Feiba 500 U Iv İnfüzyon İçin Liyofilize Toz Içere... Kısa Ürün BilgisiKan ve Kan Yapıcı Organlar » Kanama Durdurucu İlaçlar » K Vitamini ve Diğer Hemostatikler » Kan Koagülasyon (pıhtılaşma) Faktörleri » Anti hemofilik faktor viii KISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADI FEIBA 500 U IV İnfüzyon İçin Liyofilize Toz İçeren Flakon 2. KALİTATİF VE KANTİTATİF BİLEŞİMİ Etkin madde: Her bir flakon 500* ünite Faktör VIII İnhibitör Baypas Aktivitesine sahip 200-600 mg insan plazma proteini içerir. FEIBA bileşiminde aktive Faktör VII yanında büyük bölümü aktif olmayan formda Faktör II, IX ve X bulunmaktadır. Her bir ünite FEIBA, 0.1 ünite kadar Faktör VIII koagülan antijeni (F VIII C:Ag) içerir. Kallikrein-kinin sisteminin faktörleri ancak eserhj miktarlarda bulunmaktadır. * 1 ünite FEIBA içeren çözelti, yüksek titreli faktör-VIII plazmasının aktive parsiyel tromboplastin zamanını (aPTT) tampon değerin % 50'si kadar kısaltır. Yardımcı maddeler: Sodyum klorür 160 mg Sodyum sitrat 80 mg Yardımcı maddeler için 6.1'e bakınız. 3. FARMASÖTİK FORM İntravenöz infüzyonluk çözelti için kuru toz ve çözücüsü. Kuru toz beyaz, beyaza yakın veya soluk yeşil renktedir. Sulandırıldıktan sonra oluşan çözeltinin pH'sı 6.8 ile 7.6 arasındadır. 4. KLİNİK ÖZELLİKLER 4.1. Terapötik endikasyonlar - Faktör VIII'e karşı inhibitör gelişmiş hemofili A hastalarında kanamaların tedavisinde. - Faktör VIII'e karşı inhibitör gelişmiş kalıtsal hemofilisi olmayan hastalardaki kanamaların tedavisinde. - Faktör VIII'e karşı inhibitör gelişmiş ve baypas ajanı kullanımını gerektirmiş ya da yaşam kalitesinde önemli bozulmaya yol açmış ciddi bir kas-iskelet sistemi kanaması geçirmiş ve/veya yaşamı tehdit eden boyutta (örn. intrakraniyal, intraabdominal, intratorasik) bir kanaması olmuş hemofili A hastalarındaki kanamaların profilaksisinde. 4.2. Pozoloji ve uygulama şekli Tedavi koagülasyon bozuklukları konusunda uzman bir hekim tarafından başlatılmalı ve bu hekimin gözetiminde devam ettirilmelidir. Pozoloji / uygulama sıklığı ve süresi Dozaj Uygulanacak dozun miktarı, zamanlaması, uygulamanın ne kadar tekrarlanacağı ve tedavinin süresi, kanamanın şiddeti, yeri ve yayılımı yanında hastanın klinik durumuna göre değişir. Dozaj ve uygulama sıklığı her olguda klinik etkililiğe göre ayarlanmalıdır. Genel olarak 50-100 Ünite/kg FEIBA dozu önerilmektedir; kanama şiddetinin daha yüksek doz kullanımını gerektirmediği durumlarda tek uygulamada 100 Ünite/kg ve günlük olarak 200 Ünite/kg dozları aşılmamalıdır. Bkz. Bölüm 4.4. 1) Spontan kanamalar Kas eklem ve yumuşak doku kanamaları: Küçük ve orta dereceli kanamalarda önerilen doz 12 saatlik aralarla 50-75 Ünite/kg'dır. Tedaviye, ağrının kaybolması, eklem şişkinliğinin azalması ya da hareketliliğinin kazanılması gibi klinik düzelme belirtileri görülene kadar devam edilmelidir. Retroperitonal kanama gibi büyük kas ve yumuşak doku kanamalarında önerilen doz 12 saatlik aralıklarla 100 Ünite/kg'dır. Mukoza kanamaları: Hasta dikkatle izlenerek (kanama bölgesi görülerek, hemotokrit ölçümleri tekrarlanarak) 6 saatte bir 50 Ünite/kg önerilir. Kanama durmazsa, günde 200 Ünite/kg'nin üzerine çıkmamaya dikkat edilerek doz 100 Ünite/kg'a yükseltilebilir. Diğer ciddi kanamalar: Merkezi sinir sistemi kanamaları gibi ciddi kanamalarda 12 saatlik aralıklarla verilen 100 Ünite/kg dozu önerilir. Bazı hastalara FEIBA, belirgin klinik düzelme görülene kadar 6 saatlik aralarla uygulanabilir. Günlük en yüksek doz olan 200 Ünite/kg'lık doz aşılmamalıdır. 