Immunate 1000/750 Iu Iv İnfüzyonluk Toz İçeren Flakon Kısa Ürün BilgisiKISA ÜRÜN BİLGİSİV Bu ilaç ek izlemeye tabidir. Bu üçgen yeni güvenlilik bilgisinin hızlı olarak belirlenmesini sağlayacaktır. Sağlık mesleği mensuplarının şüpheli advers reaksiyonları TÜFAM'abildirmeleri beklenmektedir. Bakınız Bölüm 4.8 Advers reaksiyonlar nasıl raporlanır? 1. BEŞERİ TIBBİ ÜRÜNÜN ADIIMMUNATE 1000 IU/750 IU IV infüzyonluk toz içeren flakon Steril 2. KALİTATİF VE KANTİTATİF BİLEŞİMEtkin madde:İnsan koagülasyon faktörü VIII ve Von Willebrand Faktörü IMMUNATE 1000, kuru toz şeklinde nominal olarak 1000 IU insan koagülasyon faktörü VIII1ve 750 IU insan plazması kaynaklı von Willebrand faktörü (vWF:RCo)2 içeren bir flakon ileçözücü içerir.Ürün 10 mL steril enjeksiyonluk su ile seyreltilerek kullanıma hazır hale getirildikten sonra yaklaşık 100 IU/mL insan plazması kaynaklı koagülasyon faktörü VIII ve 75 IU/mL insanplazması kaynaklı von Willebrand faktörü içerir. Faktör VIII potensi Avrupa Farmakopesi kromojenik testiyle belirlenmiştir. IMMUNATE'in spesifik aktivitesi 70 ± 30 IU FVIII/mg protein'dir.3 vWF potensi Avrupa Farmakopesiristosetin kofaktör testiyle (vWF:RCo) belirlenmiştir. İnsan donörlerin plazmasından üretilmiştir. 1. Faktör VIII potensi, Dünya Sağlık Örgütünün faktör VIII konsantreleri için belirlediğistandartlara göre belirlenmiştir.2. von Willebrand faktörünün ristosetin kofaktör potensi Dünya Sağlık Örgütünün vonWillebrand faktör konsantresi standartlarına göre belirlenmiştir.3. Stabilizansız (albuminsiz); Oran 1:1 olduğunda faktör VIII aktivitesinin von Willebrandfaktör-antijenine maksimum spesifik aktivitesi protein başına 100 IU faktör VIII'dir.Yardımcı madde(ler):Flakon başına 19,6 mg sodyum Yardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMKuru toz ve enjeksiyonluk çözelti hazırlamak için çözücü. Beyaz veya soluk sarı toz veya ufalanabilir katı madde. 4. KLİNİK ÖZELLİKLERİ4.1. Terapötik endikasyonları- Konjenital (hemofili A) ya da edinilmiş faktör VIII eksikliğinin yol açtığı kanamaların tedavisi ve profilaksisi. - Von WiUebrand hastalığı'na karşı etkili spesifik faktör konsantresi yokluğunda ve tek başına desmopressin yanıtının olmadığı veya desmopressinin kontrendike olduğu Faktör VIII eksikliğibulunan von Willebrand hastalığı kanama tedavisinde. 4.2. Pozoloji ve uygulama şekliTedavi, kanama hastalıklarının tedavisinde deneyimli bir hekimin denetiminde başlatılmalıdır. Tedavi takibiTedavi süresince uygulanacak dozun ve tekrarlı infüzyonların sıklığına ilişkin kılavuzluk etmek için faktör VIII düzeylerinin uygun biçimde belirlenmesi tavsiye edilir. Özellikle majörcerrahi müdahaleler durumunda yerine koyma tedavisinin pıhtılaşma analizi (plazma faktörVIII aktivitesi) yoluyla kesin takibi önceliklidir. Faktör VIII'e yanıtların hastadan hastayadeğişiklik göstermesi, farklı yarılanma ömürleri ve iyileşmelere