Dacogen 50mg Tek Doz Iv Enjeksiyonluk Çözelti Içi... Kısa Ürün BilgisiAntineoplastik ve İmmünomodülatör Ajanlar » Antineoplastik İlaçlar (Kanser İlaçları) » Antimetabolitler » Pirimidin Analogları » Decitabine KISA ÜRÜN BİLGİSİ1.BEŞERİ TIBBİ ÜRÜNÜN ADIDACOGEN 50 mg, tek doz IV enjeksiyonluk çözelti için toz içeren flakon2.KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:Her flakon 50 mg desitabin içerir. 10 ml enjeksiyonluk su ile sulandırılan DACOGEN enjeksiyonluk çözelti 5 mg/ml desitabin içerir.Yardımcı maddeler:Monobazik potasyum fosfat Sodyum hidroksit
6.8 mg/ml 1.16 mg/ml Yardımcı maddeler için 6.1'e bakınız. 3.FARMASÖTİK FORMEnjeksiyonluk çözelti için toz.Beyaza yakın renkli steril liyofilize bir tozdur. 4.KLİNİK ÖZELLİKLER4.1. Terapötik endikasyonlarDACOGEN aşağıdaki hastalıkların tedavisinde endikedir: DACOGEN (desitabin) Dünya Sağlık Örgütü (WHO)'nun miyelodisplastik sendrom (MDS) sınıflandırmasına göre aşırı blast artışı gösteren dirençli anemi tip I ve tip II (RAEB-1 ve RAEB-2) erişkin hastaların tedavisinde endikedir. Ayrıca ek olarak diğer miyelodisplastik sendrom alt tiplerinde eşlik eden sitogenetik kötü risk, ağır dishematopoez, yoğun enfeksiyonla seyreden lökopeni, klinik kanamalara neden olabilecek trombositopeni ve tranfüzyonlarla düzeltilemeyen derin refrakter anemi hallerinde de kullanılır. DACOGEN, kemik iliğinde blast oranı %30'dan fazla olan, orta/kötü sitogenetik riski bulunan ve standart indüksiyon kemoterapisi için aday olmayan 70 yaş ve üstü yeni tanı konmuş akut miyeloid lösemi (AML) tedavisinde endikedir. 4.2. Pozoloji ve uygulama şekliDACOGEN, kemoterapötik ajanların kullanımı konusunda deneyimli bir hekim gözetiminde uygulanmalıdır.Uygulama sıklığı ve süresi:DACOGEN için iki farklı uygulama şeması önerilmektedir: AML tedavisi için 5 günlük tedavi şeması ve MDS tedavisi için 3 ya da 5 günlük tedavi şeması. Her iki tedavi şemasında da hastaların en az 4 siklus tedavi görmesi önerilir; ancak tam veya kısmi remisyon 4 siklusla sağlanamayabilir AML faz 3 çalışmasında ortalama yanıt süresi (tam remisyon [CR] ya da kısmi trombosit iyileşmesi [CRp] ile birlikte CR) 4.3 ay olarak bildirilmiştir. MDS'de, beş günlük tedavi şemasında faz II çalışmalarda ortalama yanıt (CR+PR) süresi 3.5 siklus olarak bildirilmiştir. MDS'de üç günlük tedavi şemasında faz III çalışmada remisyon süresi 3 siklus olarak bildirilmiştir. Hastada yanıt alındığı sürece, hasta tedaviden fayda gördüğü sürece ya da hastalık stabil kaldığı sürece, yani belirgin bir progresyon olmadığında tedaviye devam edilebilir.Dört siklus sonra hastanın hematolojik parametreleri (örn. trombosit sayısı ya da mutlak nötrofil sayısı) tedavi öncesi değerlere dönmezse ya da hastalık progresyonu görülürse (periferik blast sayısı artmaya devam eder ya da kemik iliği blast sayısı giderek kötüleşirse) hastanın tedaviye yanıt vermediğine karar verilerek, DACOGEN'e alternatif tedavi seçenekleri düşünülmelidir. Bulantı ve kusma