2) Cerrahi girişimler Cerrahi girişimlerde başlangıç dozu olarak ameliyat öncesinde 100 Ünite/kg'lık bir doz ve ameliyattan 6-12 saat sonra 50-100 Ünite/kg'lık bir doz uygulanabilir. Postoperatif idame dozu olarak 6-12 saat aralıklarla 50-100 Ünite/kg'lık dozlar uygulanabilir; doz aralıkları ve ameliyat öncesi ve sonrası tedavinin süresi, uygulanan cerrahi girişime, hastanın genel durumuna ve bireysel olarak hastada sağlanan klinik etkinliğe göre belirlenir. (200 Ünite/kg'lık maksimum günlük doz aşılmamalıdır.) 3) Profilaksi - Yüksek inhibitör titresine sahip ve başarısız olmuş ITI (İmmün tolerans tedavisi) sonrası sık kanamaları olmuş hastalarda veya ITI düşünülmeyen hastalarda kanama profilaksisi: Günaşırı 70-100 Ünite/kg'lık bir doz önerilmektedir. Duruma göre doz günde 100 Ünite/kg'a yükseltilebilir ya da giderek azaltılabilir. - Yüksek inhibitör titresine sahip hastalarda ITI (immün tolerans tedavisi) almaktayken görülen hastaların profilaksisi: FEIBA, faktör VIII inhibitör titresi <2 BU* (Bethesda Unit) oluncaya kadar günde iki kez 50-100 Ünite/kg dozunda ve faktör VIII uygulamasıyla eş zamanlı olarak kullanılabilir. * 1 Bethesda Ünitesi inkübe edilmiş (37°C'de 2 saat) plazmanın faktör VIII etkinliğinin %50'sini inhibe eden antikor düzeyi olarak tanımlanmıştır. 4) FEIBA'nın özel hasta gruplarında kullanımı Faktör IX'a karşı inhibitör gelişmiş hemofili B hastalarındaki kullanımıyla ilgili Bölüm 5.1'e bakınız. Faktör VIII inhibitörlerinin tam ve kalıcı olarak eliminasyonunu sağlamak için FEIBA, Faktör VIII konsantreleri ile kombine olarak uzun süreli tedavilerde de kullanılmıştır. Tedavinin izlenmesi FEIBA'nın etki mekanizmasının kompleks olmasından dolayı etkin maddenin doğrudan izlemi gerçekleştirilemez. Tam kan pıhtılaşma zamanı (WBCT), tromboelastogram (TEG, r değeri) ve aPTT değerlerinde genellikle ancak az bir azalma görülür ve klinik etkililik ile korelasyon göstermeyebilir. Bu nedenle FEIBA tedavisinin izlenmesinde bu testlerin önemi düşüktür. Bkz. Bölüm 4.4 Uygulama şekli: Ürünü aşağıda tarif edildiği şekilde sulandırarak kullanıma hazırlayınız ve intravenöz yoldan yavaş infüzyon yoluyla uygulayınız. FEIBA koruyucu içermediğinden uygulamadan hemen önce sulandırılmalı ve hazırlanan çözelti derhal kullanılmalıdır. Bütün içerik çözünene kadar yavaşça çalkalayınız. FEIBA'nın tamamen çözünmüş olduğundan emin olunmalıdır; aksi takdirde cihazın filtresinden daha az etkin madde geçer. Sulandırıldıktan sonra oluşan çözeltinin herhangi bir partikül içerip içermediği ve renk değişikliği olup olmadığı kontrol edilmelidir. Bulanık olan ya da tortu içeren çözeltiler kullanılmamalıdır. Sulandırmak için yalnızca ambalajında bulunan steril enjeksiyonluk su ve sulandırma cihazı kullanılmalıdır. FEIBA kutusu dışındaki bir cihaz kullanılması durumunda uygun bir filtre (en az 149 pm çapında deliği olan) kullandığınızdan emin olunuz. Liyofilize flakonun BAXJECT II Hi-Flow cihaz kullanılarak sulandırılması: 1. Gerekliyse çözücü (enjeksiyonluk su) içeren açılmamış flakonu oda sıcaklığına (15 °C -25 °C) kadar ısıtınız. Bu işlem için bir kaç dakika süreyle ılık su banyosu (maksimum 37 °C) kullanılabilir. 2. Konsantre ve çözücü flakonlarının koruyucu kapaklarını çıkarınız ve her ikisinin de lastik tıpalarını dezenfekte ediniz. Flakonları düz bir yere yerleştiriniz. 