işaret etmektedir. Vücutağırlığına dayalı doz, düşük kilolu veya aşırı kilolu hastalarda ayarlama gerektirebilir. Pozoloji/uygulama sıklığı ve süresi:Hemofiü A Hastalarında DozDoz ve uygulama süresi FVIII eksikliğinin ciddiyetine, kanamanın yerine ve yayılımına, hastanın klinik durumuna dayanır. Uygulanan FVIII'in ünite sayısı, faktör VIII ürünleri için mevcut Dünya Sağlık Örgütü standardına göre Uluslararası Ünite (IU) terimiyle ifade edilir. Plazmadaki FVIII aktivitesi, yayüzdesel olarak (normal insan plazmasına göre) ya da uluslararası ünite olarak (plazmadaki FVIII uluslararası standartlarına göre) ifade edilir. Bir Uluslararası Ünite (IU) faktör VIII aktivitesi, 1 mL normal insan plazmasındaki faktör VIII miktarına eşittir. Gereken faktör VIII dozlarının hesaplaması, vücut ağırlığının her kilogramı için 1 IU faktör VIII verildiğinde, plazma faktör VIII aktivitesinin normal aktivitenin yaklaşık %2'si kadararttığına dair ampirik bulguya dayanmaktadır. Gereken doz aşağıdaki formülle hesaplanır: Gereken ünite = vücut ağırlığı (kg) x arzulanan faktör VIII artışı (%) x 0,5Uygulanacak miktar ve uygulama sıklığı, her bir olguda ortaya çıkan klinik etkiye dayandırılarak değerlendirilmelidir. Kanamalar ve Cerrahi GirişimlerAşağıda yer alan hemorajik olaylarda, faktör VIII aktivitesi, karşılık gelen periyotta verilen plazma aktivite düzeylerinin (normale göre % olarak veya IU/dl olarak) altına düşmemelidir. Aşağıdaki tablo, kanama ataklarında ve cerrahi girişimlerde dozajın belirlenebilmesi için Bu belge Belge DckıiSvuzcouar&W fey-İ^ftiJftH^^EİRGSSaklUSHYSaklU Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys
Bazı durumlarda (örneğin düşük titreli inhibitör varlığında) formül kullanılarak hesaplanan dozdan daha fazlası gerekebilir. Uzun Süreli ProfilaksiAğır hemofili A hastalarında kanamaya karşı uzun dönem profilaktisi için, genel dozlar 2-3 günlük aralıklarda bir vücut ağırlığının her kilogramı için 20-40 IU faktör VIII'dir. Bazıvakalarda, özellikle küçük yaştaki hastalarda daha sık doz aralıkları ya da daha yüksek dozlargerekli olabilir. von Willebrand Hastalığında DozHemorajileri kontrol etmek IMMUNATE replasman tedavisinde, hemofili A için verilen tedavi kılavuzuna uyulmalıdır. IMMUNATE vWF'ye kıyasla daha yüksek miktarlarda faktör VIII içerdiğinden, tedaviyi yürüten hekim, tedavinin IMMUNATE ile sürdürülmesi durumunda faktör VIII:Cdüzeylerinin tromboz riskinde artışa yol açabilecek şekilde aşırı yükselebileceğini göz önündetutmalıdır. Pedi^yatrik popülasyonFaktör VIII ürünlerine maruziyetleri sınırlı olan 6 yaşından küçük çocuklarda bu hasta grubunda sınırlı veri mevcut olduğundan ürün dikkatle kullanılmalıdır. 