için premedikasyon rutin olarak önerilmemesine rağmen gerekiyorsa uygulanabilir. Akut Miyeloid Lösemide Tedavi ŞemasıTedavi siklusu sırasında, DACOGEN ardarda 5 gün süreyle her gün vücut yüzey alanının metrekaresi başına 20 mg dozda ve bir saatlik intravenöz infüzyon ile uygulanır (her bir siklusta toplam 5 doz). Günlük toplam doz 20 mg/m 'yi geçmemeli ve her bir tedavi siklusunda toplam 100 mg/m 'lik toplam doz aşılmamalıdır. Bu siklus hastanın klinik yanıtı ve gözlenen toksisiteye göre her 4 haftada bir tekrarlanır. Dozun atlandığı durumlarda, tedaviye mümkün olan en kısa zamanda kalındığı yerden devam edilmelidir. Bu şema ayaktan tedaviye olanak sağlar. Miyelodisplastik Sendromda Tedavi ŞemasıMDS'de 3 Günlük Tedavi ŞemasıTedavi siklusu sırasında, DACOGEN ardarda 3 gün süreyle her sekiz saatte bir, vücut yüzey alanının metrekaresi başına 15 mg sabit dozda ve üç saatlik intravenöz infüzyon ile uygulanır (her bir tedavi siklusunda toplam 9 doz). Bu siklus hastanın klinik yanıtı ve gözlenen toksisiteye bağlı olarak yaklaşık 6 haftada bir tekrarlanır. Günlük toplam doz 45 mg/m 'yi geçmemeli ve her bir tedavi siklusunda toplam 135 mg/m 'lik toplam doz aşılmamalıdır. Dozun atlandığı durumlarda, tedaviye mümkün olan en kısa zamanda kalındığı yerden devam edilmelidir. MDS'de 5 Günlük Tedavi ŞemasıTedavi siklusu sırasında, DACOGEN ardarda 5 gün süreyle her gün vücut yüzey alanının metrekaresi başına 20 mg dozda ve bir saatlik intravenöz infüzyon ile uygulanır (her bir siklusta toplam 5 doz). Bu siklus hastanın klinik yanıtı ve gözlenen toksisiteye bağlı olarak 4 haftada bir tekrarlanır. Günlük toplam doz 20 mg/m 'yi geçmemeli ve her bir tedavi siklusunda toplam 100 mg/m 'lik toplam doz aşılmamalıdır. Dozun atlandığı durumlarda, tedaviye mümkün olan en kısa zamanda kalındığı yerden devam edilmelidir. Bu şema ayaktan tedaviye olanak sağlar. Miyelosupresyon ve İlişkili KomplikasyonlarAML ve MDS'li hastaların hem tedavi görenlerinde, hem de görmeyenlerinde miyelosupresyon ve miyelosupresyonla ilişkili istenmeyen etkiler (trombositopeni, anemi, nötropeni ve febril nötropeni) yaygın olarak görülür. Miyelosupresyonun komplikasyonları arasında enfeksiyonlar ve kanama bulunmaktadır. Miyelosupresyon ve ilişkili komplikasyonların bulunduğu hastalarda tedavi aşağıda tarif edildiği şekilde modifiye edilebilir: Akut Miyeloid LösemiTedaviyi yürütmekte olan hekimin kararına göre aşağıda tarif edilen miyelosupresyonla ilişkili komplikasyonların bulunduğu hastalarda tedavi ertelenebilir: Febril nötropeni (vücut ısısı >38.5°C ve mutlak nötrofil sayısı <1,000/^L) Aktif viral, bakteriyel veya fungal enfeksiyon (intravenöz anti-infektiflerin uygulanmasını ya da yoğun destekleyici bakımı gerektiren) Kanama (trombosit sayısının <25,000/ |iL olduğu gastrointestinal, genito-üriner ve pulmoner sistem kanaması ya da merkezi sisteminde olan kanamalar) İyileşme olduğunda ya da uygun tedavilerle (anti-infektif tedavi, transfüzyonlar