Apremia 10mg + 20mg + 30mg Film Kaplı Tablet Tedaviye Başlama Paketi Kısa Ürün BilgisiKISA URUN BILGISI¡ Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERI TIBBİ ÜRÜNÜN ADIAPREMIA 10mg + 20mg + 30mg film kaplı tablet tedaviye başlama paketi 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin maddeler:APREMIA tedaviye başlama paketi içeriğinde bulunan, Apremilast 10 mg film kaplı tablet, 10 mg apremilast içerir. Apremilast 20 mg film kaplı tablet, 20 mg apremilast içerir. Apremilast 30 mg film kaplı tablet, 30 mg apremilast içerir. Yardımcı maddeler:Apremilast 10 mg film kaplı tablet, 57 mg laktoz monohidrat (sığır kaynaklı) içerir. Apremilast 20 mg film kaplı tablet, 114 mg laktoz monohidrat (sığır kaynaklı) içerir.Apremilast 30 mg film kaplı tablet, 171 mg laktoz monohidrat (sığır kaynaklı) içerir. Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMApremilast 10 mg film kaplı tablet: Turuncu renkli, yuvarlak, bikonveks film kaplı tabletler. Apremilast 20 mg film kaplı tablet: Pembe renkli, yuvarlak, bikonveks film kaplı tabletler.Apremilast 30 mg film kaplı tablet: Somon renkli, yuvarlak, bikonveks film kaplı tabletler. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlarPsöriatik artritApremilast, tek başına veya Hastalığı Modifiye Edici Antiromatizmal İlaçlarla (DMARD'lar) kombinasyon halinde önceki DMARD tedavisine yetersiz yanıt veren veya tolere edemeyenyetişkin hastalarda aktif psöriatik artritin (PsA) tedavisinde endikedir (bölüm 5.1'e bakınız). PsöriazisApremilast siklosporin, metotreksat veya psoralen ve ultraviyole-A ışığı (PUVA) dahil olmak üzere diğer sistemik tedavilere yanıt vermeyen, söz konusu tedavilere karşı kontrendikasyonuveya intoleransı bulunan yetişkin hastalarda orta ve şiddetli kronik plak tipi psöriazisintedavisinde endikedir. 4.2 Pozoloji ve uygulama şekliPozoloji/uygulama sıklığı ve süresi:Tedavi, psöriazis veya psöriatik artrit tanı ve tedavisinde deneyimli bir hekim tarafından başlatılmalı ve kontrol edilmelidir. Apremilastın önerilen dozu yemek kısıtlaması olmaksızın yaklaşık 12 saat arayla sabah ve akşam olmak üzere günde iki kez oral yoldan alınan 30 mg'dır. Aşağıdaki Tablo 1'degösterildiği şekilde bir başlangıç titrasyon programı gerekmektedir. Başlangıç titrasyonundansonra başka bir titrasyon gerekli değildir.
Hastalar bir dozu atlarsa, bir sonraki doz mümkün olan en kısa zamanda alınmalıdır. Bir sonraki dozları için zaman yaklaşmışsa atlanan doz alınmamalı ve sonraki doz normalzamanında alınmalıdır. Pivotal çalışmalar sırasında en fazla iyileşme tedavinin ilk 24 haftası içinde gözlenmiştir. Bir hasta 24 haftadan sonra terapötik fayda kanıtı göstermezse tedavi yenidendeğerlendirilmelidir. Hastanın tedaviye yanıtı düzenli olarak değerlendirilmelidir. Uygulama şekli:APREMIA ağızdan kullanım içindir. APREMIA film kaplı tabletler aç veya tok karnına bütün olarak yutulmalıdır. Özel popülasyonlara ilişkin ek bilgiler:Böbrek yetmezliği:Hafif ve orta şiddette böbrek yetmezliği olan hastalarda herhangi bir doz ayarlamasına gerek yoktur. Apremilast dozu ciddi böbrek yetmezliği (Cockcroft-Gault denklemi ile hesaplanandakikada 30 mL'den az kreatinin klirensi) olan hastalarda günde bir kez 30 mg'aazaltılmalıdır. Bu grup hastalarda başlangıç doz titrasyonu için apremilastın sadece Tablo1'de listelenen SABAH programı kullanılarak titre edilmesi ve AKŞAM dozlarının atlanmasıönerilir (bölüm 5.2'ye bakınız). Karaciğer yetmezliği:Karaciğer yetmezliği olan hastalar için herhangi bir doz ayarlamasına gerek yoktur (bölüm 5.2'ye bakınız). Pediyatrik popülasyon:0-18 yaş arasındaki çocuklarda apremilastın güvenliliği ve etkililiği belirlenmemiştir. Herhangi bir veri mevcut değildir. Geriyatrik popülasyon:Bu hasta popülasyonu için herhangi bir doz ayarlamasına gerek yoktur (bölüm 4.8 ve 5.2'ye bakınız). 4.3 Kontrendikasyonlar Etkin maddeye veya bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşıaşırı duyarlılık geliştiği durumlarda. Gebelikte (bölüm 4.6'ya bakınız) kontrendikedir. 4.4 Özel kullanım uyarıları ve önlemleriİshal, Bulantı ve KusmaPazarlama sonrası apremilast kullanımı ile ilişkili şiddetli ishal, bulantı ve kusma raporları alınmıştır. Çoğu olay tedavinin ilk birkaç haftası içinde meydana gelmiştir. Bazı durumlardahastalar hastaneye yatırılmıştır. 65 yaş veya üzerindeki hastalar komplikasyonlar açısındandaha yüksek risk taşıyabilir. Hastalarda şiddetli ishal, bulantı veya kusma gelişirse apremilastile tedavinin kesilmesi gerekebilir. Psikiyatrik bozukluklarApremilast kullanımı uykusuzluk ve depresyon gibi psikiyatrik bozuklukların artma riski ile ilişkilidir. Depresyon öyküsü olan veya olmayan hastalarda intihar da dahil olmak üzereintihar düşüncesi ve davranışı gözlemlenmiştir (bölüm 4.8'e bakınız). Hastalar öncesine aitveya mevcut psikiyatrik semptomlar bildirirlerse ya da psikiyatrik olaylara neden olması olasıdiğer tıbbi ürünlerle eşzamanlı tedavi yapılması planlanıyorsa apremilast ile tedaviyebaşlamanın veya devam etmenin riskleri ve faydaları dikkatli bir şekilde değerlendirilmelidir.Hastalara ve bakıcılarına davranış veya ruh halinde herhangi bir değişikliği veya herhangi birintihar düşüncesini reçete yazan hekimlerine bildirmeleri söylenmelidir. Hastaların yeni veyakötüleşen psikiyatrik semptomlardan muzdarip olması veya intihar düşüncesi ya da intihargirişimi tespit edildiği durumlarda apremilast ile tedavinin bırakılması önerilir.