18 yaşından küçük çocuklar ve ergenlerde hemofili A'da dozaj, vücut ağırlığına dayalıdır ve dolayısıyla genellikle yetişkinlere yönelik aynı kılavuzlara dayanmaktadır. Uygulama miktarıve sıklığı daima bireysel vakalardaki klinik etkililiğe göre yönlendirilmelidir (bkz. Bölüm4.4). Bazı vakalarda, özellikle daha küçük hastalarda daha kısa doz aralıkları veya dahayüksek dozlar gerekebilir. Uygulama şekli:İntravenöz infüzyonla uygulanır. IMMUNATE intravenöz yoldan yavaş bir şekilde uygulanmalıdır. Maksimum infüzyon hızı 2 mL/dk'yı geçmemelidir. Tıbbi ürünü kullanmadan veya uygulamadan önce alınması gereken önlemlerTıbbi ürünün uygulamadan önce sulandırılmasına ilişkin talimatlar için lütfen bölüm 6.6'ya bakınız. Özel popülasyonlara ilişkin ek bilgiler:Böbrek /Karaciğer yetmezliği:Ek bilgi bulunmamaktadır.Pediyatrik popülasyon:Doz kg başına ünite cinsinden belirlenmekte olduğundan çocuklarda özel bir kullanım şekli yoktur.Geriyatrik popülasyon:4.3. KontrendikasyonlarEtkin maddeye veya bölüm 6.1'de listelenen yardımcı maddelerden herhangi birine karşı aşırı duyarlılık. 4.4. Özel kullanım uyarıları ve önlemleriVirüs güvenliği:IMMUNATE, insan plazmasından üretilmiştir. İnsan plazmasından üretilen preparatlar, örneğin virüsler ve teorik olarak Creutzfeldt - Jakob (CJD) etkeni gibi,hastalıklara yol açabilecek infeksiyon etkenleri içerebilir. Bu durum bilinmeyen ya dayeni ortaya çıkan virüslerle diğer patojenler için de geçerlidir.İnsan kanı ya da plazmasından hazırlanan tıbbi ürünlerin kullanımından kaynaklanan enfeksiyonların önlenmesi için alınan standart önlemler arasında, donörlerin seçimi,bireysel bağışların ve plazma havuzlarının belirli enfeksiyon göstergeleri için takibi vevirüslerin inaktivasyonu/uzaklaştırılması için etkili üretim aşamalarının kullanılmasıyer almaktadır. Buna rağmen insan kanı ya da plazmasından hazırlanan tıbbi ürünleruygulandığında, enfeksiyon ajanlarının bulaşma olasılığı tam olarak ortadankaldırılamayabilir. Bu durum henüz bilinmeyen ya da yeni ortaya çıkan virüsler vediğer hastalık etkenleri için de geçerlidir.Alınan önlemlerin HIV, HBC, HCV gibi zarflı virüslerle HAV gibi zarfsız virüsler için etkili olduğu düşünülmektedir. Alınan önlemlerin Parvovirüs B19 gibi bazı zarfsızvirüsleri uzaklaştırmak ya da inaktive etmek için etkisi ise kısıtlıdır. Parvovirus B19virüsü en ciddi olarak gebe kadınları (fetusda enfeksiyona neden olabilmektedir),immün yetmezlikli hastaları veya artmış eritrosit döngüsü olan hastaları (örn. hemolitikanemi durumu) etkilemektedir.İnsan plazması kaynaklı faktör VIII ürünlerini düzenli/tekrarlayan şekilde alan hastalarda uygun aşılama (Hepatit A ve B'ye karşı) düşünülmelidir.Hasta ile ürünün parti numarası arasındaki izi sürdürebilmek açısından IMMUNATE'in her kullanımından sonra ürünün adı ve parti numarasınınkaydedilmesi önemle önerilmektedir.IMMUNATE kan grubu isoaglutininleri (anti-A ve anti-B) içerir. Kan grubu A, B ya da AB olan hastalarda tekrarlayan uygulamalardan ya da çok yüksek dozlarda uygulanmasındansonra hemoliz görülebilir. İzlenebilirlikBiyolojik