3. BAXJECT II Hi-Flow cihazının ambalajını, ambalajdaki kağıt kapağı çekerek cihazın içine dokunmadan açınız (Şekil a). Cihazı, ambalajın içinden çıkarmayınız. 4. Ambalajı ters çeviriniz ve şeffaf plastik delici kısmını çözücü flakonun tıpasına uygulayınız (Şekil b). Ambalajı kenarından tutarak BAXJECT II Hi-Flow cihazının üzerinden çıkarınız (Şekil c). BAXJECT II Hi-Flow cihazının üzerindeki mavi kapağı çıkarmayınız. 5. Çözücü flakon ve BAXJECT II Hi-Flow cihaz kombinasyonunu, çözücü flakon yukarıya gelecek şekilde çeviriniz. Diğer mor renkli plastik delici ucu, toz konsantrenin bulunduğu flakonun tıpasına uygulayınız. Çözücü vakum etkisiyle konsantrenin bulunduğu flakonun içerisine çekilecektir (Şekil d). 6. Bütün içerik çözünene kadar yavaşca çalkalayınız. FEIBA'nın tamamen çözünmüş olduğundan emin olunmalıdır; aksi takdirde, etkin madde cihazın filtresinden geçmeyecektir. 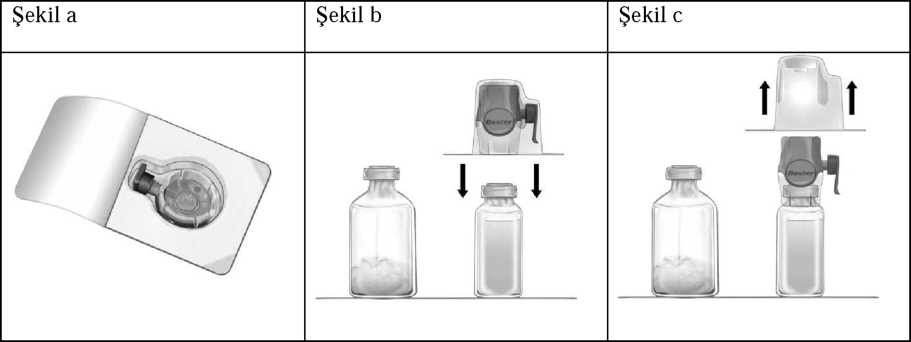 Enjeksiyon / İnfüzyon için talimatlar:
Enjeksiyon / İnfüzyon için talimatlar:1. BAXJECT II Hi-Flow cihazındaki mavi kapağı çıkarınız. Enjektörü BAXJECT II Hi-Flow cihazına iliştiriniz (ENJEKTÖRE HAVA ÇEKMEYİNİZ) (Şekil e). 2. Bileşkeyi ters çeviriniz (FEIBA flakonu üstte kalacak şekilde). Pistonunu yavaşça geriye doğru çekerek FEIBA çözeltisini enjektöre çekiniz (Şekil f). 3. Enjektördeki çözeltiyi kelebek infüzyon seti (ya da tek kullanımlık iğne) ile yavaşça intravenöz enjeksiyon şeklinde uygulayınız. 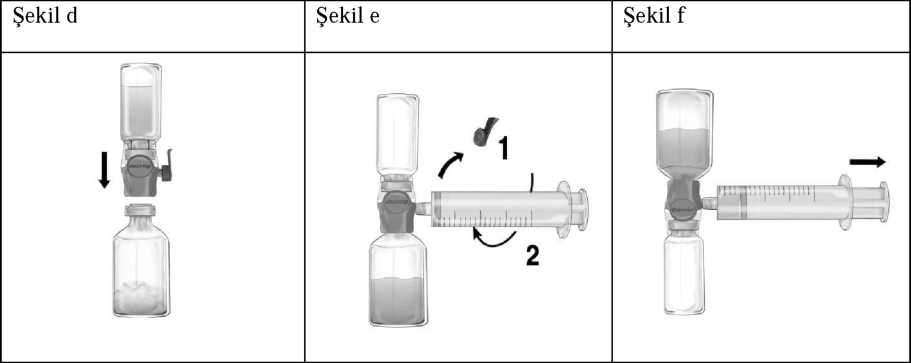 Dakikada maksimum infüzyon hızı her kilogram için 2 ünite'yi aşmamalıdır.
Dakikada maksimum infüzyon hızı her kilogram için 2 ünite'yi aşmamalıdır.Uygulama hızı: Dakikada 2 Ünite/kg infüzyon hızı aşılmamalıdır. Özel popülasyonlara ilişkin ek bilgiler: Böbrek / Karaciğer yetmezliği: Karaciğer fonksiyon testleri bozulmuş olan hastalarda aktive koagülasyon faktörlerinin eliminasyonunun uzamasından dolayı yaygın damar içi pıhtılaşma (DIC) gelişimi riski artar. Pediatrik popülasyon: 6 yaş altı çocuklardaki kullanımıyla ilgili deneyim yetersizdir. Çocuklarda klinik duruma göre erişkinlerdeki aynı doz şeması adapte edilmelidir. Geriatrik popülasyon: Y aşlılarda da erişkin dozları kullanılır. 4.3. Kontrendikasyonlar Aşağıdaki durumlarda alternatif terapötik bir tedavi varsa FEIBA kullanılmamalıdır: - Ürüne veya bileşenlerinden herhangi birine karşı aşırı duyarlılığın bulunması - Dissemine İntravasküler Koagülasyon (yaygın damar içi pıhtılaşma; DIC) Aşağıdaki durumlarda FEIBA yalnızca uygun koagülasyon faktörü konsantreleri kullanılarak yapılan tedaviye yanıt alınamadığında (örneğin çok yüksek bir inhibitör titresi varlığına bağlı olarak) kullanılabilir. 1. Kesin bir karaciğer hasarını gösteren laboratuvar ve/veya klinik semptomların bulunması: aktive koagülasyon faktörlerinin klerensinin uzamasından dolayı bu tür hastalarda DIC gelişmesi riski artmıştır. 2. Miyokard enfarktüsü, akut tromboz ve/veya embolizm: bu hastalarda FEIBA yalnızca yaşamı tehdit eden kanamalarda kullanılmalıdır. Bkz. Bölüm 4.4. 