Zayıf hastalarTedavinin başlangıcında olması gerekenden zayıf hastaların vücut ağırlıkları düzenli olarak izlenmelidir. Açıklanmayan ve klinik anlamlı kilo kaybı durumunda bu hastalar bir hekimtarafından değerlendirilmeli ve tedavinin kesilmesi düşünülmelidir. Nadir kalıtımsal galaktoz intoleransı, Lapp laktaz yetmezliği ya da glukoz-galaktoz malabsorpsiyon problemi olan hastaların bu ilacı kullanmamaları gerekir. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriGüçlü sitokrom P450 3A4 (CYP3A4) enzim indükleyicisi rifampisinin eşzamanlı uygulanması apremilastın sistemik maruziyetinde bir azalma ile sonuçlanmıştır, bu daapremilast etkinliğinin kaybıyla sonuçlanabilir. Bu nedenle güçlü CYP3A4 enzimindükleyicilerinin (örn., rifampisin, fenobarbital, karbamazepin, fenitoin ve sarı kantaron)apremilast ile kullanımı önerilmemektedir. Apremilastın birden fazla rifampisin dozu ileeşzamanlı uygulanması apremilastın konsantrasyon zaman eğrisi altındaki alan (EAA) vemaksimum serum konsantrasyonu (Cmaks) değerlerinde sırasıyla yaklaşık %72 ve %43azalma ile sonuçlanmıştır. Apremilast maruziyeti güçlü CYP3A4 indükleyicileri (örn.,rifampisin) ile eşzamanlı uygulandığında azalır ve azalmış klinik yanıtla sonuçlanabilir. Klinik çalışmalarda, apremilast topikal tedavi (kortikosteroidler, katranlı şampuan ve salisilik asit kafa derisi preparatları dahil) ve UVB fototerapisi ile eşzamanlı olarak uygulanmıştır. Ketokonazol ve apremilast arasında klinik olarak anlamlı bir ilaç-ilaç etkileşimi olmamıştır. Apremilast ketokonazol gibi potent bir CYP3A4 inhibitörü ile birlikte uygulanabilir. Psöriatik artrit hastalarında apremilast ve metotreksat arasında farmakokinetik ilaç-ilaç etkileşimi yoktur. Apremilast metotreksat ile birlikte uygulanabilir. Apremilast ve etinil östradiol ve norgestimat içeren oral kontraseptifler arasında farmakokinetik ilaç-ilaç etkileşimi olmamıştır. Apremilast oral kontraseptiflerle birlikteuygulanabilir. Özel popülasyonlara ilişkin ek bilgiler Pediyatrik popülasyon:Çocuklarda herhangi bir etkileşim çalışması yapılmamıştır. 4.6 Gebelik ve laktasyonGenel tavsiyeGebelik kategorisi: C Çocuk doğurma potansiyeli bulunan kadınlar/Doğum kontrolü (Kontrasepsiyon)Tedaviye başlamadan önce hastanın gebe olup olmadığı mutlaka kontrol edilmelidir. Çocuk doğurma potansiyeli olan kadınlar tedavi sırasında gebeliği önlemek için etkili bir doğumkontrol yöntemi kullanmalıdır. Gebelik dönemiGebe kadınlarda apremilast kullanımı ile ilgili veriler sınırlıdır. Apremilast gebelik sırasında kontrendikedir. Apremilastın gebelik üzerindeki etkileri halihazırda önerilen en yüksek insan dozundan daha yüksek dozlarda fareler ve maymunlardaembriyofetal kaybı ve farelerde azalmış fetüs ağırlığı ve gecikmiş kemikleşmeyi içermiştir.Klinik maruziyetin 1.3 katı maruziyetle hayvanlarda bu tip etkiler gözlenmemiştir (bölüm5.3'e bakınız). Laktasyon dönemiApremilast laktasyondaki farelerin sütünde tespit edilmiştir (bölüm 5.3'e bakınız). Apremilastın ya da metabolitlerinin insanlarda anne sütüne geçip geçmediği bilinmemektedir.Emzirilen bebekler için risk dışlanamaz, bu nedenle apremilast emziren annelerdekullanılmamalıdır. Üreme yeteneği/Fertiliteİnsanlarda fertilite verisi yoktur. Farelerde yürütülen hayvan çalışmalarında klinik maruziyetin 3 katı maruziyet düzeylerinde erkeklerde ve klinik maruziyetin 1 katı maruziyetdüzeylerinde dişilerde hiçbir advers etki gözlenmemiştir. Klinik öncesi fertilite verileri içinbölüm 5.3'e bakınız. 4.7 Araç ve makine kullanımı üzerindeki etkilerApremilastın araç ve makine kullanma becerisi üzerinde bir etkisi yoktur ya da ihmal edilebilir düzeydedir. 4.8 İstenmeyen etkilerGüvenlilik profilinin özetiPsA ve PSOR'da apremilastın en yaygın bildirilen yan etkileri, ishal (%15.7) ve mide bulantısı (%13.9) dahil olmak üzere gastrointestinal (GI) bozukluklardır. En sık bildirilendiğer yan etkiler arasında üst solunum yolu enfeksiyonları (%8.4), baş ağrısı (%7.9) vegerilim tipi baş ağrısı (%7.2) yer alır ve çoğunlukla hafif ila orta şiddettedir. Gastrointestinal yan etkiler genellikle tedavinin ilk 2 haftasında meydana gelir ve genellikle 4 hafta içinde düzelir. Aşırı duyarlılık reaksiyonları apremilast klinik çalışmalarında nadiren gözlenmiştir (bölüm 4.3'e bakınız). Apremilast ile tedavi edilen hastalarda ortaya çıkan istenmeyen etkiler Tablo 2'de MedDRA organ sistemine göre sıralanmıştır. Advers ilaç reaksiyonları apremilast klinik geliştirme programından veriler temelinde belirlenmiştir. Advers ilaç reaksiyonlarının sıklıkları psöriatik artritte yürütülen dört Faz IIIçalışmanın (n=1945) veya psöriaziste yürütülen iki Faz III çalışmanın (n=1184) apremilastkollarında bildirilenlerdir (Tablo 2'de her iki veri havuzuna ait en yüksek sıklıkgösterilmektedir). Advers ilaç reaksiyonları aşağıda tanımlanan sıklığa göre listelenmiştir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek (<1/10.000); bilinmiyor (eldeki verilerden hareketletahmin edilemiyor) olarak sınıflandırılır.
