmış en iyi genel yanıt açısından vemurafenibin tedavi yararını göstermiştir. V600E ve V600K dışındaki BRAF V600 mutasyonlara sahip melanomlu hastalara ilişkin veri bulunmamaktadır. Daha önce en az bir tedavinin başarısız olduğu hastalarda yapılan faz II çalışmanın (NP22657) bulguları Gerçek zamanlı polimeraz zincir reaksiyonu tayini testine göre BRAF V600E mutasyon-pozitif metastatik melanomu olan ve önceden en az bir tedavi uygulanmış 132 hastada faz II tek kollu, çok merkezli, çok uluslu bir çalışma yapılmıştır. Hastaların %19'u 65 yaşın üzerinde olmak üzere, medyan yaş 52'dir. Hastaların büyük bölümü erkektir (%61), beyaz ırktandır (%99) ve M1c hastalık mevcuttur (%61). Kırk dokuz hastada, daha önceki >2 tedavi başarısız olmuştur. 12.9 aylık medyan takiple (0.6 ila 20.1 aralığı), bağımsız bir değerlendirme kurulu (IRC) tarafından değerlendirildiği üzere doğrulanmış en iyi genel yanıt oranı (TY + KY) şeklinde birincil sonlanım noktası %53 olmuştur (%95 GA: %44, %62). Medyan genel sağkalım 15.9 ay olmuştur (%95 GA: 11.6, 18.3). Genel sağkalım oranı, 6 ayda %77 (%95 GA: %70, %85) ve 12 ayda %58 (%95 GA: %49, %67) olmuştur. NP22657'de yer alan 132 hastadan dokuzunda, retrospektif Sanger sekansa göre V600K mutasyon-pozitif tümörler belirlenmiştir. Bu hastalardan 3'ü KY, 3'ü SH ve 2'si PH elde etmiş, bir hasta değerlendirilememiştir. 5.2. Farmakokinetik özelliklerGenel özelliklerBiyofarmasötikleri Sınıflandırma Sistemi'nde (BCS) açıklanan kriterlere göre vemurafenib Sınıf IV bir maddedir (düşük çözünürlük ve permeabilite). Vemurafenib için farmakokinetik parametreler; faz I ve faz III çalışmalarda kompartmanlı olmayan analiz kullanılarak (günde iki kez 960 mg doz uygulaması yapılan 15 gün ardından 20 hasta ve 22. günde kararlı durumda olan 204 hasta) ve yanısıra, 458 hastaya ait toplu verilerin kullanıldığı popülasyon farmakokinetik analizi yoluyla belirlenmiştir. Bu hastalardan 457'si beyaz ırktandır.Emilim:Vemurafenib 240 mg tabletin mutlak biyoyararlanımı bilinmemektedir. Günde iki kez 960 mg dozda vemurafenib, yaklaşık 4 saatlik medyan Tmaks ile emilmektedir. Vemurafenib, hastalar arasında büyük değişkenlik göstermektedir. Faz II çalışmada, 1. gündeki EAA0-8 saat ve Cmaks sırasıyla 22.1 ± 12.7 |igsaat/mL ve 4.1 ± 2.3 |ig/mL olmuştur. Vemurafenibin günde iki kez çoklu doz uygulamasıyla birikme meydana gelir. Kompartmanlı olmayan analizde, günde iki kez 960 mg vemurafenib uygulamasından sonra kararlı durum koşullarında 15. Gün / 1. Gün oranı EAA için 15 ila 17 kat ve Cmaks için 13 ila 14 kat aralığında olmuş ve EAA0-8 saat ve Cmaks sırasıyla 380.2 ± 143.6 |igsaat/mL ve 56.7 ± 21.8 |ig/mL olmuştur. Yiyecekler (fazla yağlı yemek) vemurafenibin 960 mg'lık tek dozunun bağıl biyoyararlanımını artırır. Tokluk ve açlık hali arasındaki geometrik ortalama oranlar Cmaks ve EAA için sırasıyla 2.6 ve 4.7 katı olmuştur. Tek bir vemurafenib dozu yiyeceklerle birlikte alındığında medyan Tmaks, 4 saatten 8 saate çıkmıştır. Yiyeceklerin kararlı durumda vemurafenib maruziyeti üzerindeki etkisi şu an bilinmemektedir. Sürekli olarak aç karnına vemurafenib alımı, yemekle birlikte veya yemekten kısa bir süre sonra sürekli vemurafenib alımı ile kıyaslandığında anlamlı oranda daha düşük bir kararlı durum maruziyetine yol açabilir. Bununla birlikte vemurafenibin ara sıra aç karnına alımının, kararlı durumda vemurafenibin yüksek düzeyde birikmesinden dolayı kararlı durum maruziyeti üzerindeki etkisinin sınırlı olması beklenmektedir. Tek başına veya yiyeceklerle birlikte vemurafenib alan hastalardan pivotal çalışmalarda güvenlilik ve etkililik verileri toplanmıştır. Gastrointestinal sıvı içeriği, hacim, pH, motilite ve geçiş süresi ile safra bileşimindeki farklılıklara bağlı olarak da maruziyette değişkenlik görülebilir. Kararlı durumda