4.4. Özel kullanım uyarıları ve önlemleri Virüs güvenliği FEIBA insan plazmasından elde edilmektedir. İnsan plazmasından elde edilen ilaçlar, virüsler, ve teorik olarak Varyant Creutzfeldt-Jacob (v-CJD) gibi, çeşitli hastalıklara yol açabilen enfeksiyon yapıcı ajanlar içerebilirler. FEIBA'da Varyant Creutzfeldt-Jacob hastalığının bulaşma riski teorik olarak minimumken, klasik Creutzfeldt-Jacob hastalığının bulaşma riski hiçbir kanıtla desteklenmez. Alınan önlemlere rağmen, bu tür ürünler halen potansiyel olarak hastalık bulaştırabilir. Bu tip ürünlerin enfeksiyon yapıcı ajanları bulaştırma riski, plazma verenlerin belirli virüslere önceden maruz kalıp kalmadığının izlenmesi, belirli virüs enfeksiyonlarının halihazırda varlığının test edilmesi ve belirli virüslerin yok edilmesi ve/veya inaktivasyonu ile azaltılmıştır. Bütün bu önlemlere rağmen, bu ürünler hala potansiyel olarak hastalık bulaştırabilirler. Ayrıca, henüz bilinmeyen enfeksiyon yapıcı ajanların bu ürünlerin içersinde bulunma ihtimali mevcuttur. HIV, HBV, HCV gibi zarflı virüsler ve HAV gibi zarflı olmayan virüslerin etkisi için önlemlerin alınmasına, dikkat edilmelidir. Parvovirus B19 gibi zarflı olmayan virüslere karşı alınan tedbirler sınırlı sayıda olabilir. Parvovirüs B19 enfeksiyonu, gebelikte (fetal infeksiyon) ve immün yetmezlik ya da kırmızı kan hücre üretiminde artış olan hastalarda tehlikeli olabilir (hemolitik anemi gibi).Doktor, bu ilacı hastaya reçete etmeden veya uygulamadan önce hastası ile risk ve yararlarını tartışmalıdır. Trombotik ve Tromboembolik Olay Riski FEIBA tedavisi sırasında yaygın damar içi pıhtılaşma (DIC), venöz tromboz, pulmoner embolizm, miyokard enfarktüsü ve inme dahil olmak üzere trombotik ve tromboembolik olaylar oluşmuştur. FEIBA'nın yüksek dozda kullanılmasıyla trombotik ve tromboembolik olay riski artabilir. Bu olayların çoğu 200 Ünite/kg/gün'den yüksek dozlarla ya da tromboembolik olayların olduğu diğer risk faktörlerinin bulunduğu hastalarda görülmüştür. Her zaman konjenital ve kazanılmış hemofilisi olan hastalarda bu türden risk faktörlerinin bulunabileceği dikkate alınmalıdır. Trombotik ve tromboembolik olayların ilk işaretleri görülür görülmez infüzyon hemen durdurulmalı ve uygun tanısal ve terapötik önlemler uygulanmaya başlanmalıdır. Aşağıdaki durumlarda FEIBA yalnızca uygun koagülasyon faktörü konsantreleri kullanılarak yapılan tedaviye yanıt beklenmediğinde, örneğin çok yüksek bir inhibitör titresi varlığı ve yaşamı tehdit edebilen boyutta (örn. travma veya ameliyat sonrası) bir kanama veya kanama riski varsa kullanılabilir: - Yaygın damar içi pıhtılaşma (DIC): laboratuvar bulgularının ve/veya klinik semptomların bulunması. - Karaciğer hasarı: aktive koagülasyon faktörlerinin klerensinin uzamasından dolayı karaciğer işlevleri bozulmuş hastalarda DIC gelişmesi riski artmıştır. - Koroner kalp hastalığı, akut tromboz ve/veya embolizm. Alerjik Aşırı Duyarlılık Reaksiyonları İntravenöz yoldan uygulanan herhangi bir protein ürününde olduğu gibi, alerjik tipte aşırı duyarlılık reaksiyonları görülebilir. Hastalar eritem, deri döküntüsü, yaygın ürtiker, kaşıntı, solunum zorluğu/dispne, göğüste sıkışma hissi, genel bir keyifsizlik durumu, baş dönmesi/sersemlik hali, hafif bir düşmeden alerjik şoka değişen kan basıncı azalmaları gibi aşırı duyarlılık reaksiyonlarının erken belirtileri konusunda bilgilendirilmelidir. Hastalara, bu belirtiler görülür görülmez uygulamayı durdurarak hemen hekimlerine başvurmaları söylenmelidir. Şok, modern şok tedavisi ilkelerine uyularak tedavi edilir. Ürüne veya bileşenlerinden herhangi birine karşı aşı duyarlılığı olduğu bilinen ya da kuşkulanılan hastalarda FEIBA'yı yeniden kullanmadan önce, hastadaki bilinen ya da kuşkulanılan aşırı duyarlılığın tipi (alerjik ya da alerjik olmayan) ile potansiyel düzeltici ve/veya önleyici tedaviler ya da alternatif terapötik ajanlar göz önünde bulundurularak yeniden kullanımdan sağlanacak faydalarla olası risk dikkatle karşılaştırılmalıdır. Tedavinin İzlenmesi Tek uygulamada 100 Ünite/kg'lık ve günlük olarak 200 Ünite/kg'lık dozlar aşılmamalıdır. 