tıbbi ürünlerin izlenebilirliğinin artırılması için uygulanan ürünün ismi ve seri numarası açıkça kaydedilmiş olmalıdır. Aşırı duyarlılık:IMMUNATE ile alerjik tipte aşırı duyarlılık reaksiyonları gelişmesi olasılığı vardır. Hastalara, aşırı duyarlılık semptomları gelişirse, ilacın kullanılmasına derhal son vererekhekimlerine başvurmaları söylenmelidir. Hastalar, kurdeşen, yaygın ürtiker, döküntü, yüz veboyunda kızarma (flushing), kaşıntı, ödem (yüz ve gözkapağı ödemi dahil), göğüste sıkışmahissi, hırıltılı solunum, dispne, göğüste ağrı, taşikardi, hipotansiyon ve alerjik şoka kadarilerleyebilecek anafilaksinin de dahil olduğu aşırı duyarlılık reaksiyonlarının erken bulgularıaçısından bilgilendirilmelidjrei geŞokniige^§ffle§Âza dusufflMflda, standart tıbbi şok tedavisi Belge Dcğyıgulanmalidir? SHY3aklURG83RG83aklUSHY3aklU Belge Takip Adresi:https://www.turkiye.gov.tr/saglik-titck-ebys  İnhibitörler (Hemofili A hastaları):Faktör VIII'e karşı nötralize edici antikor (inhibitörler) oluşumu, hemofili A hastalarının tedavisinde bilinen bir komplikasyondur. Bu inhibitörler genellikle faktör VIII prokoagülanaktiviteye yönelik olan IgG immünoglobülinleridir ve modifiye tetkik kullanılarak her mlplazmada Bethesda Ünitesi (BU) olarak ölçülür. İnhibitör gelişme riski, faktör VIII'emaruziyetin yanı sıra hastalığın şiddeti ile ilişkilidir ve bu risk ilk 50 maruziyet gününde enyüksek seviyededir; ancak risk yaygın görülmemesine rağmen yaşam boyu devam eder. İnhibitör gelişiminin klinik önemi inhibitör titresine bağlı olacaktır; düşük titrenin teşkil ettiği yetersiz klinik yanıt riski, yüksek titreli inhibitörlere kıyasla daha az olacaktır. Genel olarak, koagülasyon faktörü VIII ürünü ile tedavi edilen tüm hastalar, uygun klinik gözlem ve laboratuvar testleri ile inhibitör gelişimi açısından dikkatliceizlenmelidir. Eğer beklenen faktör VIII plazma aktivite düzeyine ulaşılamazsa veyayeterli doz ile kanama kontrol altına alınamazsa faktör VIII inhibitörü varlığı açısındantest yapılmalıdır. İnhibitör düzeyleri yüksek olan hastalarda faktör VIII tedavisi etkiliolmayabilir ve diğer tedavi seçenekleri düşünülmelidir. Böyle hastaların tedavisi hemofilive faktör VIII inhibitörleri tedavisi konusunda deneyimli hekimler tarafındanyönlendirilmelidir. Kardiyovasküler olaylarMevcut kardiyovasküler risk faktörleri olan hastalarda FVIII ile yerine koyma tedavisi, kardiyovasküler riski artırabilir. İnhibitörler (von Willebrand hastalığı olan hastalar):Özellikle tip 3 hastaları olmak üzere von Willebrand hastalığı olan hastalarda, von Willebrand faktörüne karşı nötralizan antikorlar (inhibitörler) gelişebilmektedir. İstenen plazmavWF:RCo aktivitesine erişilemezse veya yeterli doza rağmen kanama kontrol altınaalınamamışsa, olası bir von Willebrand faktör inhibitörü varlığını araştırmak için uyguntestler yapılmalıdır. Yüksek inhibitör seviyesine