Immunate 500 Iu Kısa Ürün BilgisiKan ve Kan Yapıcı Organlar » Kanama Durdurucu İlaçlar » K Vitamini ve Diğer Hemostatikler » Kan Koagülasyon (pıhtılaşma) Faktörleri » Anti hemofilik faktor viii KISA ÜRÜN BİLGİSİ 1. BEŞERİ TIBBİ ÜRÜNÜN ADIIMMUNATE 500 IU IV infüzyon için liyofilize toz içeren flakon 2. KALİTATİF VE KANTİTATİF BİLEŞİM Etkin madde: İnsan kanı koagülasyon faktörü VIII IMMUNATE 500, kuru toz şeklinde nominal olarak 500 IU insan koagülasyon faktörü VIII1 ve 375 IU insan plazması kaynaklı von Willebrand faktörü (vWF:RCo) içeren bir flakon ile çözücü çözelti içerir. Ürün 5 mL steril enjeksiyonluk su ile seyreltilerek kullanıma hazır hale getirildikten sonra yaklaşık 100 IU/mL insan plazması kaynaklı koagülasyon faktörü VIII ve 75 IU/mL insan plazması kaynaklı von Willebrand faktörü içerir. Faktör VIII potensi Avrupa Farmakopesi kromojenik testiyle belirlenmiştir. IMMUNATE'in o spesifik aktivitesi 70 ± 30 IU FVIII/mg protein'dir. vWF potensi Avrupa Farmakopesi ristosetin kofaktör testiyle (vWF:RCo) belirlenmiştir. 1. Faktör VIII potensi, Dünya Sağlık Örgütünün faktör VIII konsantreleri için belirlediği standartlara göre belirlenmiştir.2. von Willebrand faktörünün ristosetin kofaktör potensi Dünya Sağlık Örgütünün von Willebrandfaktör konsantresi standartlarına göre belirlenmiştir.3. Stabilizansız (albuminsiz); Oran 1:1 olduğunda faktör VIII aktivitesinin von Willebrand faktör-antijenine maksimum spesifik aktivitesi protein başına 100 IUfaktör VIII'dir.Yardımcı madde(ler): Bir flakon yaklaşık 9.8 mg sodyum içerir. Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORM Enjeksiyonluk çözelti hazırlamak için liyofilize toz içeren flakon. Beyaz veya soluk sarı toz veya gevrek katı madde. 4. KLİNİK ÖZELLİKLERİ 4.1 Terapötik endikasyonları - Konjenital (hemofili A) ya da edinilmiş faktör VIII eksikliğinin yol açtığı kanamaların tedavisi ve profilaksisi. - Faktör VIII yetmezliğinin bulunduğu von Willebrand hastalığı. 4.2 Pozoloji ve uygulama şekli Tedavi, hemofili tedavisinde deneyimli bir hekimin denetiminde başlatılmalıdır. Pozoloji/uygulama sıklığı ve süresi: Doz ve uygulama süresi FVIII eksikliğinin ciddiyetine, kanamanın yerine ve yayılımına, hastanın klinik durumuna dayanır. Uygulanan FVIII'in ünite sayısı, faktör VIII ürünleriyle ilgili Dünya Sağlık Örgütünün belirlemiş olduğu standart Uluslararası Ünite (IU) terimiyle ifade edilir. Plazmadaki FVIII aktivitesi, ya yüzdesel olarak (normal insan plazmasına göre) ya da uluslarası ünite olarak (Dünya Sağlık Örgütü tarafından belirlenmiş F VIII uluslararası standartlarına göre) ifade edilir. 1 Uluslararası Ünite (IU) faktör VIII aktivitesi, 1 mL normal insan plazmasındaki faktör VIII aktivitesine eşittir. Hemofili A Hastalarında DozAşağıda belirtilen gerekli faktör VIII dozları, vücut ağırlığının her kilogramı için 1 IU faktör VIII verildiğinde, plazma faktör VIII aktivitesinin %1.5 - %2 kadar arttığının ampirik olarak gözlenmesine dayanılarak hesaplanmıştır. Hasta plazmasının başlangıçtaki faktör VIII aktivitesi göz önüne alınarak, gereken doz aşağıdaki formülle hesaplanır: IMMUNATE dozu (IU F VIII) = vücut ağırlığı (kg) x arzulanan faktör VIII artışı (%) x 0.5Tedavi seyrinin izlenmesi ve uygun idame dozlarının hesaplanması, her olguda ortaya çıkan klinik etkiye dayandırılarak ayrıca değerlendirilmelidir. Kanamalar ve Cerrahi Girişimlerde DozAşağıdaki tabloda, hemorajilerin tedavisi için ya da cerrahi profilakside gereken faktör VIII plazma düzeyleri ve bu düzeylerin ne kadar süre ile korunması gerektiği verilmiştir.