100 Ünite/kg'lık dozların uygulandığı hastalar dikkatle, özellikle de DIC ya da akut koroner iskemi semptomlarının gelişimi açısından izlenmelidir. Yüksek dozlarda FEIBA ancak kesin olarak gerektiği süre boyunca - kanama durana kadar - kullanılmalıdır. Kan basıncı ile kalp atım hızında klinik olarak anlamlı değişiklikler, solunum zorluğu, öksürük ya da göğüs ağrısı ortaya çıkarsa infüzyon derhal durdurulmalı, uygun tanı ve tedavi metodları uygulanmalıdır. Fibrinojen düzeylerinde bir azalma olması, trombosit sayısında bir azalma olması ve/veya fibrin/fibrinojen yıkım ürünlerinin (FDP) ortaya çıkması DIC açısından anlamlı laboratuvar parametreleridir. Diğer DIC göstergeleri arasında trombin zamanı, protrombin zamanı ve aPTT'nin açık bir şekilde uzaması bulunur. İnhibitörlü hemofili hastalarında veya faktör VIII, faktör IX ve/veya faktör XI'e karşı kazanılmış inhibitörü olan hastalarda aPTT altta yatan hastalığa bağlı olarak uzayabilir. FEIBA tedavisi uygulanan inhibitörlü hemofili hastalarında veya koagülasyon faktörlerine karşı kazanılmış inhibitörleri olan hastalarda aynı zamanda kanamaya eğilim ve tromboz riski artabilir. Laboratuvar testleri ve klinik etkililik Etkililiğin kanıtı olabilecek aPTT, tam kan pıhtılaşma zamanı (WBCT) ve tromboelastogram (TEG) gibi in vitro testlerle klinik tablonun korelasyon göstermesi şart değildir. Bu nedenle bu değerleri normale döndürmek için FEIBA dozu arttırılmamalıdır; aksine olası bir aşırı doza bağlı DIC olayını tetikleyebilme riski nedeniyle kesinlikle dozun arttırılmaması gerekir. Trombosit sayısının önemi FEIBA'nın etkili olabilmesi için hastada yeterli sayıda ve fonksiyonel olarak sağlam trombosit bulunması gerektiğinden FEIBA ile yürütülen tedaviye yanıt yetersizse bir trombosit sayımı yapılması önerilir. Enfeksiyöz ajanların bulaşmasını önleyici önlemler: İnsan kanı ya da plazmasından hazırlanan tıbbi ürünlerin kullanımından kaynaklanan enfeksiyonların önlenmesi için alınan standart önlemler arasında, donörlerin seçimi, bireysel bağışların ve plazma havuzlarının belirli enfeksiyon göstergeleri için takibi ve virüslerin inaktivasyonu/uzaklaştırılması için etkili üretim aşamalarının kullanılması yer almaktadır. Buna rağmen insan kanı ya da plazmasından hazırlanan tıbbi ürünler uygulandığında, enfeksiyon ajanlarının bulaşma olasılığı tam olarak ortadan kaldırılamayabilir. Bu durum henüz bilinmeyen ya da yeni ortaya çıkan virüsler ve diğer hastalık etkenleri için de geçerlidir. Alınan önlemlerin HIV, HBC, HCV gibi zarflı virüslerle HAV gibi zarfsız virüsler için etkili olduğu düşünülmektedir. Alınan önlemlerin Parvovirüs B19 gibi bazı zarfsız virüsleri uzaklaştırmak ya da inaktive etmek için etkisi ise kısıtlıdır. Parvovirus B19 virüsü en ciddi olarak gebe kadınları (fetusda enfeksiyona neden olabilmektedir), immün yetmezlikli hastaları veya artmış eritrosit döngüsü olan hastaları (örn. hemolitik anemi durumu) etkilemektedir. İnsan plazması kaynaklı faktör VIII inhibitörü ürünlerini düzenli/tekrarlayan şekilde alan hastalarda uygun