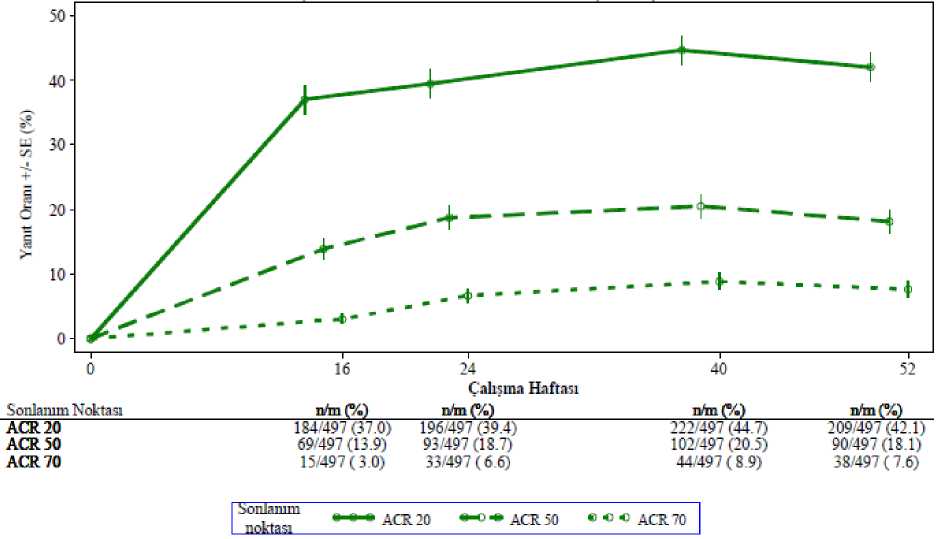
Seçili advers reaksiyonların tanımı Psikolojik bozukluklar#Klinik çalışmalarda ve pazarlama sonrası deneyimde, yaygın olmayan intihar düşüncesi ve davranışı vakaları bildirilirken, pazarlama sonrasında gerçekleştirilmiş intihar bildirilmiştir.Hastalara ve bakıcılarına herhangi bir intihar düşüncesini hekimlerine bildirmelerisöylenmelidir (bölüm 4.4'e bakınız). Kilo kaybıHasta kilosu klinik çalışmalarda rutin olarak ölçülmüştür. Apremilast ile 52 haftaya kadar tedavi edilen hastalarda ortalama gözlenen kilo kaybı 1.99 kg'dır. Apremilast alan hastalarıntoplamda %14.3'ünde %5-10 arası kilo kaybı gözlenirken, %5.7'sinde %10'dan fazla kilokaybı gözlenmiştir. Bu hastaların hiçbirinde kilo kaybına bağlı aşikar klinik sonuçlargelişmemiştir. Apremilast ile tedavi edilen hastaların toplamda %0.1'i azalmış kilo adversreaksiyonu nedeniyle ilacı bırakmıştır. Tedavinin başlangıcında zayıf olan hastalar için bölüm 4.4'teki ilave uyarıya bakınız. Özel popülasyonlara ilişkin ek bilgiler:Geriyatrik popülasyon:Pazarlama sonrası deneyime göre 65 yaş ve üstündeki hastalar şiddetli ishal, bulantı ve kusma komplikasyonları açısından daha yüksek risk taşıyabilir. Karaciğer yetmezliği:Karaciğer yetmezliği olan psöriatik artritli veya psöriazisli hastalarda apremilastın güvenliliği değerlendirilmemiştir. Böbrek yetmezliği:Psöriatik artrit veya psöriazis klinik çalışmalarında, hafif böbrek yetersizliği olan hastalarda gözlenen güvenlilik profili normal böbrek fonksiyonuna sahip hastalardakine benzerdir.Klinik çalışmalarda orta şiddette veya şiddetli böbrek yetersizliği olan psöriatik artritli veyapsöriazisli hastalarda apremilastın güvenliliği değerlendirilmemiştir. Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir (www.titck.gov.tr:4.9 Doz aşımı ve tedavisiApremilast sağlıklı gönüllülerde 4.5 gün boyunca 100 mg'lık (50 mg BID olarak verilen) maksimum toplam günlük dozda doz kısıtlayıcı toksisite kanıtı görülmeksizin araştırılmıştır.Bir doz aşımı durumunda hastanın advers etki bulgu ve belirtileri açısından izlenmesi veuygun semptomatik tedavinin başlatılması önerilir. Doz aşımı durumunda semptomatik vedestekleyici bakım tavsiye edilir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özelliklerFarmakoterapötik grup: Selektif immünosüpresantlar ATC kodu: L04AA32 Etki MekanizmasıBir oral küçük moleküllü fosfodiesteraz 4 (PDE4) inhibitörü olan apremilast bir pro-inflamatuar ve anti-inflamatuar mediyatör ağını düzenlemek üzere intraselüler olarak iş görür. PDE4, siklik adenozin monofosfata (cAMP) özgü bir PDE'dir ve inflamatuar hücrelerdeki baskın olan PDE'dir. PDE4 inhibisyonu intraselüler cAMP düzeylerini artırır, bu da TNF-a,IL-23, IL-17 ve diğer inflamatuar sitokinlerin ortaya çıkmasını düzenleyerek inflamatuaryanıtı azaltır. Siklik AMP aynı zamanda IL-10 gibi anti-inflamatuar sitokinlerin düzeylerinide düzenler. Bu pro-inflamatuar ve anti-inflamatuar mediyatörlerin psöriatik artrit vepsöriaziste işe karıştıkları gösterilmiştir. Farmakodinamik EtkilerPsöriatik artritli hastalarda yürütülen klinik çalışmalarda, apremilast IL-1a, IL-6, IL-8, MCP-1, MIP-1B, MMP-3 ve TNF-a'nın plazma protein düzeylerini tamamen inhibe etmemekle birlikte önemli ölçüde düzenlemiştir. Apremilast ile 40 haftalık tedaviden sonra IL-17 ve IL-23'ün plazma protein düzeylerinde bir azalma ve IL-10'da bir artış görülmüştür. Psöriazislihastalarda yürütülen klinik çalışmalarda apremilast lezyonlu deri epidermal kalınlığı,inflamatuar hücre infiltrasyonu ve indüklenebilir nitrik oksit sentaz (iNOS), IL-12/IL-23p40,IL-17A, IL-22 ve IL-8 için olanlar dahil pro-inflamatuar genlerin ekspresyonunu azaltmıştır.50 mg BID'e varan dozlarda uygulanan apremilast sağlıklı gönüllülerde QT aralığınıuzatmamıştır. Klinik çalışmalarPsöriatik ArtritApremilastın güvenliliği ve etkililiği, küçük moleküllü veya biyolojik DMARD'larla önceki tedaviye rağmen aktif psöriatik artritli (>3 şiş eklem ve >3 hassas eklem) yetişkin hastalardabenzer tasarıma sahip 3 çok merkezli, randomize, çift kör, plasebo kontrollü çalışmada(PALACE 1, PALACE 2 ve PALACE 3 Çalışmaları) değerlendirilmiştir. Toplamda 1493hasta randomize edilmiş ve plasebo, apremilast 20 mg ya da apremilast 30 mg ile günde ikikez oral yolla tedavi edilmiştir. Bu çalışmalardaki hastalar en az 6 aylık psöriatik artrit tanısına sahipti. PALACE 3'te ayrıca niteleyici bir psöriazis deri lezyonu da (en az 2 cm çapında) gerekliydi. Apremilast küçükmoleküllü DMARD'ların stabil dozları ile kombinasyon halinde (%65.2) veya monoterapi(%34.8) olarak kullanılmıştır. Hastalar şunların biri veya daha fazlası ile kombinasyonhalinde apremilast almıştır: metotreksat (MTX, < 25 mg/hafta, %54.5), sülfasalazin (SSZ, < 2g/gün, %9.0) ve leflunomid (LEF; < 20 mg/gün, %7.4). TNF blokerleri dahil olmak üzerebiyolojik DMARD'larla eşzamanlı tedaviye izin verilmemiştir. Simetrik poliartrit (%62.0),asimetrik oligoartrit (%26.9), distal interfalangeal (DIP) eklem artriti (%6.2), artritis mutilans(%2.7) ve baskın olarak spondilit (%2.1) dahil her bir psöriatik artrit alt tipindeki hastalar bu3 çalışmaya kaydedilmiştir. Önceden mevcut entezopati (%63) veya daktilit (%42) görülenhastalar kaydedilmiştir. Hastaların toplamda %76.4'ü daha önce sadece küçük moleküllüDMARD'larla tedavi edilirken, hastaların %22.4'ü daha önce biyolojik DMARD'larla tedaviedilmişti (bunların %7.8'inde önceki biyolojik DMARD ile terapötik başarısızlık vardı).Medyan psöriatik artrit hastalık süresi 5 yıldı. Çalışma tasarımına dayalı olarak, hassas ve şiş eklem sayımları 16. haftada en az %20 iyileşme göstermemiş hastalar yanıt vermeyenler olarak kabul edilmiştir. Yanıt vermeyenlerolarak kabul edilen plasebo hastaları 1:1 oranında körlenmiş bir şekilde ya günde iki kezapremilast 20 mg ya da günde iki kez 30 mg'a yeniden randomize edilmiştir. 