veya büyüme faktörleri) hastanın durumu stabil hale geldiğinde DACOGEN tedavisi yeniden başlatılabilir. Klinik çalışmalarda, DACOGEN alan hastaların yaklaşık üçte birinde doz ertelenmesi gerekti. Doz azaltılması önerilmez. Miyelodisplastik Sendrom5 Günlük Tedavi ŞemasıHastanın tedaviden yararını optimumda tutabilmek için bu klinik durumda dozda azaltma önerilmez, doz aşağıda belirtildiği şekilde ertelenmelidir: İlk 3 Tedavi Siklusunda Doz Şeması DeğişiklikleriTedavinin ilk sikluslarında Evre 3 ve 4 sitopeni yaygındır ve bu durum MDS progresyonu anlamına gelmeyebilir. Üçüncü siklustan sonrasına kadar sitopeni durumlarının önceden tedavisi işe yaramayabilir. İlk üç siklus için, orta şiddette nötropeni durumunda (mutlak nötrofil sayısı <1000/^l), hastadaki faydayı optimize etmek için standart tedavi siklusları sırasında tam dozla devam edilmesine çalışılmalıdır. Granülosit düzeyi 500/^l üzerine çıkana kadar, tedaviyi yürüten kuruluşun kılavuzları doğrultusunda ek antimikrobiyal profilaksi uygulanabilir. Klinisyenler, bu dönemde MDS'li hastalardaki enfeksiyonların önlenmesi ya da tedavisi için büyüme faktörlerinin erken uygulanmasının gerekip gerekmediğini değerlendirmelidir. Benzer şekilde orta şiddette trombositopeni durumunda (trombosit sayısı <25.000/^l), hastadaki faydayı optimize etmek için standart tedavi siklusları sırasında tedaviye tam dozla devam edilmesine çalışılmalıdır; kanama olayları görüldüğünde eş zamanlı trombosit transfüzyonları yapılmalıdır. Üçüncü Siklustan Sonraki Doz Şeması DeğişiklikleriAşağıda belirtilen toksisitelerin tedaviyle ilişkisi en azından olası olarak değerlendirildiği durumlarda dozun uygulanması geciktirilmelidir: - Kemik iliği baskılanmasıyla ilişkili şiddetli komplikasyonlar (uygun anti-infektif tedaviye rağmen düzelmeyen enfeksiyonlar, uygun tedaviye rağmen düzelmeyen kanamalar) - Bir tedavi kürüne başlanmasından 6 hafta ya da daha uzun süreyle hastalıkta progresyon olmamasıyla beraber hiposelüler kemik iliği olarak (sellüleritenin %5 ya da daha düşük olması) tanımlanan uzamış kemik iliği baskılanması Eğer düzelme (mutlak nötrofil sayısı >1.000/^l ve trombosit sayısı >50.000/^l) için 8 haftadan uzun süre gerekirse hastada ilaç tedavisi kesilerek, bu 8 haftadan sonraki 7 gün içinde hastalık progresyonu açısından değerlendirilmelidir (kemik iliği aspirasyonuyla). En az 6 siklus tedavi görmüş ve tedaviden yarar görmeye devam eden hastalar için, progresyon olmadığı durumlarda hekimin kararına göre 8 haftadan uzun bir gecikmeye izin verilebilir. 