aşılama (Hepatit A ve B'ye karşı) düşünülmelidir. Hasta ile ürünün partisi arasındaki izi sürdürebilmek açısından FEIBA'nın her kullanımından sonra ürünün adı ve parti numarasının kaydedilmesi önemle önerilmektedir. ÖNLEMLER Hastaya özel faktörler nedeniyle baypas yapan bir ajana yanıt değişik olabilmekte ve belirli bir kanama durumunda bir ajanla yetersiz yanıt alınan hastalarda, başka bir ajanla yanıt alınabilmektedir. Bypass yapan bir ajanla yetersiz yanıt alınması durumunda, diğer bir ajanın kullanılması düşünülmelidir. İnhibitörlü hastalarda FEIBA uygulanması başlangıçta inhibitör düzeylerinde anamnestik bir yükselmeye yol açabilmektedir. FEIBA uygulamaya devam edildiğinde, inhibitörler zamanla azalabilir. Klinik ve yayınlanmış veriler FEIBA'nın etkililiğinin azalmadığını öngörmektedir. Hemofili hastalarındaki kanamanın profilaksisinde FEIBA uygulamasıyla ilişkili olarak ancak kısıtlı klinik veri bulunmaktadır. FEIBA 500 U her flakonda yaklaşık 80 mg sodyum içerir. Bu durum düşük sodyum diyeti almakta olan hastalarda dikkate alınmalıdır. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleri Epsilon-amino kaproik asit gibi antifibrinolitiklerin FEIBA ile birlikte uygulanması önerilmez. Hem epsilon-amino kaproik asit gibi antifibrinolitiklerin, hem de FEIBA'nın uygulanmasının endike olduğu durumlarda, iki ürünün uygulanması arasında en az 6 saatlik bir süre olmalıdır. 4.6. Gebelik ve laktasyon Genel tavsiye: Gebelik Kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (kontrasepsiyon): FEIBA'nın çocuk doğurma potansiyeli bulunan kadınlarda üreme kapasitesini etkileyip etkilemediği bilinmemektedir. Hasta hamile kaldığında veya hamilelik hamilelik kararı aldığında doktorunu bilgilendirmesi gerektiği husunda uyarılmalıdır Bilinen herhangi bir etkisi yoktur. Gebelik dönemi Hayvanlar üzerinde yapılan araştırmalar, gebelik / ve-veya / embriyonal / fetal gelişim / ve-veya / doğum / ve-veya / doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. bölüm 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. FEIBA'nın insanlarda gebelik ya da emzirme döneminde kullanımdaki güvenilirliği kanıtlanmamıştır. Bu nedenle reçetelemeden önce her bir hasta için risk ve faydaları karşılaştırılmalıdır. Gebelik ve postpartum dönemde tromboz riski artar ve gebelikte ortaya çıkan birçok komplikasyon DIC riskinde artış ile ilişkilidir. Laktasyon döne miFEIBA emziren annelerde kullanılmamalıdır. Üreme yeteneği / Fertilite FEIBA ile hayvan üreme çalışmaları yapılmamıştır. İnsanlardaki üreme yeteneği/fertiliteyi etkileyip etkilemediği bilinmemektedir. 4.7. Araç ve makine kullanımı üzerindeki etkiler Araç ve makine kullanım becerisi üzerine FEIBA'nın etkisi bilinmemektedir. Araç ve makine kullanırken dikkatli olunmalıdır. 4.8. İstenmeyen etkiler Aşağıda klinik çalışmalarda ya da pazarlama sonrasında görülen istenmeyen etkiler listelenmiştir: Advers ilaç reaksiyonlarının sıklık sınıflandırması şu şekildedir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000), çok seyrek, izole raporlar dahil (<1/10.000); bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Kan ve lenfatik sistem hastalıkları Bilinmiyor: Yaygın damar içi pıhtılaşma (DIC) İnhibitör düzeyinde yükselme (anamnestik yanıt) (a) Bağışıklık sistemi hastalıkları Bilinmiyor: Aşırı duyarlılık reaksiyonları Ürtiker Anafilaktik reaksiyon Sinir sistemi hastalıkları Bilinmiyor: Parestezi