Özellikle başlangıç dozunda olmak üzere, bazı durumlarda hesaplanan dozdan daha fazlası gerekebilir. 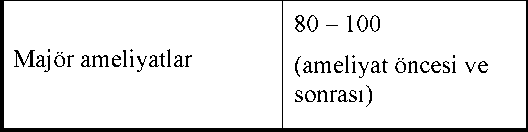 Tedavi süresince, doz uygulamasına rehber olması için F VIII seviyesi ölçülmesi önerilmektedir. Özellikle majör cerrahi girişimlerde, koagülasyon analizleriyle (plazma faktör VIII aktivitesi) yerine koyma tedavisinin sıkı takibi gereklidir. Farklı yarılanma zamanı ve in vivo yanıt nedeniyle, faktör VIII'e cevap kişiden kişiye değişebilir. 6 yaşından küçük çocuklarda IMMUNATE uygulamasıyla ilgili sınırlı veri mevcut olduğundan preparat dikkatle uygulanmalıdır. Uzun Süreli Profilakside DozAğır hemofili A hastalarının profilaktik tedavisinde, 2-3 günde bir vücut ağırlığının her kilogramı için 20 - 40 IU faktör VIII verilir. Bazı vakalarda, özellikle küçük yaştaki hastalarda kanamayı önlemek için daha sık doz aralıkları ya da daha yüksek dozlar gerekli olabilir. Faktör VIII'e Karşı İnhibitör Gelişmiş Hemofili Hastalarında DozHastalar faktör VIII inhibitör gelişimi açısından takip edilmelidir. İstenen F VIII plazma aktivitesine erişilemezse veya yeterli uygulamaya rağmen kanama kontrol altına alınamamışsa, faktör VIII inhibitör varlığını araştırmak için uygun testler yapılmalıdır. İnhibitör titresi her mL'de 10 Bethesda Ünitesinin (BU) altındaysa, ilave insan faktör VIII uygulamasıyla inhibitör nötralize edilebilir. İnhibitör titresi 10 BU veya daha yüksek olan hastalarda veya anamnestik yanıtı yüksek olan hastalarda faktör VIII uygulamasıyla hemostaz sağlanması olanaklı ya da uygulanabilir değildir. Dolayısıyla özel bir anti-inhibitör tedavisi yürütülmelidir. Bu tedaviler hemofili konusunda deneyimli hekimler tarafından yönetilmelidir (Bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri). Faktör VIII Eksikliği Bulunan von Willebrand Hastalığında DozIMMUNATE, faktör VIII aktivitesinin azaldığı von Willebrand hastalarının faktör VIII replasman tedavisinde endikedir. Hemorajileri kontrol etmek ve cerrahi girişimlere bağlı kanamaları önlemek için IMMUNATE replasman tedavisinde, hemofili A için verilen tedavi kılavuzuna uyulmalıdır. Uygulama şekli: Preparat, bölüm 6.6'da belirtildiği şekilde hazırlanmalı ve intravenöz yoldan yavaş bir şekilde uygulanmalıdır. 2 mL/dk'dan daha hızlı bir şekilde uygulanmaması önerilmektedir. Preparat, uygulamadan hemen önce sulandırılmalıdır. Hazırlanan çözelti hemen kullanılmalıdır (preparat koruyucu madde içermez). Bulanık ya da partikül içeren çözeltiler kullanılmamalıdır. Kullanılmayan çözeltiler uygun bir şekilde atılmalıdır. Özel popülasyonlara ilişkin ek bilgiler: Böbrek /Karaciğer yetmezliği: Ek bilgi bulunmamaktadır. Pediyatrik popülasyon: Doz kg başına ünite cinsinden belirlenmekte olduğundan çocuklarda özel bir kullanım şekli yoktur. Geriyatrik popülasyon: Yaşlılarda kullanım gerekirse doz bireysel olarak belirlenmelidir. 