sahip hastalarda, von Willebrand faktörütedavisi etkili olmayabilir ve diğer terapötik yaklaşımlar dikkate alınmalıdır. Trombotik olaylarÖzellikle klinik ya da laboratuvar risk faktörleri olduğu bilenen hastalarda trombotik olayların gelişmesi riski bulunmaktadır. Bu nedenle hastalar trombozun erken işaretleri açısındanizlenmelidir. Venöz tromboemboliye karşı mevcut öneriler doğrultusunda profilaksibaşlatılmalıdır. IMMUNATE vWF'ye kıyasla daha yüksek miktarlarda faktör VIIIiçerdiğinden, tedaviyi yürüten hekim, devam tedavisinin faktör VIII:C düzeylerinin aşırıyükselmeye neden olabileceğini göz önünde bulundurmalıdır. IMMUNATE alan hastaların, plazma faktör VIII:C düzeylerinin trombotik olay riskinde bir artışa neden olabilecek şekilde ı111 1 Bu beJge, güvenli elektronik knza ile imzaJanmışürT=xTxıı ı ı* ı*Belge DoaefekalfflMafi§kisdenlıÇınfflaliçin plazm^afalörAVeIIIttC :düzeyAer^e.izlenmelidiE.ebys
Sodyum içeriğiBu tıbbi ürün her flakonda, DSÖ'ye göre bir yetişkine yönelik 2 g sodyum tavsiye edilen maksimum günlük alımının yaklaşık %1'ine eş değer olan 19,6 mg sodyum içerir. Pediyatrik PopülasyonFaktör VIII ürünlerine sınırlı maruziyeti olan 6 yaşından küçük çocuklarda bu hasta grubunda sınırlı klinik veri mevcut olduğundan ürün dikkatle kullanılmalıdır. Listelenen uyarılar ve önlemler hem yetişkinler hem pediyatrik hastalar için geçerlidir. 4.5. Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriIMMUNATE ile herhangi bir etkileşim çalışması yapılmamıştır. İnsan koagülasyon faktör VIII ürünleri ile diğer tıbbi ürünler arasında bildirilmiş herhangi bir etkileşim bildirilmemiştir. Özel popülasyonlara ilişkin ek bilgilerHerhangi bir etkileşim çalışması yapılmamıştır. Pediyatrik popülasyonHerhangi bir etkileşim çalışması yapılmamıştır. 4.6. Gebelik ve laktasyonGenel tavsiye:Gebelik Kategorisi: C. Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon)Herhangi bir veri mevcut değildir. Gebelik dönemiHayvanlar üzerinde yapılan çalışmalar, gebelik /ve-veya/ embriyonal/fetal gelişim /ve-veya/ doğum /ve-veya/ doğum sonrası gelişim üzerindeki etkiler bakımından yetersizdir (bkz. kısım5.3). İnsanlara yönelik potansiyel risk bilinmemektedir. Hemofili A kadınlarda seyrek rastlanan bir hastalık olduğundan, gebelik döneminde faktör VIII kullanımıyla ilgili bir bilgi bulunmamaktadır. Bu nedenle, IMMUNATE, gebelikdöneminde ancak zorunlu ise kullanılabilir. Parvovirus B19 enfeksiyonu hakkında bilgi için Bölüm 4.4'e bakınız. Laktasyon dönemiHemofili A kadınlarda seyrek rastlanan bir hastalık olduğundan, emzirme döneminde faktör VIII kullanımıyla ilgili bir bilgi bulunmamaktadır. Emziren kadınlarda IMMUNATE'in annesütüne geçip geçmediği bilinmemektedir. Bu nedenle emzirme döneminde, ancak zorunlu iseIMMUNATE kullanılabilir. Üreme yeteneği / fertiliteIMMU^ATE'in üreme yeteneği / fertilite üzerindeki etkisini araştıran bir çalışma bulunmamaktadır. 