Hipoestezi Trombotik inme Embolik inme Başağrısı. Uykuya meyil Baş dönmesi / sersemlik hali Tat alma bozukluğu Kardiyak hast alıklarBilinmiyor: Miyokard enfarktüsü Taşikardi Vasküler hastalıklar Bilinmiyor: Embolizm (tromboembolik komplikasyonlar) Hipotansiyon Hipertansiyon Yüz ve boyunda kızarma (flushing) Solunum, göğüs bozuklukları ve mediastinal hastalıklar Bilinmiyor: Pulmoner embolizm Bronkospazm Hırıltılı soluk alıp verme Öksürük Zorlu soluk alıp verme (dispne) Gastrointestinal hastalıklar Bilinmiyor: Kusma Diyare Karında rahatsızlık hissi Bulantı Deri ve deri altı dokunun hast alıklarıBilinmiyor: Yüzde hissizlik Anjiyoödem Ürtiker Kaşıntı Döküntü Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklar: Bilinmiyor: Enjeksiyon yerinde ağrı Halsizlik Sıcaklık hissi Titreme Ateş yükselmesi Göğüste ağrı Göğüste rahatsızlık hissi Araştırmalar Bilinmiyor: Kan basıncında düşme (a) MedDRA sınıflandırma sisteminde yer almayan inhibitör düzeyinde yükselme (anamnestik yanıt) FEIBA uygulanmasından sonra mevcut inhibitör düzeylerinde olan yükselmedir. Bkz Bölüm 4.4. Hızlı intravenöz infüzyon kan basıncında bir düşme yanında bıçak saplanır gibi bir baş ağrısı ile yüzde ve bacaklarda hissizliğe neden olabilir. Maksimum günlük dozun üzerindeki dozların kullanımından ve/veya uzun süreli uygulamalardan sonra tromboembolik olaylar görülebilir (Bkz. Bölüm 4.4). Kan yoluyla bulaşma açısından (virüs güvenliği) Bölüm 4.4'e bakınız. 4.9. Doz aşımı ve tedavisi FEIBA doz aşımı tromboembolizm, DIC ya da miyokard enfarktüsü gibi istenmeyen olay riskini arttırır (Bkz Bölüm 4.4). 5. FARMAKOLOJİK ÖZELLİKLER 5.1. Farmakodinamik özellikler Farmakoterapötik Grubu: Kan koagülasyon faktörleri ATC kodu: B02BD03 FEIBA yetmişli yılların başlarında geliştirilmiş ve hem in vitro,in vivoolarak Faktör VIII'i baypas edici etkinliği gösterilmiş olmasına rağmen, etki mekanizması hala bilimsel tartışma konusudur. Buna rağmen, yakın tarihli bilimsel çalışmalar, FEIBA'nın etki mekanizmasında aktive protrombin kompleksinin spesifik bileşenleri, protrombin (Faktör II) ve aktive Faktör X'un (Faktör Xa) rolü bulunduğuna işaret etmektedirler.Faktör IX inhibitörü olan hemofili B hastalarındaki deneyim kısıtlıdır. Faktör IX inhibitörü olan hemofili B hastalarındaki kanama ataklarının tedavisinde ve profilaksisinde FEIBA kullanılan 40 vaka raporu bulunmaktadır. Bu 40 hastadan 3'ünde tedavi sırasında anafilaktik reaksiyon bildirilmiştir. FEIBA'nın faktör X, XI ve XIII'e karşı kazanılmış inhibitörleri olan hastalarda kullanımıyla ilgili izole raporlar da bulunmaktadır. 5.2. Farmakokinetik özellikler FEIBA'nın etki mekanizması halen kesin olarak bilinmediğinden, farmakokinetik özellikleri hakkında kesin yargılarda bulunmak mümkün değildir. 5.3. Klinik öncesi güvenlilik verileri FEIBA'nın normal farelere, FVIII knockout farelere ve sıçanlara, insanlarda önerilen günlük maksimum dozun üzerinde uygulandığı (>200 Ünite/kg) akut toksisite çalışmalarına göre, FEIBA ile ilişkili yan etkilerin büyük oranda ürünün farmakolojik özelliklerinden kaynaklanan hiperkoagülasyonun sonucunda ortaya çıktığı sonucuna varılabilir. Hayvanlarda, heterolog proteinlere karşı gelişen antikorların sonucu etkilemesi nedeniyle, tekrarlayan doz toksisite çalışmaları mümkün değildir. İnsan kanındaki pıhtılaşma faktörlerinin karsinojen ya da mutajen olarak görülmemelerinden dolayı, özellikle heterolog türlerde deneysel çalışmaların yapılmasına gerek duyulmamıştır. 