4.3 Kontrendikasyonlar Aktif maddeye veya içeriğindeki diğer maddelere karşı aşırı duyarlılık. 4.4 Özel kullanım uyarıları ve önlemleri Virüs güvenliği: IMMUNATE, insan plazmasından üretilmiştir. insan plazmasından üretilen preparatlar, örneğin virüsler ve teorik olarak Creutzfeldt - Jakob (CJD) etkeni gibi, hastalıklara yol açabilecek infeksiyon etkenleri içerebilir. Bu durum bilinmeyen ya da yeni ortaya çıkan virüslerle diğer patojenler için de geçerlidir. insan kanı ya da plazmasından hazırlanan tıbbi ürünlerin kullanımından kaynaklanan enfeksiyonların önlenmesi için alınan standart önlemler arasında, donörlerin seçimi, bireysel bağışların ve plazma havuzlarının belirli enfeksiyon göstergeleri için takibi ve virüslerin inaktivasyonu/uzaklaştırılması için etkili üretim aşamalarının kullanılması yer almaktadır. Buna rağmen insan kanı ya da plazmasından hazırlanan tıbbi ürünler uygulandığında, enfeksiyon ajanlarının bulaşma olasılığı tam olarak ortadan kaldırılamayabilir. Bu durum henüz bilinmeyen ya da yeni ortaya çıkan virüsler ve diğer hastalık etkenleri için de geçerlidir. Alınan önlemlerin HIV, HBC, HCV gibi zarflı virüslerle HAV gibi zarfsız virüsler için etkili olduğu düşünülmektedir. Alınan önlemlerin Parvovirüs B19 gibi bazı zarfsız virüsleri uzaklaştırmak ya da inaktive etmek için etkisi ise kısıtlıdır. Parvovirus B19 virüsü en ciddi olarak gebe kadınları (fetusda enfeksiyona neden olabilmektedir), immün yetmezlikli hastaları veya artmış eritrosit döngüsü olan hastaları (örn. hemolitik anemi durumu) etkilemektedir.insan plazması kaynaklı faktör VIII ürünlerini düzenli/tekrarlayan şekilde alan hastalarda uygun aşılama (Hepatit A ve B'ye karşı) düşünülmelidir. Hasta ile ürünün partisi arasındaki izi sürdürebilmek açısından IMMUNATE'in her kullanımından sonra ürünün adı ve parti numarasının kaydedilmesi önemle önerilmektedir. Diğer protein içeren intravenöz preparatlarda da olduğu gibi, alerjik tipte aşırı duyarlılık reaksiyonları gelişmesi olasılığı vardır. Faktör VIII ve von Willebrand faktörünün yanısıra, preparatın içeriğinde insan kaynaklı az miktarda başka proteinler de vardır. Hastalar, alerjik şoka kadar ilerleyebilecek, kaşıntı, genel ürtiker, göğüste sıkışma hissi, hırıltılı solunum, hipotansiyon gibi potansiyel aşırı duyarlılık reaksiyonlarının erken bulguları açısından bilgilendirilmelidir. Eğer bu semptomlar gelişirse, preparatın uygulanmasına derhal son verilmeli ve hekime danışılmalıdır. Şok gelişmesi durumunda, güncel şok tedavisi ilkelerine uyulmalıdır. Hemofili A tedavisinde, faktör VIII antikorlarının (inhibitörlerin) oluşması bilinen bir komplikasyondur. Bu antikorlar F VIII prokoagülan aktivitesine karşı oluşmuş IgG tipi antikorlardır, plazmada Modifiye Bethesda Ünitesi (BU) ile ölçülürler. Antikor oluşma riski anti-hemofilik faktör VIII'e maruz kalma ile orantılıdır, tedavinin ilk 20 gününde risk en yüksektir. Nadiren ilk 100 günden sonra oluşurlar. İnsan koagülasyon faktörü F VIII ile tedavi edilen hastalar, uygun laboratuvar testleri ve klinik gözlemlerle inhibitör antikor oluşumu açısından dikkatle kontrol edilmelidir (Bkz. Bölüm 4.8. İstenmeyen etkiler). Preparat, F VIII'le henüz kısıtlı karşılaşmış 6 yaş altındaki çocuklarda dikkatle kulanılmalıdırlar. Maksimum günlük doz uygulandığında alınan günlük sodyum miktarı 200 mg'ı aşabileceğinden, bu durum düşük sodyum diyeti alan hastalarda dikkate alınmalıdır. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri Bilinen bir etkileşimi yoktur. Özel popülasyonlara ilişkin ek bilgiler Hiçbir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyon Hiçbir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyon Genel tavsiye: Gebelik Kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon) Herhangi bir veri mevcut değildir. Gebelik dönemi Hayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. kısım 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. IMMUNATE gerekli olmadıkça gebelik döneminde kullanılmaz. IMMUNATE'in gebelerde kullanımının güvenliliği kontrollü klinik çalışmalarda saptanmamıştır. Hayvanlar üzerinde yapılan deneysel çalışmalar, üreme, embriyo ya da fetusun gelişmesi, gestasyon dönemi, perinatal ve postnatal gelişme açısından güvenli olduğu yargısını oluşturmaya elverişli değildir. Bu nedenle, IMMUNATE, gebelik ve emzirme döneminde ancak zorunlu ise kullanılabilir. Laktasyon dönemi Emziren kadınlarda IMMUNATE'in anne sütüne geçip geçmediği bilinmemektedir; bu nedenle emziren bir kadına IMMUNATE uygulanırken dikkatli olunmalı, her bir hasta için tedavinin potansiyel riskleriyle olası yararı dikkatle gözönünde bulundurulmalıdır. Üreme yeteneği / fertilite IMMUNATE'in üreme yeteneği / fertilite üzerindeki etkisini araştıran bir çalışma bulunmamaktadır. 4.7 Araç ve makine kullanımı üzerindeki etkiler Araç ve makine kullanımı üzerinde herhangi bir etki bildirilmemiştir. 4.8 İstenmeyen etkiler İnsan plazması kaynaklı faktör VIII ürünleriyle bildirilen istenmeyen etkiler: Aralarında anjiyonörotik ödem, infüzyon bölgesinde yanma ve batma, titreme, yüz ve boyun bölgesinde ani sıcaklık hissi, yaygın ürtiker, başağrısı, kurdeşen, kan basıncında düşme, letarji, bulantı, huzursuzluk, taşikardi, göğüste sıkışma hissi, karıncalanma, kusma, hırıltılı solunum gibi aşırı duyarlılık ya da alerjik reaksiyonlar. Bazı vakalarda bu istenmeyen etkiler şiddetli anafilaksiye kadar ilerleyebilir (şok dahil). Hastalara bu tür reaksiyonlar gördüklerinde hekimlere başvurmaları önerilmelidir (Bkz. Bölüm 4.4). Nadiren vücut ısısında yükselme görülebilir. Hemofili A hastalarında FVIII'e karşı antikor (inhibitör) gelişebilir. İnhibitör oluşumu, tedaviye yanıt alınamaması şeklinde ortaya çıkabilir. Bu gibi durumlarda özel hemofili merkezleriyle bağlantı kurulmalıdır. Yüksek dozların uygulanmasından sonra A, B, AB kan grubu olan hastalarda hemoliz gelişebilir. Kan transfüzyonu ile bulaşabilen hastalıklarla ilgili ürün güvenliliği için bölüm 4.4'e bakınız. IMMUNATE ile yapılan klinik çalışmalar ve pazarlama sonrası deneyimlerde bildirilen istenmeyen etkiler: Advers etkilerin görülme