24. haftada, tümgeri kalan plasebo ile tedavi edilen hastalar ya apremilast 20 ya da 30 mg BID'ye geçirilmiştir. 52 haftalık tedaviden sonra hastalar PALACE 1, PALACE 2 ve PALACE 3 çalışmalarının uzun vadeli uzatılması kapsamında açık etiketli apremilast 20 mg veya 30 mg'a5 yıla varan toplam tedavi süresince (260 hafta) devam edebilmiştir. Birincil sonlanım noktası 16. haftada Amerikan Romatoloji Derneği (ACR) 20 yanıtını elde eden hastaların yüzdesiydi. Apremilast ile tedavi 16. haftada plaseboya kıyasla ACR 20 yanıt kriterleri ile değerlendirilen psöriatik artrit bulgu ve semptomlarında anlamlı iyileşmelerle sonuçlanmıştır. 16. haftadagünde iki kez 30 mg apremilast için ACR 20/50/70 elde eden hastaların oranı (PALACE 1,PALACE 2 ve PALACE 3 Çalışmalarındaki yanıtlar ve PALACE 1, PALACE 2 ve PALACE3 çalışmaları için birleştirilmiş veriler) Tablo 3'te gösterilmektedir. ACR 20/50/70 yanıtları24. haftada devam etmiştir. Başlangıçta günde iki kez 30 mg apremilast tedavisine randomize edilmiş hastalar arasında ACR 20/50/70 yanıt oranları birleştirilmiş PALACE 1, PALACE 2 ve PALACE 3Çalışmalarında 52. haftaya kadar korunmuştur (Şekil 1). Tablo 3. 16. haftada PALACE 1, PALACE 2 ve PALACE 3 çalışmaları ve birleştirilmiş çalışmalarda ACR yanıtlarına sahip hastaların oranı
*NRI: Yanıt vermeyen. Zaman noktasından önce ayrılan ve zaman noktasında yanıt durumunun kesin bir şekilde belirlenmesi için yeterli veriye sahip olmayan denekler yanıtvermeyenler olarak sayılmaktadır. Başlangıçta günde iki kez 30 mg apremilasta randomize edilmiş 497 hasta içerisinden 375 hasta (%75) 52. haftada halen bu tedaviye devam etmekteydi. Bu hastalarda 52. haftadakiACR 20/50/70 yanıtları sırasıyla %57, %25 ve %11'di. Başlangıçta günde iki kez 30 mgapremilasta randomize edilen 497 hasta arasında 375 hasta (%75) uzun vadeli uzatmaçalışmalarına girmiş olup, bunlardan 221 hasta (%59) 260. haftada halen bu tedaviyialmaktaydı. ACR yanıtları uzun vadeli açık etiketli uzatma çalışmalarında 5 yıla kadarkorunmuştur. Apremilast ile tedavi edilen grupta gözlenen yanıtlar MTX dahil eşzamanlı DMARD kullanan ve kullanmayan hastalarda benzerdi. Daha önce DMARD'lar veya biyolojiklerle tedaviedilmiş apremilast alan hastalar 16. haftada plasebo alan hastalardan daha yüksek ACR 20yanıtı elde etmiştir. DIP dahil farklı psöriatik artrit alt tiplerinin görüldüğü hastalarda benzer ACR yanıtları gözlenmiştir. Artritis mutilans ve baskın spondilit alt tipleri görülen hastaların sayısı anlamlıdeğerlendirilebilmek için çok küçüktü. PALACE 1, PALACE 2 ve PALACE 3'te Hastalık Aktivitesi Ölçeği (DAS) 28 C-reaktif protein (CRP) ve modifiye psöriatik artrit yanıt kriterlerine (PsARC) elde eden hastalarınoranındaki iyileşmeler 16. haftada plaseboya kıyasla apremilast grubunda daha yüksekti(nominal p-değeri sırasıyla p<0.0017, p<0.0004). Bu iyileşmeler 24. haftada devam etmiştir. Çalışmanın başlangıcında randomize edildikleri apremilast tedavisini sürdüren hastalar arasında DAS28(CRP) skoru ve PsARC yanıtı 52. haftaya kadar korunmuştur. 16 ve 24. haftalarda apremilast ile tedavi edilen hastalarda psöriazis artritin periferik aktivite özelliğine ilişkin parametrelerde (örn., şiş eklem sayısı, ağrılı/hassas eklem sayısı, daktilit veentezit) ve psöriazisin deri bulgularında iyileşmeler görülmüştür. Çalışmanın başlangıcındarandomize edildikleri apremilast tedavisini sürdüren hastalarda bu iyileşmeler 52. haftayakadar korunmuştur. Açık etiketli uzatma çalışmalarında 5 yıla varan tedavi boyunca psöriazisin deri bulgularında ve aynı periferik aktivite parametrelerindeki klinik yanıtlar korunmuştur. Fiziksel fonksiyon ve sağlıkla ilişkili yaşam kalitesiApremilast ile tedavi edilen hastalar PALACE 1, PALACE 2 ve PALACE 3 çalışmalarında ve birleştirilmiş çalışmalarda 16. haftada plaseboya kıyasla sağlık değerlendirme anketininengellilik indeksinde (HAQ-DI) başlangıca göre değişiklik değerlendirildiğinde fizikselfonksiyonda istatistiksel olarak anlamlı bir iyileşme göstermiştir. HAQ-DI skorlarındakiiyileşme 24. haftada devam etmiştir. Başlangıçta günde iki kez 30 mg apremilast tedavisine randomize edilmiş hastalar arasında, PALACE 1, PALACE 2 ve PALACE 3 çalışmalarının açık etiketli fazının birleştirilmişanalizinde 52. haftadaki HAQ-DI skorunda başlangıca göre değişiklik günde iki kez 30 mgapremilast grubunda -0.333 idi. PALACE 1, PALACE 2 ve PALACE 3 çalışmalarında, 16 ve 24. haftalarda plaseboya kıyasla apremilast ile tedavi edilen hastalarda Kısa Form Sağlık Anketi versiyon 2'nin (SF-36v2)fiziksel işlevsellik (PF) domeninde ve Fonksiyonel Kronik Hastalık Tedavisi Değerlendirmesi- Yorgunluk (FACIT-yorgunluk) skorlarında başlangıca göre değişiklikte sağlıkla ilişkiliyaşam kalitesinde anlamlı iyileşmeler gösterilmiştir. Çalışmanın başlangıcında randomizeedildikleri apremilast tedavisini sürdüren hastalar arasında fiziksel fonksiyon ve FACIT-yorgunluktaki iyileşme 52. haftaya kadar korunmuştur. HAQ-DI ve SF36v2PF domeni ve FACIT-yorgunluk skorları ile değerlendirildiğinde iyileşmiş fiziksel fonksiyon açık etiketli uzatma çalışmalarında 5 yıllık tedavi boyuncakorunmuştur. PsöriazisApremilastın güvenliliği ve etkililiği, orta şiddette ila şiddetli plak psöriazisli, (> %10 vücut yüzey alanı (BSA) tutulumu görülen, Psöriazis Alan ve Ciddiyet İndeks (PASI) skoru > 12olan, statik Hekim Genel Değerlendirmesi (sPGA) > 3 (orta şiddette veya şiddetli) vefototerapi ya da sistemik tedaviye aday, toplamda 1257 hastanın kaydedildiği iki çokmerkezli, randomize, çift kör, plasebo kontrollü çalışmada (ESTEEM 1 ve ESTEEM 2Çalışmaları) değerlendirilmiştir. Bu çalışmalar 32. haftaya kadar benzer tasarıma sahiptir. Her iki çalışmada da hastalar 2:1 oranında 16 hafta boyunca apremilast 30 mg BID veya plaseboya (plasebo kontrollü faz)randomize edilmiş ve 16-32. haftalar arası tüm hastalar apremilast 30 mg BID (idame fazı)almıştır. Randomize Tedavinin Geri Çekilme Fazı sırasında (32-52. haftalar) orijinal olarakapremilasta randomize edilmiş, PASI skorlarında %75 azalma (PASI-75) (ESTEEM 1) veyaPASI skorlarında %50 azalma (PASI-50) (ESTEEM 2) elde etmiş hastalar 32. haftadayeniden plasebo veya apremilast 30 mg BID'ye randomize edilmiştir. Plaseboya yenidenrandomize edilmiş ve 32. haftada PASI-75 yanıtını (ESTEEM 1) kaybetmiş veya PASIiyileşmesinin %50'sini kaybetmiş hastalar (ESTEEM 2) apremilast 30 mg BID ile yenidentedavi edilmiştir. 32. haftaya kadar belirlenen PASI yanıtına ulaşmamış veya başlangıçtaplaseboya randomize edilmiş hastalar, 52. haftaya kadar apremilast tedavisinde kalmıştır.Çalışmalarda yüz, koltukaltı ve kasıkta düşük potensli topikal kortikosteroid, katranlışampuan ve/veya salisilik asit saç derisi preparatlarının kullanılmasına izin verilmiştir. Bunailaveten, 32. haftada, ESTEEM 1'de PASI-75 yanıtı veya ESTEEM 2'de PASI-50 yanıtı eldeetmemiş deneklerin apremilast 30 mg BID tedavisine ek olarak topikal psöriazis tedavilerive/veya fototerapi kullanmalarına izin verilmiştir. 52 haftalık tedaviden sonra, hastalar ESTEEM 1 ve ESTEEM 2 çalışmalarının uzun vadeli uzatma çalışmalarıyla 5 yıla kadar toplam tedavi süresi boyunca (260 hafta) açık etiketliapremilast 30 mg kullanmaya devam edebilmiştir. Her iki çalışmada da birincil sonlanım noktası 16. haftada PASI-75 elde eden hastaların oranıdır. Majör ikincil sonlanım noktası 16. haftada temiz (0) ya da neredeyse temiz (1) sPGAskoru elde etmiş hastaların oranıdır. Ortalama başlangıç PASI skoru 19.07 (medyan 16.80) olup, başlangıçta sPGA skoru 3 (orta şiddette) ve 4 (şiddetli) olan hastaların oranı %25.19'luk ortalama BYA tutulumu (medyan%21.0) ile sırasıyla %70.0 ve %29.8 idi. Tüm hastaların yaklaşık %30'u psöriazis tedavisiiçin daha önce fototerapi ve %54'ü daha önce konvansiyonel sistemik ve/veya biyolojiktedavi görmüş olup (tedavi başarısızlıkları dahil), %37'si daha önce konvansiyonel sistemiktedavi ve %30'u daha önce biyolojik tedavi görmüştü. Hastaların yaklaşık üçte biri daha öncefototerapi, konvansiyonel sistemik veya biyolojik tedavi görmemişti. Hastaların toplamda%18'i psöriatik artrit öyküsüne sahipti. PASI-50, -75 ve -90 yanıtları elde eden ve sPGA skoru temiz (0) veya neredeyse temiz (1) olan hastaların oranı aşağıdaki Tablo 4'te sunulmaktadır. Apremilast ile tedavi plaseboyakıyasla 16. haftada PASI-75 yanıtına sahip hastaların oranı ile gösterildiği üzere orta şiddetteila şiddetli plak tipi psöriaziste anlamlı iyileşme ile sonuçlanmıştır. 16. haftada sPGA, PASI-50 ve PASI-90 yanıtları ile ölçülen klinik iyileşme gösterilmiştir. Buna ilaveten apremilastkaşıntı, tırnak hastalığı, saç derisi tutulumu ve yaşam kalitesi ölçümleri dahil olmak üzereçeşitli psöriazis bulgularında tedavi faydası göstermiştir. Tablo 4. ESTEEM 1 ve ESTEEM 2 çalışmalarında 16. haftadaki klinik yanıt (FAS a, LOCFb)