6. FARMASÖTİK ÖZELLİKLERİ 6.1. Yardımcı maddelerin listesi Sodyum klorür Sodyum sitrat Enjeksiyonluk steril su (çözücü) 6.2. Geçimsizlikler Etkililiğinin bozulmaması ya da bir geçimsizlik oluşmaması için FEIBA, bölüm 6.6'da bahsedilen çözücüsü dışında başka ilaçlarla karıştırılmamalıdır. Tüm kan pıhtılaşma faktör konsantrelerinde geçerli olduğu gibi, başka ilaçlarla karıştırılması etkinlik ve toleransta bozulmalara neden olabilir. Uygulama diğer ilaçların da kullanıldığı bir venöz uygulama setinden yapılıyorsa, FEIBA infüzyonu öncesi ve sonrası venöz uygulama setinin, izotonik sodyum klorür gibi uygun çözeltilerle yıkanması önerilmektedir. İnsan plazmasından elde edilen koagülasyon faktörleri belli enjeksiyon/infüzyon cihazlarının iç yüzeyleri tarafından adsorbe edilebilirler. Bu durum tedavinin başarısız olması ile sonuçlanabilir. Bu nedenle FEIBA ile sadece onaylanmış plastik infüzyon cihazları kullanılmalıdır. 6.3. Raf ömrü 2 yıl. Sulandırılarak kullanıma hazır hale getirilmiş çözelti oda sıcaklığında kimyasal ve fiziksel olarak 3 saat süreyle stabil kalır. Mikrobiyolojik açıdan, sulandırma işlemi mikrobiyal kontaminasyon olasılığını bertaraf edecek şekilde (kontrollü ve valide edilmiş aseptik koşullarda) yapılmamışsa, sulandırılarak kullanıma hazır hale getirilmiş çözelti hemen kullanılmalıdır. Kullanıma hazır çözelti hemen kullanılmazsa, saklama koşulları ve süresinden kullanıcı sorumludur. Sulandırılmış çözelti buzdolabında saklanmamalıdır. 6.4. Saklamaya yönelik özel tedbirler 25°C'nin altındaki oda sıcaklığında saklanmalıdır. Dondurulmamalıdır. Işıktan korumak amacıyla ambalajı içerisinde saklanmalıdır. Sulandırılarak kullanıma hazır hale getirilmiş ilacın saklanması için bkz. Bölüm 6.3. 6.5. Ambalajın niteliği ve içeriği FEIBA kuru toz ve sulandırıcı çözeltiler flakon içinde sunulmaktadır (kuru toz içeren flakon: yüzeyi soda-lime ile muamele edilmiş Tip II hidrolitik cam; sulandırıcı içeren flakon: yüzeyi soda-lime ile muamele edilmiş Tip I hidrolitik cam). Flakonlar, bütil lastik tıpa ve koruyucu kapakla kapalıdır. Ambalaj içeriği: - 1 adet lastik tıpalı liyofilize toz içeren flakon - 1 adet 20 mL enjeksiyonluk su içeren çözücü flakon - 1 adet Baxject II Hi-Flow cihazı (iki flakondaki ürünün birbiriyle karıştırılarak bir enjektöre alınmasını sağlayan iğnesiz transfer cihazı) - 1 adet tek kullanımlık enjektör - 1 adet tek kullanımlık iğne - 1 adet klempli kelebek iğne (enjeksiyon için kanatlı set). 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemler Ambalajı açılmış ürün yeniden kullanılmamalıdır. Steril bariyeri bozulan, ambalajı hasar görmüş ya da bozulma belirtisi gösteren ürünü kullanmayınız. Kullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİAdı : Eczacıbaşı-Baxter Hastane Ürünleri Sanayi ve Ticaret A.Ş. Adresi : Cendere Yolu, Pırnal Keçeli Bahçesi 34390 Ayazağa-İSTANBUL Tel : (0.212) 329 62 00 Faks : (0.212) 289 92 75 Üretim yeri : Baxter AG, Viyana Avusturya 8. RUHSAT NUMARASI 30.01.2013-87 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİ İlk ruhsat tarihi: 30.01.2013 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ 14 |
İlaç BilgileriFeiba 500 U Iv İnfüzyon İçin Liyofilize Toz Içere...Etken Maddesi: Faktör Viii Inhibitör By-pass Aktivitesine Sahip ... Atc Kodu: B02BD03 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.