plazmadaki ortalama vemurafenib maruziyeti, sabah dozundan önce ve sabah dozundan 2-4 saat sonra plazma konsantrasyonları arasındaki 1.13'lük ortalama oran ile gösterildiği gibi, 24 saatlik ara boyunca stabildir. Oral doz uygulamasının ardından metastatik melanom hastalarından oluşan popülasyon için emilim hızı sabiti 0.19 saat-1 saat (%101'lik hastalar arası değişkenlik ile) olarak tahmin edilmektedir. Dağılım:Metastatik melanomlu hastalarda vemurafenibin görünen dağılım hacmi 91 L olarak hesaplanmıştır (hastalar arası değişkenlik %64.8). In vitro,insan plazma proteinlerine yüksek oranda bağlıdır (>%99).Biyotransformasyon:Vemurafenibin ve metabolitlerinin göreceli oranları, oral yolla tek doz 14C-işaretli vemurafenib uygulanmasıyla insanlara ilişkin bir kütle denge çalışmasında belirlenmiştir. CYP3A4, vemurafenibin in vitrometabolizmasından sorumlu birincil enzimdir. İnsanlarda konjugasyon metabolitleri de (glukuronidasyon ve glikozilasyon) belirlenmiştir. Bununla birlikte, plazmadaki baskın bileşen (%95) ana bileşik olmuştur. Metabolizmanın, plazmada önemli miktarda metabolite yol açmadığı düşünülmektedir ancak atılım açısından metabolizmanın önemi göz ardı edilemez.Eliminasyon:Metastatik melanomlu hastalarda, popülasyonda görünen vemurafenib klirensi 29.3 L/gün olarak hesaplanmıştır (hastalar arası değişkenlik %31.9). Vemurafenib için popülasyon PK analizi yoluyla hesaplanan popülasyon eliminasyon yarılanma ömrü 51.6 saattir (bireysel yarılanma ömrü tahminlerinin 5. ve 95. persentil aralığı 29.8-119.5 saattir). İnsanlarda yapılan vemurafenibin oral yolla uygulandığı kütle denge çalışmasında, dozun ortalama %95'i 18 gün içinde geri kazanılmıştır. Vemurafenible ilişkili maddenin büyük bölümü (%94) dışkıda ve <%1'i idrarda belirlenmiştir. Değişikliğe uğramamış bileşiğin biliyer atılımı önemli bir atılım yolu olabilir. Bununla birlikte, mutlak biyoyararlanımın bilinmemesi nedeniyle, ana bileşik vemurafenibin klirensi açısından hepatik ve renal atılımın önemi belirsizdir. Vemurafenib, P-gp'nin in vitrosubstratı ve inhibitörüdür.Doğrusallık/ doğrusal olmayan durum:Vemurafenib farmakokinetiğinin, günde iki kez alınan 240 ila 960 mg arasında dozla orantılı olduğu gösterilmiştir, popülasyon farmakokinetik analizi de vemurafenib farmakokinetiğinin lineer olduğunu doğrulamaktadır. Hastalardaki karakteristik özelliklerYaşlı hastalar (65 yaş veya 65 yaşın üstünde):Popülasyon farmakokinetik analizleri temelinde, yaşın vemurafenib farmakokinetikleri üzerinde istatistiksel anlamlı bir etkisi bulunmamaktadır. Çocuklar ve adolesanlar (18 yaşa kadar):Pediyatrik hastalarda vemurafenibin farmakokinetiklerini incelemek üzere çalışma yapılmamıştır. Cinsiyet:Popülasyon farmakokinetik analizi, kadınlara kıyasla erkeklerde görünen klirensin (CL/F) %17 daha fazla ve görünen dağılım hacminin (V/F) %48 daha yüksek olduğunu göstermiştir. Bunun cinsiyetin mi, yoksa vücut yüzey alanının mı etkisi olduğu bilinmemektedir. Bununla birlikte maruz kalınan miktardaki farklılıklar, vücut yüzey alanı veya cinsiyet temelinde doz ayarlaması gerektirecek kadar büyük olmamıştır. Böbrek yetmezliği:Metastatik melanomlu hastalarda yapılan klinik çalışmalara ait verilerin kullanıldığı popülasyon farmakokinetik analizinde, hafif ve orta düzeyde böbrek yetmezliği vemurafenibin görünen klirensini etkilememiştir (kreatinin klirensi >40 mL/dakika). Şiddetli böbrek yetmezliği olan hastalara ilişkin veri bulunmamaktadır (bkz. bölüm 4.2 ve 4.4). Karaciğer yetmezliği:Klinik öncesi veriler ve insanlara ilişkin kütle denge çalışması temelinde, vemurafenibin büyük bölümü karaciğer yoluyla elimine edilmektedir. Metastatik melanomlu hastalarda yapılan klinik çalışmalara ait verilerin kullanıldığı popülasyon farmakokinetik analizinde, normal üst limitin üç katına kadar AST ve ALT yükselmeleri vemurafenibin görünen klirensini etkilememiştir. Karaciğerin metabolik veya ekskretuar yetmezliğinin vemurafenib farmakokinetikleri üzerindeki etkisini belirlemek açısından veriler yetersizdir (bkz. bölüm 4.2 ve 4.4). 