sıklığı, izleyen kriterler kullanılarak değerlendirilmiştir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Kan ve lenf sistemi hastalıkları Bilinmiyor: Pıhtılaşma bozuklukları; Faktör VIII inhibisyonu Bağışıklık sistemi hastalıkları Yaygın olmayan: Aşırı duyarlılık* Sinir sistemi hastalıkları Bilinmiyor: Baş dönmesi/sersemlik hali; Baş ağrısı Göz hastalıkları Bilinmiyor: Konjonktivit Kardiyak hastalıkları Bilinmiyor: Çarpıntı Vasküler hastalıkları Bilinmiyor: Hipotansiyon Solunum, göğüs bozuklukları ve mediastinal hastalıkları Bilinmiyor: Öksürük, Dispne Gastrointestinal hastalıkları Bilinmiyor: Bulantı Deri ve deri altı doku hastalıkları Bilinmiyor: Eritem; Ekzantem; Nörodermatit; Kaşıntı; Döküntü; Eritematöz döküntü; Papüler döküntü; Ürtiker Kas-iskelet bozukluklar, bağ dokusu ve kemik hastalıkları Bilinmiyor: Miyalji Genel bozukluklar ve uygulama bölgesine ilişkin hastalıkları Bilinmiyor: Titreme; Enjeksiyon yerinde iritasyon; Ağrı; Ateş * Klinik çalışmalarda bildirilmiştir. 4.9 Doz aşımı ve tedavisi Doz aşımında ortaya çıkan özel bir istenmeyen etkisi bilinmemektedir. 5. FARMAKOLOJİK ÖZELLİKLER 5.1 Farmakodinamik özellikler Farmakoterapötik Grubu: Saflaştırılmış İnsan Koagülasyon Faktör VIII Konsantresi ATC kodu: B02BD06 Faktör VIII / von Willebrand faktör kompleksi farklı fizyolojik fonksiyonları olan 2 molekülden (F VIII ve vWF) oluşmuştur. Aktive faktör VIII, aktive faktör IX için kofaktör görevi görür ve faktör X'un aktive faktör X'a dönüşümünü hızlandırır. Aktive olan faktör X ise protrombini trombine dönüştürür. Trombin ise fibrinojeni fibrine dönüştürür ve pıhtı oluşumu gerçekleşebilir. Hemofili A, faktör VIII C düzeylerinin düşük seyrettiği cinsiyete bağlı herediter bir kan pıhtılaşma bozukluğudur; spontan veya travma sonrası eklemlere, kaslara ve iç organlara olan bol miktarda kanama ile seyreder. Yerine koyma tedavisiyle F VIII plazma düzeyleri yükseltilerek, faktör eksikliğinde ve kanama eğiliminde geçici bir düzelme sağlanabilir. von Willebrand faktörü (vWF), F VIII'i koruyucu bir protein olması yanında vasküler yaralanma bölgesinde trombosit adezyonuna aracılık eder, trombosit agregasyonunda rol alır ve von Willebrand hastalığında yerine koyma tedavisinde vazgeçilmez bir faktördür. 5.2 Farmakokinetik özellikler Emilim:Enjeksiyon sonrası plazmada ölçülen F VIII aktivitesi, beklenen plazma F VIII aktivitesinin %80-120'si kadar olur. Yapılan bir farmakokinetik çalışmada IMMUNATE enjeksiyonu sonrası in vivo ortalama FVIII düzeylerinin %100 olduğu gösterilmiştir. Dağılım:Uygulanan faktör VIII, normal faktör VIII'le aynı şekilde dağılıma uğrar. Biyotransformasyon:Plazmada IMMUNATE'den gelen faktör VIII, normal faktör VIII'le aynı şekilde tüketilerek biyotransformasyona uğrar. Plazma FVIII aktivitesi iki fazlı eksponansiyel düşüş göstermektedir. Başlangıç fazında, intravasküler ve diğer kompartmanlar arası dağılım (vücut sıvıları), 3-6 saatlik plazma eliminasyon yarılanma süresine göre gerçekleşmekte; FVIII'in yaklaşık olarak % - % kadarı dolaşımda kalmaktadır. İkinci ve yavaş fazda ise (muhtemelen FVIII tüketimini yansıtan faz) yarılanma süresi 8-20 saat arasında değişmektedir (ortalama 12 saat). Bu süre, biyolojik yarılanma süresi ile uyumludur. IMMUNATE ile gerçekleştirilen bu çalışmada, faktör VIII yarılanma süreleri modele bağlı ve bağlı olmayan yöntemlerle belirlenmiştir. Her iki durumda da 11 saatlik ortalama değerler bulunmuştur. Eliminasyon:Faktör VIII normal yollardan tüketilir ve elimine edilir. 