3 Günlük Tedavi Şeması İlk 3 Tedavi Siklusunda Doz Şeması DeğişiklikleriTedavinin ilk sikluslarında Evre 3 ve 4 sitopeni yaygındır ve bu durum MDS progresyonu anlamına gelmeyebilir. Üçüncü siklustan sonrasına kadar sitopeni durumlarının önceden tedavisi işe yaramayabilir. İlk üç siklus için, orta şiddette nötropeni durumunda (mutlak nötrofil sayısı <1000/^l), hastadaki faydayı optimize etmek için standart tedavi siklusları sırasında tam dozla devam edilmesine çalışılmalıdır. Granülosit düzeyi 500/^l üzerine çıkana kadar, tedaviyi yürüten kuruluşun kılavuzları doğrultusunda ek antimikrobiyal profilaksi uygulanabilir. Klinisyenler, bu dönemde MDS'li hastalardaki enfeksiyonların önlenmesi ya da tedavisi için büyüme faktörlerinin erken uygulanmasının gerekip gerekmediğini değerlendirmelidir. Benzer şekilde orta şiddette trombositopeni durumunda (trombosit sayısı <25.000/^l), hastadaki faydayı optimize etmek için standart tedavi siklusları sırasında tedaviye tam dozla devam edilmesine çalışılmalıdır; kanama olayları görüldüğünde eş zamanlı trombosit transfüzyonları yapılmalıdır. Üçüncü Siklustan Sonraki Doz Şeması DeğişiklikleriKalıcı sitopeninin ilaç uygulamasıyla ilişkili kabul edildiği durumlarda hematolojik düzelme için (mutlak nötrofil sayısı >1.000/^l ve trombosit sayısı >50.000/^l) gereken süre 6 haftayı geçtiğinde, bir sonraki DACOGEN siklusu geciktirilebilir ve doz aşağıdaki algoritmaya göre azaltılabilir. Yapılacak daha sonraki sikluslarda bu azaltılmış dozlarla devam edilmeli; doz yeniden ayarlanmamalıdır. - Düzelme için gereken sürenin 6 haftadan uzun, 8 haftadan az olduğu durumlarda, DACOGEN uygulaması 2 hafta daha geciktirilebilir ve her sekiz saatte bir intravenöz 2 2 2 infüzyonla uygulanan 11 mg/m 'ye (33 mg/m /gün, 99 mg/m /siklus) düşürülebilir. - Düzelme için gereken sürenin 8 haftadan uzun, 10 haftadan az olduğu durumlarda, DACOGEN uygulaması 2 haftaya kadar geciktirilebilir ve her sekiz saatte bir intravenöz 2 2 2 infüzyonla uygulanan 11 mg/m 'ye (33 mg/m /gün, 99 mg/m /siklus) düşürülür ve daha sonraki sikluslarda bu dozlarla devam edilebilir. - Düzelme için gereken sürenin 10 haftadan uzun olduğu durumlarda ilaç tedavisi kesilerek, bu 10 haftadan sonraki 7 gün içinde hastalık progresyonu değerlendirilmelidir (kemik iliği aspirasyonuyla). Ancak en az 6 siklus tedavi görmüş ve tedaviden yarar görmeye devam eden hastalar için, progresyon olmadığı durumlarda hekimin kararına göre 10 haftadan uzun bir gecikmeye izin verilebilir. Uygulama şekli :DACOGEN, intravenöz infüzyon yoluyla uygulanır. Uygulama için bir santral venöz katetere gerek yoktur. DACOGEN, 10 ml steril enjeksiyonluk su ile seyreltilir. Seyreltilen çözelti, uygulama için daha sonra%%5 Dekstroz ya da Laktatlı Ringer çözeltisi kullanılarak daha seyreltik hale getirilmelidir.Kullanım ve hazırlamayla ilgili talimatlar için bölüm 6.6'ya bakınız. Bulantı ve kusmayı önlemek amacıyla premedikasyon rutin olarak önerilmemesine rağmen, istenirse uygulanabilir. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Böbrek yetmezliği olan hastalarda çalışma yapılmamıştır. Böbrek yetmezliği olan hastalarda doz ayarlamasına gerek olup olmadığı konusu araştırılmamıştır (bkz. Bölüm 4.4 ve 5.2).Karaciğer yetmezliği:Karaciğer yetmezliği olan hastalarda doz ayarlamasına gerek olup olmadığı konusu araştırılmamıştır. Karaciğer fonksiyonlarında kötüleşme olursa, hastalar dikkatle izlenmelidir (bkz. Bölüm 4.4 ve 5.2).Pediyatrik popülasyon:18 yaşın altındaki hastalarda etkinlik ve güvenlilik kanıtlanmamıştır.Geriyatrik popülasyon:Yeterli ve iyi kontrollü klinik çalışmalara dahil edilmiş geriyatrik hastalar, genellikle daha genç yetişkin hastalarla aynı seviyede dozlanmışlardır. Toksisite için doz azaltımı, genel popülasyon için belirlenen şekilde gerçekleştirilmelidir.Faz 3 çalışmasına katılan 83 hastadan 61'i 65 yaş ve üzerinde, 21'i ise 75 yaş ve üzerindeydi. Hem bu çalışmada, hem de diğer rapor edilen klinik deneyimlere göre bu yaşlı hastalarla, genç hastalar arasında etkinlik ve emniyet açısından bir farklılık olmamasına rağmen, bazı yaşlı bireylerin duyarlılığının daha fazla olabileceği göz ardı edilmemelidir. 4.3 KontrendikasyonlarDesitabin ya da yardımcı maddelerinden herhangi birine karşı aşırı duyarlılığı olduğu bilinen hastalarda kontrendikedir (bkz. Bölüm 6.1).Laktasyon döneminde kontrendikedir (bkz. Bölüm 4.6). 4.4 Özel kullanım uyarıları ve önlemleriMiyelosupresyonDACOGEN tedavisi ile MDS'li ya da AML'li hastalarda kemik iliği baskılanması ve bu baskılanmaya bağlı enfeksiyon ve kanama dahil komplikasyonlarda alevlenme görülebilir. AML klinik çalışmalarında, hastaların çoğunda başlangıçta Evre 3/4 kemik iliği baskılanması vardı. Başlangıçta Evre 2 kemik iliği baskılanması olan hastalarda, kemik iliği baskılanmasının kötüleşmesi hastaların çoğunda görüldü ve başlangıçta Evre 1 veya 0 olan hastalardan daha sıktı. DACOGEN ile oluşan kemik iliği baskılanması geri dönüşlüdür. Klinik açıdan gerekli olduğunda ve her bir tedavi siklusu öncesinde tam kan ve trombosit sayımı yapılmalıdır. Kemik iliği baskılanması ya da baskılanmaya bağlı komplikasyonların görülmesi durumunda, Bölüm 4.2 ve 4.8'de önerildiği şekilde DACOGEN tedavisi kesilebilir, doz azaltılabilir ya da destekleyici önlemler alınabilir. Karaciğer yetmezliğiKaraciğer yetmezliği olan hastalarda doz ayarlamasına gerek olup olmadığı konusu araştırılmamıştır. Karaciğer fonksiyonlarında kötüleşme olursa, hastalar dikkatle izlenmelidir (bkz. Bölüm 4.2 ve 5.2). Böbrek yetmezliğiDACOGEN şiddetli böbrek yetmezliği olan hastalarda çalışılmamıştır. DACOGEN'i şiddetli böbrek yetmezliği olan hastalarda (Kreatin klerensi [CrCl] <30 ml/dak) kullanırken dikkatli olunmalı ve hastalar yakından izlenmelidir (bkz. Bölüm 4.2). Kalp hastalığıAğır konjestif kalp yetmezliği ya da klinik olarak unstabil kalp hastalığı hikayesi olan hastalar klinik çalışmalara alınmamıştır ve bu nedenle DACOGEN'in bu hastalardaki etkinlik ve güvenliliği kanıtlanmamıştır. DACOGEN her ml'sinde 1 mmol (23 mg)'dan daha az sodyum ihtiva eder. Sodyuma bağlı herhangi bir etki gözlenmez. DACOGEN her ml'sinde 1 mmol (39 mg)'dan daha az potasyum ihtiva eder. Enjeksiyon yerinde ağrıya neden olabilir. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriDesitabinle gerçekleştirilen klinik ilaç etkileşim çalışması bulunmamaktadır.Ardışık fosforilasyon ile aktive olan (intraselüler fosfokinaz aktivitesi yoluyla) ve/veya desitabinin inaktivasyonunda rol alan enzimlerle (sitidin deaminaz gibi) metabolize olan diğer ajanlar ile ilaç -ilaç etkileşimine girme olasılığı vardır. Bu yüzden bu ajanlar DACOGEN ile kombine edilirken dikkatli olunmalıdır. Birlikte uygulanan ilaçların desitabin üzerine etkisi:Desitabinin metabolizması CYP450 enzim sistemi tarafından değil de oksidatif deaminasyon yoluyla olduğundan CYP450 tarafından yönetilen metabolik ilaç etkileşimleri beklenmez. Desitabin, in vitroIn vitroveriler desitabinin zayıf bir P-glikoprotein (P-gp) substratı olduğunu ve bu nedenle P-gp inhibitörleriyle etkileşime yatkın olmadığını göstermiştir.Desitabinin birlikte uygulanan ilaçlar üzerindeki etkileri: In vitroIn vitroçalışmalar desitabinin belirlenmiş maksimum terapötik dozlarının (Cmaks) 20 katından yüksek dozlarda CYP 450 enzimlerini inhibe etmediğini ya da uyarmadığını göstermiştir. Bu nedenle, CYP tarafından yönlendirilmiş metabolik ilaç etkileşimleri beklenmez ve ilacın bu metabolik yolla metabolize olan ajanlarla etkileşimi beklenmemektedir.Desitabin, P-gp aracılı in vitrotaşıma olayının zayıf bir inhibitörüdür ve bu nedenle de birlikte uygulanan ilaçların P-gp aracılı taşınmasını etkilemesi beklenmez (Bkz Bölüm 5.2).4.6 Gebelik ve laktasyon Genel tavsiyeGebelik Kategorisi: DLiteratürde hayvanlarda desitabinin, fertilite, embriyo-fetal ve post-natal gelişim dahil üreme siklusunun her döneminde üreme toksisitesine yol açtığı gösterilmiştir (bkz. Bölüm 5.3). Çocuk doğurma potansiyeli bulunan kadınlar/doğum kontrolü(Kontrasepsiyon)Çocuk doğurma potansiyeli olan kadın hastaların DACOGEN ile tedaviye başlamadan önce oosit kriyoprezervasyonu konusunda bir uzmandan destek almaları önerilmektedir.Gebelik dönemiDesitabin'in gebelik ve/veya fetus/yeni doğan üzerinde zararlı farmakolojik etkileri bulunmaktadır. DACOGEN gerekli olmadıkça gebelik döneminde kullanılmamalıdır. DACOGEN kullanmakta olan doğurganlık çağındaki kadınlara kontraseptif yöntemler kullanmaları ve gebe kalmamaları önerilmelidir. DACOGEN kullanımından ne kadar süre sonra hamile kalınması güvenlidir bilinmemektedir. DACOGEN'in gebe kadınlarda kullanımına ilişkin yeterli veri yoktur. Yapılan çalışmalar desitabinin fare ve sıçanlarda teratojen etkili olduğunu göstermiştir (bkz kısım 5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. Hayvanlarda gerçekleştirilen çalışmalar ve etki mekanizması nedeniyle DACOGEN gebelik döneminde gerekli görülmedikçe kullanılmamalıdır.Gebelikte kullanımı veya hastanın DACOGEN kullanırken gebe kalması durumunda hasta fetusa olabilecek hasar konusunda bilgilendirilmelidir. Laktasyon dönemiDesitabin ya da metabolitlerinin süte geçip geçmedikleri bilinmemektedir. DACOGEN laktasyon döneminde kontrendikedir; bu nedenle bu dönemde DACOGEN tedavisine gerek duyulursa emzirmeye son verilmelidir (bkz. Bölüm 4.3).