* Sırasıyla p=0.0042 ve p=0.0078 değerleri ile ESTEEM 2 PASI 90 ve SF-36 MCS'deki Değişiklik haricinde plaseboya karşı apremilast için p<0.0001 a FAS = Tam Analiz Seti b LOCF = İleri Taşınan Son Gözlem c PASI = Psöriazis Psöriazis Alan ve Ciddiyet İndeksi d sPGA = Statik Hekim Genel Değerlendirmesi e BSA = Vücut Yüzey Alanı fVS = Görsel Analog Ölçek; o = en iyi, 100 = en kötü g DLQI = Dermatoloji Yaşam Kalitesi İndeksi; o = en iyi, 30 = en kötüh SF-36 MCS = Medikal Sonuç Çalışması Kısa Form 36-Maddeli Sağlık Anketi, RuhsalBileşen Özeti Apremilastın klinik faydası başlangıç demografikleri ve başlangıç klinik hastalık özellikleri ile tanımlanan (psöriazis hastalığı süresi ve psöriatik artrit öyküsü olan hastalar dahil) çoksayıda alt grup arasında gösterilmiştir. Apremilastın klinik faydası aynı zamanda öncekipsöriazis ilacı kullanımı ve önceki psöriazis tedavilerine verilen yanıta bakılmaksızıngösterilmiştir. Tüm kilo aralıklarında benzer yanıt oranları gözlenmiştir. Apremilasta yanıt, 2. hafta itibariyle plaseboya kıyasla PASI, deride rahatsızlık/ağrı ve kaşıntı dahil psöriazis bulgu ve semptomlarında anlamlı olarak daha fazla iyileşmelerle hızlıydı.Genelde PASI yanıtları 16. hafta itibariyle elde edilmiş ve 32. haftaya kadar devam etmiştir.Her iki çalışmada da PASI'da başlangıca göre ortalama yüzde iyileşme 32. haftadaapremilasta yeniden randomize edilmiş hastalar için Randomize Tedaviden Çekilme Fazısırasında stabil kalmıştır (Tablo 5). Tablo 5. 0. Haftada APR 30 BID'ye randomize edilmiş ve 32. hafta ila 52. haftada APR 30 BID'ye yeniden randomize edilmiş deneklerdeki etkinin kalıcılığı
a Değerlendirilen çalışma haftasında başlangıç değeri ve başlangıç sonrası değere sahip 32. haftada APR 30 BID'ye yeniden randomize edilmiş denekleri içerir. b N, 32. haftada APR 30 BID'ye yeniden randomize edilmiş başlangıçta orta şiddette veya daha fazla saç derisi psöriazisi olan deneklere dayanmaktadır. Verileri eksik denekler yanıtvermeyenler olarak sayılmıştır. ESTEEM 1 Çalışmasında, 32. haftada apremilasta yeniden randomize edilmiş hastaların yaklaşık %61'i 52. haftada PASI-75 yanıtına sahipti. Randomize Tedaviden Geri ÇekilmeFazı sırasında 32. haftada plaseboya yeniden randomize edilmiş en azından PASI-75 yanıtınasahip hastalardan %11.7'si 52. haftada PASI-75 yanıtı veren hastalardı. Plaseboya yenidenrandomize edilmiş hastalar arasında PASI-75 yanıtı kaybına kadar geçen medyan süre 5.1haftaydı. ESTEEM 2 Çalışmasında, 32. haftada apremilasta yeniden randomize edilmiş hastaların yaklaşık %80.3'ü 52. haftada PASI-50 yanıtına sahipti. 32. haftada plaseboya yenidenrandomize edilmiş en az PASI-50 yanıtına sahip hastalardan %24.2'si 52. haftada PASI-50yanıtı veren hastalardı. 32. hafta PASI iyileşmelerinde %50 kayba kadar geçen medyan süre12.4 haftaydı. 32. haftada randomize tedaviden geri çekilme sonrasında, ESTEEM 1 Çalışmasındaki hastaların yaklaşık %70'i ve ESTEEM 2 Çalışmasındaki hastaların %65.6'sı apremilasttedavisinin yeniden başlatılmasından sonra PASI-75 (ESTEM 1) veya PASI-50 (ESTEEM 2)yanıtlarını yeniden kazanmıştır. Çalışma tasarımından dolayı yeniden tedavinin süresideğişken olup, 2.6 ile 22.1 hafta arasında değişmiştir. ESTEEM 1 Çalışmasında, çalışmanın başlangıcında apremilasta randomize edilmiş 32. haftada PASI-75 yanıtı elde etmemiş hastaların, 32 ve 52. haftalar arasında eşzamanlı topikaltedaviler ve/veya UVB fototerapisi kullanmalarına izin verilmiştir. Bu hastalardan %12'si 52.haftada apremilast artı topikal tedavi ve/veya fototerapi ile PASI-75 yanıtı elde etmiştir. ESTEEM 1 ve ESTEEM 2 Çalışmalarında, 16. haftada plasebo ile tedavi edilen hastalara kıyasla apremilast alan hastalarda Tırnak Psöriazis Şiddet İndeksinde (NAPSI) başlangıcagöre ortalama değişiklik yüzdesi ile ölçüldüğü üzere tırnak psöriazisinde anlamlı iyileşmeler(azalmalar) gözlenmiştir (sırasıyla p<0.0001 ve p=0.0052). Apremilast ile tedavi edilmeyedevam eden hastalarda 32. haftada tırnak psöriazisinde ilave iyileşmeler gözlenmiştir. ESTEEM 1 ve ESTEEM 2 Çalışmalarında plasebo ile tedavi edilen hastalara kıyasla apremilast alan hastalarda 16. haftada Saç Derisi Psöriazisi Hekimin Genel Değerlendirmesi(ScPGA) temiz (0) ya da minimum (1) skoru elde etmiş hastaların oranı ile ölçüldüğü üzereen az orta şiddette (>3) saç derisi psöriazisinde anlamlı iyileşmeler gözlenmiştir. İyileşmelerapremilasta yeniden randomize edilmiş deneklerde 32. haftadan 52. haftaya genelliklekorunmuştur (Tablo 5). ESTEEM 1 ve ESTEEM 2 Çalışmalarında, plasebo ile tedavi edilen hastalara kıyasla apremilast alan hastalarda Dermatoloji Yaşam Kalitesi İndeksi (DLQI) ve SF-36v2MCS ileölçülen yaşam kalitesinde anlamlı iyileşmeler görülmüştür (Tablo 4). DLQI'daki iyileşmeler32. haftada apremilasta yeniden randomize edilmiş deneklerde 52. haftaya kadar korunmuştur(Tablo 5). Buna ilaveten, ESTEEM 1 Çalışmasında, plaseboya kıyasla apremilast alanhastalarda İş Kısıtlamaları Anketinde (WLQ-25) anlamlı iyileşme elde edilmiştir. Başlangıçta günde iki kez 30 mg apremilasta randomize edilmiş 832 hasta arasında 443 hasta (%53) ESTEEM 1 ve ESTEEM 2'nin açık etiketli uzatma çalışmalarına girmiş olup,bunlardan 115 hasta (%26) 260. haftada halen tedavi görmekteydi. ESTEEM 1 ve ESTEEM 2çalışmalarının açık etiketli uzatmasında apremilasta devam eden hastalar için PASI skoru,etkilenmiş BSA, kaşıntı, tırnak ve yaşam kalitesi ölçümlerinde iyileşmeler genellikle 5 yılakadar korunmuştur. Psöriatik artrit ve psöriazis görülen hastalarda günde iki kez 30 mg apremilastın uzun dönem güvenliliği 5 yıla varan toplam tedavi süresince değerlendirilmiştir. Apremilast ile açıketiketli uzatma çalışmalarında uzun dönemli deneyim genellikle 52 haftalık çalışmalardakinebenzer olmuştur. 