5.3 Klinik öncesi güvenlilik verileri IMMUNATE'in içerisinde yer alan insan koagülasyon faktörü VIII, insan plazmasının normal bileşenlerinden birisidir ve endojen faktör VIII gibi davranır. Yüksek doz uygulamalar, aşırı doz yüklenmesi yaratacağı için, tek doz toksisite testleri yapılması uygun değildir. Kilogram başına önerilen insan dozlarından birkaç kat daha yüksek dozlar dahi laboratuvar hayvanları üzerinde toksik etkilere neden olmamıştır. Hayvanlarda tekrarlayan doz toksisitesi testleri, heterolog proteine karşı gelişen antikorların engellemesi nedeniyle uygulanamamaktadır. Klinik deneyimlerde, insan plazma proteini faktör VIII'in mutajenik bir etkiye ya da tümör oluşumuna yol açtığına dair herhangi bir bulguya rastlanmadığı için, heterolog deney hayvanlarında deneysel çalışmalara gerek olmadığı düşünülmüştür. 6. FARMASÖTİK ÖZELLİKLERİ 6.1 Yardımcı maddelerin listesi Kuru toz içeren flakon: - İnsan albümini - Glisin - Lizin-HCl - Sodyum klorür - Trisodyum sitrat-2H2O - Kalsiyum klorür-2H2O Çözücü: - Steril Enjeksiyonluk Su 6.2 Geçimsizlikler Diğer tıbbi ürünlerle bilinen bir etkileşimi yoktur. Ürünün etkisini ve güvenliğini bozabileceği için, diğer kan pıhtılaşma faktörü konsantrelerinde olduğu gibi, uygulama öncesi herhangi bir preparatla karıştırılmamalıdır. IMMUNATE infüzyonu öncesi ve sonrası venöz uygulama setinin izotonik sodyum klorür çözeltisiyle yıkanması önerilmektedir. 6.3 Raf ömrü 24 aydır. Sulandırılmış IMMUNATE çözeltisinin oda sıcaklığında yaklaşık olarak 3 saat kimyasal ve fiziksel stabilitesini koruduğu gösterilmiştir. Mikrobiyolojik nedenlerle, sulandırıldıktan hemen sonra kullanılması önerilir. Hemen kullanılmadığı durumlarda, saklanma koşullarından kullanıcı sorumludur. Rekonstitüsyondan sonra tekrar buzdolabında saklanmamalıdır. Ürün raf ömrü içindeyken yalnızca bir dönem olmak üzere 25°C altı oda sıcaklığında 6 ay süreyle tutulabilir. Böyle saklanıyorsa lütfen ürünün karton ambalajı üzerine oda sıcaklığında saklanmaya başladığı tarihi not ediniz. Oda sıcaklığında saklanmaya başlanan ürün tekrardan buzdolabında saklanmaz. Altı aylık saklama süresi dolduğunda kullanılmazsa atılmalıdır. 6.4 Saklamaya yönelik özel tedbirler 2-8°C arasında saklayınız ve transfer ediniz. Dondurmayınız. Işıktan korumak amacıyla ambalajı içerisinde saklanmalıdır. Sulandırılarak kullanıma hazır hale getirilmiş ilacın saklanması için bkz. Bölüm 6.3. 6.5 Ambalajın niteliği ve içeriği Her kutu bir adet IMMUNATE cam flakon içerir. Toz halindeki preparat ve 5 mL'lik çözücü, kauçuk tıpalı tek dozluk cam flakonlardadır. Her ambalaj ayrıca, rekonstitüsyon ve enjeksiyon kiti içerir. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. Çevreyi korumak amacıyla, kullanılmayan IMMUNATE şehir suyuna veya çöpe atılmamalıdır. Uygulama Öncesi Hazırlanması: Aseptik teknik kullanılmalıdır. 