Üreme yeteneği /FertiliteErkeklerde kullanım: Erkeklere, DACOGEN kullanırken ve tedaviyi tamamladıktan sonra 3 ay süreyle çocuk sahibi olmamaları önerilir (bkz. Bölüm 5.3). DACOGEN tedavisine bağlı infertilite olasılığı nedeniyle, erkek hastalar, tedavi öncesi spermlerini saklama konusunda danışmanlık almalıdır.4.7 Araç ve makine kullanımı üzerindeki etkilerDACOGEN'in araç ve makina kullanımı üzerinde hafif etkisi olabilir. Hastalar tedavi sırasında anemi gibi olumsuz etkilerin görülebileceği konusunda bilgilendirilmelidir. Bu nedenle araç ve makine kullanımı sırasında dikkatli olmaları önerilmelidir.4.8 İstenmeyen etkilerHem 3-günlük hem de 5-günlük tedavi şemalarında en önemli ve en sık görülen advers ilaç reaksiyonları kemik iliği baskılanması ve buna bağlı reaksiyonlardır.Klinik çalışmalardan elde edilen veriler Bu bölümde yan etkiler rapor edilmiştir. Advers reaksiyonlar, mevcut advers olay bilgilerin kapsamlı değerlendirmesine dayanan DACOGEN kullanımı ile ilişkili olduğu düşünülen advers olaylardır. Bireysel vakalarda DACOGEN ile güvenli nedensel bir ilişki tespit edilemez. Ayrıca, klinik çalışmalar değişen koşullar altında yapıldığından, klinik çalışmalarda gözlenen bir ilaca ait advers etki oranlar, diğer bir ilaca ait oranlar ile karşılaştırılamaz ve klinik uygulamada gözlenen oranları yansıtmayabilir. Advers ilaç reaksiyonları DACOGEN'in güvenilirliği, AML ve MDS'li 682 hastada gerçekleştirilen klinik çalışmalarla (D-0007, DACO-016, DACO-017, DACO-020, EORTC-06011 ve ID03-0180 çalışmaları) araştırılmıştır. Bu klinik çalışmalarda DACOGEN 5 günlük ya da 3 günlük tedavi şemalarıyla uygulanmıştır. Bu klinik çalışmalarda bildirilen advers ilaç reaksiyonları aşağıdaki Tablo 1'de özetlenmiştir. Aşağıdaki sıklık grupları kullanılmıştır: Çok yaygın, >1/10; yaygın, >1/100 ila <1/10; yaygın olmayan, >1/1000 ila <1/100; seyrek, >1/10.000 ila <1/1000; çok seyrek, <1/10.000. Enfeksiyon ve enfestasyonlarÇok yaygın : Pnömoni*, idrar yolu enfeksiyonu*Yaygın : Septik şok*, sepsis*, sinüzit Kan ve lenf sistemi hastalıklarıÇok yaygın : Febril nötropeni*, nötropeni*, trombositopenia*, anemi, lökopeni Yaygın : Pansitopeni*Bağışıklık sistemi hastalıklarıYaygın : Anafilaktik reaksiyon dahil hipersensitivitebSinir sistemi hastalıklarıÇok yaygın : Baş ağrısıSolunum sistemi, , göğüs bozuklukları ve mediastinal hastalıklarÇok yaygın : EpistaksisGastrointestinal hastalıklarÇok yaygın : Diyare, kusma, bulantı Yaygın: StomatitDeri ve deri altı doku hastalıklarıYaygın olmayan : Akut febril nötrofilik dermatoz (Sweet's sendromu)Genel sistem hastalıkları ve uygulamayla ilgili reaksiyonlarÇok yaygın : AteşabHematolojik advers ilaç reaksiyonları
| Parametre
| DACOGEN (n=99)
| Toplam yanıt oranı (CR + mCR + PR)
| 33 (%33)
| Tam remisyon (CR)
| 17 (%17)
| Kemik iliği tam remisyon (mCR)
| 16 (%16)
| Toplam iyileşme oranı (CR + mCR + PR + HI)
| 51 (%52)
| CR = tam remisyon; mCR = kemik iliği tam remisyon;PR = kısmi remisyon; HI = hematolojik iyileşme.Kaynak: DACO-020 CSRFaz-III Çalışma (D-0007): 3 Günlük Tedavi Seması