5.2 Farmakokinetik özelliklerGenel ÖzelliklerEmilimApremilast yaklaşık 2.5 saatlik medyan sürede (tmaks) meydana gelen pik plazma konsantrasyonları (Cmaks) eşliğinde yaklaşık %73 mutlak oral biyoyararlanım ile iyi düzeydeemilmektedir. Apremilast farmakokinetiği doğrusal olup, günlük 10 ile 100 mg doz aralığındasistemik maruziyette dozla orantılı bir artış söz konusudur. Birikim apremilast günde bir kezuygulandığında minimum ve günde iki kez uygulandığında sağlıklı gönüllülerde yaklaşık%53 ve psöriazisli gönüllülerde %68'dir. Gıdalarla birlikte uygulama biyoyararlanımıdeğiştirmediğinden apremilast aç veya tok karnına uygulanabilir. DağılımApremilastın insan plazma proteinine bağlanması yaklaşık %68'dir. Ortalama görünür dağılım hacmi (Vd) 87L olup, ekstravasküler dağılıma işaret eder. BiyotransformasyonApremilast oksidasyon, hidroliz ve konjugasyon dahil yaygın olarak hem CYP aracılı hem de CYP dışı yolaklarla metabolize edilir, bu da tek bir klirens yolağının inhibisyonunun belirginilaç-ilaç etkileşimine neden olmasının muhtemel olmadığını düşündürür. Apremilastınoksidatif metabolizmasına başlıca CYP3A4 aracılık ederken CYP1A2 ve CYP2A6'dan minörkatkılar söz konusudur. Apremilast oral uygulamayı takiben dolaşımdaki majör bileşendir.Apremilast, uygulanan ana bileşiğin sırasıyla sadece % 3 ve % 7'sinin idrar ve dışkıda gerikazanılmasıyla yoğun bir metabolizmaya maruz kalır. Dolaşımdaki majör inaktif metabolit O-demetile apremilastın glukuronid konjugatıdır (M12). Apremilastın bir CYP3A4 substratıolması ile tutarlı olarak apremilast maruziyeti güçlü bir CYP3A4 indükleyicisi olan rifampisinile eşzamanlı uygulandığında azalır. İn vitro apremilast sitokrom P450 enzimlerinin inhibitörü ya da indükleyicisi değildir. Bu nedenle CYP enzimlerinin substratları ile birlikte uygulanan apremilastın CYP enzimleri ilemetabolize edilen etkin maddelerin klirensini ve maruziyetini etkilemesi olası değildir. İn vitro apremilast bir P-glikoprotein substratı ve zayıf inhibitörüdür (IC50>50^M), bununla birlikte P-gp aracılığıyla klinik açıdan ilgili ilaç etkileşimlerinin meydana gelmesibeklenmemektedir. İn vitro apremilast Organik Anyon Taşıyıcısı (OAT)1 ve OAT3, Organik Katyon Taşıyıcısı (OKT)2, Organik Anyon Taşıyıcısı Polipeptid (oATP)1B1 ve OATP1B3 veya meme kanseridirenç proteini (BCRP) üzerinde inhibe edici etkiye sahip değildir veya çok az etki sözkonusudur ve bu taşıyıcılar için bir substrat değildir. Bu nedenle apremilast bu taşıyıcılarınsubstratları veya inhibitörleri ile birlikte uygulandığında klinik açıdan ilgili ilaç-ilaçetkileşimleri olası değildir. EliminasyonApremilastın plazma klirensi yaklaşık 9 saatlik bir terminal eliminasyon yarılanma ömrü ile sağlıklı kişilerde ortalama yaklaşık 10 L/saattir. Radyo-etiketli apremilastın oral yollauygulanmasını takiben radyoaktivitenin yaklaşık %58 ve %39'u sırasıyla idrar ve feçeste gerikazanılırken, radyoaktif dozun sırasıyla %3'ü ve %7'si idrar ve feçeste apremilast olarak gerikazanılmıştır. Özel popülasyonlar:Geriyatrik popülasyon:Apremilast genç ve yaşlı sağlıklı gönüllülerde araştırılmıştır. Yaşlı gönüllülerde (65 ila 85 yaş) maruziyet genç gönüllülere (18 ila 55 yaş) kıyasla apremilast için EAA'da yaklaşık %13daha yüksek ve Cmaks'ta yaklaşık %6 daha yüksektir. Klinik çalışmalarda 75 yaş üzerindekigönüllülerde farmakokinetik veriler sınırlıdır. Yaşlı hastalar için herhangi bir doz ayarlamasıgerekli değildir. Böbrek yetmezliği:Hafif veya orta şiddette böbrek yetmezliği olan gönüllüler ve eşleştirilmiş sağlıklı gönüllüler arasında apremilastın FK'sında anlamlı bir fark yoktur (her biri N=8). Bulgular hafif ve ortaşiddette böbrek yetmezliği olan hastalarda doz ayarlaması gerekmediğini desteklemektedir.Şiddetli böbrek yetmezliği (eGFR <30 mL/dakika/1.73 m2 veya CLcr < 30 mL/dakika) olanhastalarda apremilast dozu günde bir kez 30 mg'a azaltılır. 30 mg'lık tek doz apremilastuygulanan şiddetli böbrek yetmezliği olan 8 gönüllüde apremilastın EAA ve Cmaks değerlerisırasıyla yaklaşık %89 ve %42 artmıştır. Karaciğer yetmezliği:Apremilast ve majör metaboliti M12'nin farmakokinetiği orta şiddette veya şiddetli karaciğer yetmezliğinden etkilenmez. Karaciğer yetmezliği olan hastalar için herhangi bir dozayarlaması gerekli değildir. 5.3 Klinik öncesi güvenlilik verileriKlinik dışı veriler konvansiyonel güvenlilik farmakolojisi ve tekrarlanan doz toksisitesi çalışmaları temelinde insanlar için özel bir tehlikeyi ortaya koymamıştır. İmmünotoksikpotansiyel, dermal irritasyon veya fototoksik potansiyel için kanıt yoktur. Fertilite ve erken embriyonik gelişimBir erkek fare fertilite çalışmasında 1, 10, 25 ve 50 mg/kg/günlük oral dozlarda apremilast erkek fertilitesi üzerinde bir etkiye yol açmamıştır; erkek fertilitesi için advers etkigözlenmeyen düzey (NOAEL) 50 mg/kg/günden fazladır (klinik maruziyetin 3 katı). 