5.3. Klinik öncesi güvenlilik verileriVemurafenibin klinik öncesi güvenlilik profili sıçanlarda, köpeklerde ve tavşanlarda değerlendirilmiştir.Tekrarlı doz toksikoloji çalışmalarında, karaciğer ve kemik iliği köpeklerdeki hedef organlar olarak belirlenmiştir. Köpeklerde yapılan 13 haftalık bir çalışmada, beklenen klinik maruziyetin (EAA karşılaştırmaları temelinde) altındaki ilaç maruziyetlerinde, karaciğerde geri döndürülebilir toksik etkiler (hepatoselüler nekroz ve dejenerasyon) kaydedilmiştir. Erken sonlandırılan 39 haftalık BID köpek çalışmasındaki bir köpekte, beklenen klinik maruziyete (EAA karşılaştırmaları temelinde) benzer ilaç maruziyetlerinde fokal kemik iliği nekrozu kaydedilmiştir. In vitrokemik iliği sitotoksisite çalışmasında; klinik ilgili konsantrasyonlarda bazı sıçan, köpek ve insan lenfo-hematopoietik hücre popülasyonlarında hafif sitotoksisite gözlenmiştir.450 mg/kg/güne kadar dozlarda (beklenen klinik maruziyetin (EAA karşılaştırmaları temelinde) altındaki ilaç maruziyetlerinde) yapılan bir sıçan çalışmasında, vemurafenibin kültüre edilmiş mürin fibroblastlarında UVA ışın uygulamasından sonra in vivoin vitrofototoksik olduğu gösterilmiştir. Fertilite üzerindeki etkiyi değerlendirmek üzere vemurafenible spesifik çalışma yapılmamıştır. Bununla birlikte, tekrarlı doz çalışmalarında 450 mg/kg/güne kadar dozlarda (EAA karşılaştırmaları temelinde beklenen klinik maruziyetin altındaki ilaç maruziyetlerinde) erkek ve dişi sıçanların ve köpeklerin üreme organları ile ilgili histopatolojik bulgu saptanmamıştır. Sıçanlarda ve tavşanlarda, beklenen klinik maruziyetin altındaki ilaç maruziyetlerine yol açan (EAA karşılaştırması temelinde) sırasıyla 250 mg/kg/gün ve 450 mg/kg/gün dozlara kadar embriyo-fetal gelişim çalışmalarında teratojenisite gözlenmemiştir. Bununla birlikte embriyo-fetal gelişim çalışmalarındaki maruziyet, EAA karşılaştırması temelinde klinik maruziyetin altında olmuştur ve bu nedenle, bu bulguların ne ölçüde insanlara uyarlanabileceğini belirlemek güçtür. Sonuç olarak vemurafenibin fetüs üzerindeki etkisi göz ardı edilemez. Pre- ve postnatal gelişim ile ilgili çalışma yapılmamıştır.In vitroin vivosıçan kemik iliği mikronükleus testi yapılmamıştır.Vemurafenib ile karsinojenisite çalışmaları yapılmamıştır. 6. FARMASÖTİK ÖZELLİKLER6.1. Yardımcı maddelerin listesiKroskarmeloz sodyum Kolloidal anhidröz silika Magnezyum stearat Hidroksipropilselüloz Polivinil alkol Titanyum dioksit (E171)Makrogol 3350 Talk Demir oksit kırmızı (E172) 6.2. GeçimsizliklerGeçerli değil.6.3. Raf ömrü24 ay.6.4. Saklamaya yönelik özel tedbirler30°C'nin altındaki oda sıcaklığında saklayınız.Nemden korumak için orijinal ambalajında saklayınız. 6.5. Ambalajın niteliği ve içeriğiOPA/Al/PVC blister filmi ve Alüminyum/Isı ile kapatılmış blister folyosu.Perfore birim doz blisterler. Ambalaj boyutu: 56 x 1 film kaplı tablet (8 x 1 tabletlik 7 blister) 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller ''Tıbbi Atıkların Kontrolü Yönetmeliği'' ve ''Ambalaj Atıklarının Kontrolü Yönetmelik''lerine uygun olarak imha edilmelidir.7. RUHSAT SAHİBİRoche Müstahzarları Sanayi Anonim Şirketi,Eski Büyükdere Caddesi No: 13 Güney Plaza 34398 Maslak/İstanbul Tel: (0212) 366 90 00 Faks: (0 212) 285 22 00 8. RUHSAT NUMARASI(LARI)2014/939. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 13 Şubat 2014 Ruhsat yenileme tarihi:10. KÜB'ÜN YENİLENME TARİHİ |
İlaç BilgileriZelboraf 240 Mg Film Kaplı TabletEtken Maddesi: Vemurafenib Atc Kodu: L01XE15 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.