|
| Tablo 4: Tipik bir Hastadaki Popülasyon Farmakokinetiği Analizinin Özeti (5-Günlük ve 3-Günlük Tedavi Şeması)
|
| 5-Günlük Şema
|
| 3-Günlük Şema
|
| Parametre
| Beklenen Değer
| %95 GA
| Beklenen Değer
| AG59ox
| Cmaks (ng/ml)
| 107
| 88.5 - 129
| 42.3
| 35.2 - 50.6
| AUCcum (ng.h/ml)
| 580
| 480 - 695
| 1161
| 972 - 1390
| t(min)
| 68.2
| 54.2 - 79.6
| 67.5
| 53.6 - 78.8
| Vdss (L)
| 116
| 84.1 - 153
| 49.6
| 34.9 - 65.5
| CL (L/h)
| 298
| 249 - 359
| 201
| 168 - 241 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
DACOGEN |
Destek bakımı | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
IPSS alt grubu |
Toplam yanıt |
AML ya da ölüm |
Toplam yanıt |
AML ya da ölüm | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
oranı |
için geçen ortalama |
oranı |
için geçen ortalama | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
(CR + PR) |
zaman (gün) |
(CR + PR) |
zaman (gün) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tüm hastalar |
15/89 (%17) |
340 |
0/81 |
219 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Int-2 ve yüksek riskli |
11/61 (%18) |
335 |
0/57 |
189 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Int-2 |
7/38 (%18) |
371 |
0/36 |
263 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Yüksek riskli |
4/23 (%17) |
260 |
0/21 |
79 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Tablo 4: DAC0-016 çalışmasında Diğer Etkinlik Sonlanma Noktaları (ITT popülasyonu) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sonuçlar |
DACOGEN n = 242 |
TC (birleştirilmiş grup) n = 243 |
p-değeri | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
CR + CRp |
43 (%17.8) |
19 (%7.8) |
0.0011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
OR=2.5 (1.40; 4.78) b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
CR |
38 (%15.7) |
18 (%7.4) |
- | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EFSa |
3.5 |
2.1 |
0.0025 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
(2.5; 4.1)b |
(1.9; 2.8) b |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
HR=0.75 (0.62; 0.90)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
PFSa |
3.7 |
2.1 |
0.0031 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
(2.7; 4.6) b |
(1.9; 3.1)b |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
HR=0.75 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
(0.62; 0.91) b |
|
İlaç BilgileriDacogen 50mg Tek Doz Iv Enjeksiyonluk Çözelti Içi...Etken Maddesi: Desitabin Atc Kodu: L01BC08 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
İlgili İlaçlar |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
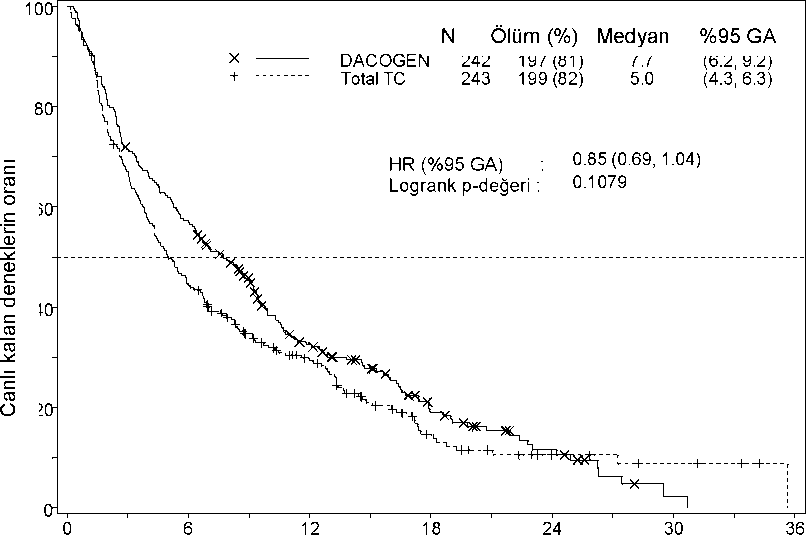 Risk altındaki hasta sayısı Süre (ay)
Risk altındaki hasta sayısı Süre (ay)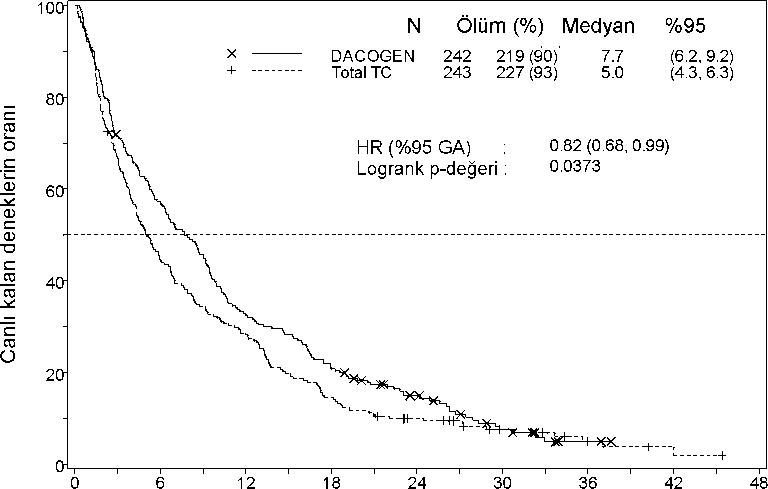 DACOGEN 242 Total TC 243
DACOGEN 242 Total TC 243Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2026 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.