10, 20 ,40 ve 80 mg/kg/günlük oral dozajlarla birleştirilmiş bir dişi fare fertilitesi ve embriyo-fetal gelişimsel toksisite çalışmasında 20 mg/kg/gün ve üzerinde östrus sikluslarda uzama ve çiftleşmeye kadar geçen sürede artış gözlenmiştir; buna rağmen farelerin tümü çiftleşmiş vegebelik oranları etkilenmemiştir. Dişi fertilitesi için advers etki gözlenmeyen düzey (NOEL)10 mg/kg/gündür (klinik maruziyetin 1.0 katı). Embriyo-fetal gelişim10, 20, 40 ve 80 mg/kg/günlük oral dozajlarla birleştirilmiş bir dişi fare fertilitesi ve embriyo-fetal gelişimsel toksisite çalışmasında maternal hayvanların mutlak ve/veya nispi kalp ağırlıkları 20, 40 ve 80 mg/kg/günde artmıştır. Artmış erken rezorpsiyon sayısı ve azalmışkemikleşmiş tarsal sayısı 20, 40 ve 80 mg/kg/günde gözlenmiştir. 40 ve 80 mg/kg/gündesupraoksipital kafatası kemiğinin osifikasyonunda gecikme ve azalmış fetal ağırlıklargözlenmiştir. Farede maternal ve gelişimsel NOEL 10 mg/kg/gündür (klinik maruziyetin 1.3katı). Bir maymun embriyo-fetal gelişimsel toksisite çalışmasında 20, 50, 200 ve 1000 mg/kg/günlük oral dozajlar 50 mg/kg/gün ve üzerindeki dozajlarda prenatal kayıpta (düşük)dozla ilişkili bir artışla sonuçlanmıştır; 20 mg/kg/günde (klinik maruziyetin 1.4 katı) prenatalkayıpta test maddesi ile ilişkili bir etki gözlenmemiştir. Pre-natal ve post-natal gelişimBir pre-natal ve post-natal çalışmada apremilast gebe dişi farelere 10, 80 ve 300 mg/kg/gün dozajlarında 6. gestasyon gününden (GG) laktasyonun 20. gününe kadar oral yollauygulanmıştır. 300 mg/kg/günde maternal vücut ağırlığı ve kilo artışında azalmalar veyavruların doğurulmasında güçlükle ilişkili bir ölüm gözlenmiştir. Yavruların doğumu ileilişkili maternal toksisitenin fiziksel belirtileri aynı zamanda 80 ve 300 mg/kg/günün herbirinde bir farede gözlenmiştir. >80 mg/kg/günde (klinik maruziyetin >4.0 katı) artmış prenatal ve postnatal yavru ölümleri ve laktasyonun ilk haftası sırasında azalmış yavru vücutağırlıkları gözlenmiştir. Gebelik süresi, gestasyon periyodunun sonunda gebe farelerin sayısı,yavru doğuran farelerin sayısı üzerinde apremilast ile ilişkili etkiler veya doğum sonrası 7.gün ötesinde yavrularda herhangi bir gelişimsel etkiler söz konusu değildir. Postnatalperiyodun ilk haftası sırasında gözlenen yavru gelişimsel etkileri muhtemelen apremilast ileilişkili yavru toksisitesi (azalmış yavru kilosu ve canlılığı) ve/veya maternal bakım eksikliği(yavruların midesinde süt olmaması açısından daha yüksek insidans) ile ilişkilidir. Tümgelişimsel etkiler postnatal periyodun ilk haftası sırasında gözlenmiştir; sütten kesme öncesive sonrası periyotların geri kalanında cinsel olgunlaşma, davranış, çiftleşme, fertilite ve uterin parametreleri dahil olmak üzere apremilastla ilişkili hiçbir etki görülmemiştir. Maternal toksisite ve F1 jenerasyonu için faredeki NOEL 10 mg/kg/gündür (klinik EAA'mn 1.3 katı). Karsinojenisite çalışmalarıFareler ve sıçanlarda karsinojenisite çalışmaları apremilast tedavisi ile ilişkili karsinojenisite kanıtı göstermemiştir. Genotoksisite çalışmalarıApremilast genotoksik değildir. Apremilast Ames analizinde mutasyonları veya metabolik aktivasyon varlığında veya yokluğunda kültürlenmiş insan periferik kan lenfositlerindekromozom anomalilerini indüklememiştir. Apremilast 2000 mg/kg/güne varan dozlarda invivo fare mikronukleus analizinde klastojenik bulunmamıştır. Diğer çalışmalarİmmünotoksik, dermal iritasyon veya fototoksik potansiyele dair kanıta rastlanmamıştır. 6.FARMASOTIK ÖZELLİKLER6.1 Yardımcı maddelerin listesiAPREMİLAST 10 mg film kaplı tabletTablet çekirdeğiMikrokristalin selülozLaktoz monohidrat (sığır kaynaklı)Kroskarmelloz sodyumSilikon dioksitMagnezyum stearatFilm kaplama maddesiPolivinil alkolTitanyum dioksit (E171)Makrogol 3350 Talk Demir oksit kırmızı (E172) Demir oksit sarı (E172) APREMİLAST 20 mg film kaplı tabletTablet çekirdeğiMikrokristalin selülozLaktoz monohidrat (sığır kaynaklı)Kroskarmelloz sodyumSilikon dioksitMagnezyum stearatFilm kaplama maddesiPolivinil alkolTitanyum dioksit (E171)Makrogol 3350 Talk Demir oksit kırmızı (E172) Demir oksit sarı (E172) APREMİLAST 30 mg film kaplı tabletTablet çekirdeğiMikrokristalin selüloz Laktoz monohidrat (sığır kaynaklı) Kroskarmelloz sodyum Silikon dioksitMagnezyum stearat Film kaplama maddesiPolivinil alkolTitanyum dioksit (E171)Makrogol 3350 Talk Demir oksit kırmızı (E172) Demir oksit sarı (E172) Demir oksit siyah (E172) 6.2 GeçimsizliklerGeçerli değil. 6.3 Raf ömrü24 ay 6.4 Saklamaya yönelik özel tedbirler25°C altındaki oda sıcaklığında saklayınız. 6.5 Ambalajın niteliği ve içeriğiTedaviye başlama paketi 27 film kaplı tablet içerir (4x10 mg, 4x20 mg, 19x30 mg). Film kaplı tabletler şeffaf PVC/PE/PVdC - alüminyum folyo blister ambalajlarda kullanma talimatı ile birlikte sunulmaktadır. 6.6 Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliğine uygun olarak imha edilmelidir. 7. RUHSAT SAHİBİADEKA İlaç Sanayi ve Ticaret A.Ş. 55020 - İlkadım / SAMSUN 8. RUHSAT NUMARASI2023/145 9. İLK RUHSAT TARİHİ/RUHSAT YENİLEME TARİHİİlk ruhsatlandırma tarihi: 25.04.2023 Ruhsat Yenileme Tarihi: 10. KÜB'ÜN YENİLENME TARİHİ |
İlaç BilgileriApremia 10mg + 20mg + 30mg Film Kaplı Tablet Tedaviye Başlama PaketiEtken Maddesi: Apremilast Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2024 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.