1. Çözücü içeren (enjeksiyonluk steril su) kapalı flakonu oda sıcaklığına kadar ısıtınız (maksimum 37°C). 2. Konsantre koagülasyon faktörü flakonunun ve çözücü flakonunun koruyucu kapaklarını çıkarınız (Şekil A) ve her ikisinin de lastik tıpalarını dezenfekte ediniz. 3. Transfer setinin dalgalı kenarını çözücü flakonunun üzerine yerleştiriniz ve bastırınız (Şekil B). 4. Transfer setinin koruyucu kapağını, açıkta kalan kısımlarına temas etmemeye dikkat ederek diğer tarafından çıkarınız. 5. Çözücü flakonuna takılı haldeyken transfer setini ters çevirerek, iğnesini konsantre flakonunun tıpasına batırınız (Şekil C). Çözücü, konsantre, flakonunun vakumu sayesinde emilecektir. 1 dakika kadar bekleyiniz. 6. Transfer setinin bağlı olduğu çözücü flakon ile konsantre flakonunu ayırınız (Şekil D). Preparat kolaylıkla çözündüğünden, flakonu sadece gerekiyorsa hafifçe sallayınız. FLAKON İÇERİĞİNİ ÇALKALAMAYINIZ. İÇERİĞİNİ ENJEKTÖRE ÇEKMENİN HEMEN ÖNCESİNE KADAR, FLAKONU TERS ÇEVİRMEYİNİZ. IMMUNATE gibi parenteral preparatlar, uygulama öncesinde içeriğinde partikül olup olmadığı ya da rekonstitüsyondan sonra renk değişimi olup olmadığı açısından gözle kontrol edilmelidir. Rekonstitüsyon için gerekli uygulamalar doğru bir şekilde yapılsa dahi, birkaç küçük partikül gözle görülebilir. Ambalaj içeriğinde bulunan filtreli set, bu partikülleri uzaklaştıracaktır. Ambalaj etiketinde belirtilen farmasötik olarak etkili madde içeriğinde azalma olmayacaktır. Uygulama: Aseptik teknik kullanılmalıdır. 1. Tıpadan kopan lastik parçalarının uygulanmasına engel olmak için (mikroemboli riski), ve çözünmüş preparatı çekmek için, ambalajdaki filtreli seti kullanınız. Filtreli seti, ambalajdaki tek kullanımlık enjektöre takınız ve lastik tıpaya batırınız (Şekil E). 2. Enjektörü bir an için filtreli setten ayırınız. Konsantreyi içeren flakona hava girecek ve oluşmuş hava kabarcıkları kaybolacaktır. Bundan sonra, filtreli set aracılığıyla çözeltiyi enjektöre çekiniz (Şekil F). 3. Enjektörü filtreli setten ayırınız ve ambalajdaki infüzyon seti (ya da tek kullanımlık iğne) ile çözeltiyi yavaş olarak (enjeksiyon hızı dakikada 2 mL'yi aşmamalıdır) intravenöz enjeksiyon şeklinde uygulayınız. 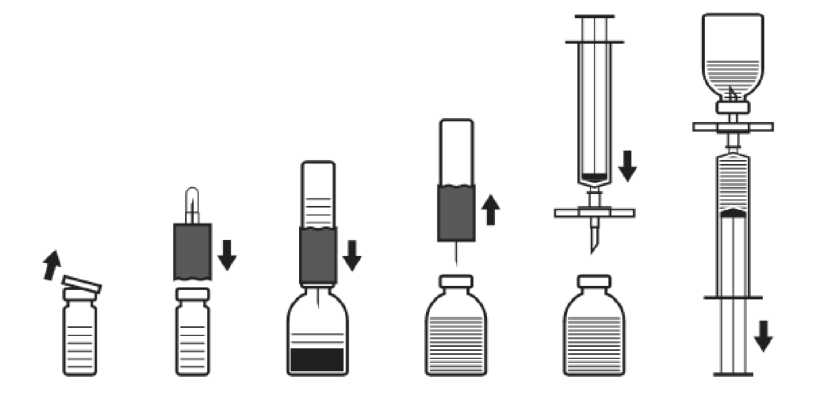 Şek. A
Şek. AŞek. B Şek. C Şek. D Şek. E
Şek. F Uygulamadan sonra, ambalaj içeriğindeki enjektörle ve/veya infüzyon setiyle beraber, kullanılmış olan bütün iğneler atılmalıdır. 7. RUHSAT SAHİBİ Eczacıbaşı-Baxter Hastane Ürünleri Sanayi ve Ticaret A.Ş. Cendere Yolu, Pırnal Keçeli Bahçesi 34390 Ayazağa-İSTANBUL (0.212) 329 62 00 (0.212) 289 92 75
Adı Adresi Tel Faks Üretim yeri : Baxter AG, Industriestrasse 72-Lange Allee 24-B Viyana, AVUSTURYA 8. RUHSAT NUMARA SI(LARI) 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİ İlk ruhsat tarihi: 24/05/2013 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ 11 |
İlaç BilgileriImmunate 500 IuEtken Maddesi: Insan Koagülasyon Faktörü Viii Ve Von Willebrand ... Atc Kodu: B02BD06 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.