4.7. Araç ve makine kullanımı üzerindeki etkilerIMMU^ATE'in araç ve makine kullanımı üzerindeki etkileri hakkında herhangi bir bilgi bulunmamaktadır. 4.8. İstenmeyen etkilerİnsan plazması kaynaklı faktör VIII ürünleriyle bildirilen istenmeyen etkiler:Güvenlilik profili özetiAşırı duyarlılık ya da alerjik reaksiyonlar (bunlar arasında anjiyoödem, infüzyon bölgesinde yanma ve batma, titreme, sıcak basması, yaygın ürtiker, baş ağrısı, kurdeşen, kan basıncındadüşme, letarji, bulantı, huzursuzluk, taşikardi, göğüste sıkışma hissi, karıncalanma, kusma,hırıltılı solunum yer alabilir) nadiren gözlenmiştir ve bazı olgularda şiddetli anafilaksiye kadarilerleyebilir (şok dahil). Hastalara, bu semptomlar ortaya çıkarsa doktorlarıyla temasageçmeleri tavsiye edilmelidir (bkz. Bölüm 4.4). IMMUNATE de dahil olmak üzere faktör VIII ile tedavi edilmiş hemofili A hastalarında nötralize edici antikorlar (inhibitörler) gelişebilir. Bu tür inhibitörler oluşursa, durum, yetersizklinik yanıt şeklinde kendini gösterebilir. Bu gibi durumlarda uzman hemofili merkezleriylebağlantı kurulması önerilmektedir. Çok seyrek olarak özellikle tip 3 olmak üzere von Willebrand hastalığı olan hastalarda, von Willebrand faktörüne karşı nötralizan antikorlar (inhibitörler) gelişebilmektedir. Bu türinhibitörler oluşmuşsa, durum kendini tedaviye yetersiz klinik yanıt vererek ortayaçıkaracaktır. Bu tür antikorlar anafilaktik reaksiyonlarla yakın ilişkili olarak oluşabilir. Bunedenle anafilaktik reaksiyon görülen hastaların olası bir inhibitör varlığı açısındandeğerlendirilmesi gerekir. Bu gibi durumlarda özel hemofili merkezleriyle bağlantıkurulmalıdır. Yüksek dozların uygulanmasından sonra A, B veya AB kan grubu olan hastalarda hemoliz gelişebilir.  IMMUNATE ile yapılan klinik çalışmalar ve pazarlama sonrası deneyimlerde bildirilen istenmeyen etkiler:Aşağıda verilen liste, MedDRA sistem organ sınıflandırmasına (SOC ve Tercihli Terim Düzeyi) uygundur. Görülme sıklıkları şu yaklaşıma göre değerlendirilmiştir: Çok yaygın (>1/10); yaygın (>1/100 ila <1/10); yaygın olmayan (>1/1.000 ila <1/100); seyrek (>1/10.000 ila <1/1.000); çok seyrek(<1/10.000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor). Advers reaksiyonlar her bir sıklık grubunda azalan ciddiyet sırasına göre verilmiştir. Bağışıklık sistemi hastalıklarıYaygın olmayan: Aşırı duyarlılık** Kan ve lenf sistemi hastalıklarıÇok yaygın: *Faktör VIII inhibisyonu (HTGH) Yaygın olmayan: *Faktör VIII inhibisyonu (TGH) * Sıklık, şiddetli hemofili A hastalarının yer aldığı, tüm FVIII ürünleri ile yapılmış çalışmalara dayanmaktadır. HTGH (Daha önce hiç tedavi görmemiş hastalar) TGH (Dahaönce tedavi görmüş hastalar)
Bilinmiyor:
Pıhtılaşma bozuklukları Psikiyatri hastalıklarıBilinmiyor: Huzursuzluk Sinir sistemi hastalıklarıBilinmiyor: Parestezi, baş dönmesi/sersemlik hali, baş ağrısı Göz hastalıklarıBilinmiyor: Konjonktivit Kardiyak hastalıklarıBilinmiyor: Taşikardi, çarpıntı Vasküler hastalıklarıBilinmiyor: Hipotansiyon, yüz ve boyunda kızarma (flushing), solukluk Solunum, göğüs bozuklukları ve mediastinal hastalıklarıBilinmiyor: Dispne, öksürük Gastrointestinal hastalıklarıBilinmiyor: Kusma, bulantı  Deri ve deri altı doku hastalıklarıBilinmiyor: Ürtiker, döküntü (Eritematöz ve papüler-döküntü dahil), kaşıntı, eritem, hiperhidrozis, nörodermatit Kas-iskelet bozukluklar, bağ dokusu ve kemik hastalıklarıBilinmiyor: Miyalji Genel bozukluklar ve uygulama bölgesine ilişkin hastalıklarıBilinmiyor: Göğüste ağrı, göğüste rahatsızlık hissi, ödem (periferik, göz kapağı ve yüzde ödem dahil), ateş, titreme, enjeksiyon yeri reaksiyonları (yanma dahil), ağrı ** Bir klinik çalışmada yer alan 5 hastada uygulanan 329 infüzyonda görülen bir adet aşırı duyarlılık reaksiyonu Şüpheli advers reaksiyonların raporlanmasıRuhsatlandırma sonrası şüpheli ilaç advers reaksiyonlarının raporlanması büyük önem taşımaktadır. Raporlama yapılması, ilacın yarar/risk dengesinin sürekli olarak izlenmesineolanak sağlar. Sağlık mesleği mensuplarının herhangi bir şüpheli advers reaksiyonu TürkiyeFarmakovijilans Merkezi (TÜFAM)'ne bildirmeleri gerekmektedir. (www.titck.gov.tr; e-posta: [email protected]; tel: 0 800 314 00 08; faks: 0 312 218 35 99) 4.9. Doz aşımı ve tedavisiHerhangi bir doz aşımı vakası rapor edilmemiştir. Tromboembolik olaylar oluşabilir. Bkz. Bölüm 4.4. A, B ve AB kan grubu olan hastalarda hemoliz gelişebilir. Bkz. Bölüm 4.4. 5. FARMAKOLOJIK ÖZELLIKLER5.1. Farmakodinamik özellikler
kombine von Willebrand faktörü ve
Farmakoterapötik Grubu: Anti-hemorajikler: koagülasyon faktörü VIII ATC kodu: B02BD06 Etki mekanizmasıFaktör VIII / von Willebrand faktör kompleksi farklı fizyolojik fonksiyonları olan 2 molekülden (faktör VIII ve von Willebrand faktörü) oluşmuştur. Hemofilisi olan bir hastayainfüze edildiğinde, faktör VIII hastanın dolaşımında von Willebrand faktörüne bağlanır.Aktive faktör VIII, aktive faktör IX için kofaktör görevi görür ve faktör X'un aktive faktörX'a dönüşümünü hızlandırır. Aktive olan faktör X ise protrombini trombine dönüştürür.Trombin ise fibrinojeni fibrine dönüştürür ve pıhtı oluşumu gerçekleşebilir. Hemofili A,faktör VIII C düzeylerinin düşük seyrettiği cinsiyete bağlı herediter bir kan pıhtılaşmabozukluğudur; spontan veya kaza veya cerrahi travma sonrası eklemlere, kaslara ve içorganlara olan bol miktarda kanama ile seyreder. Yerine koyma tedavisiyle F VIII plazmadüzeyleri yükseltilerek, faktör eksikliğinde ve kanama eğiliminde geçici bir düzelmesağlanabilir.  von WiUebrand faktörü (vWF), F VIII'i koruyucu bir protein olması yanında vasküler yaralanma bölgesinde trombosit adezyonuna aracılık eder, trombosit agregasyonunda rol alır. 5.2. Farmakokinetik özelliklerGenel özelliklerIMMUNATE'e yönelik tüm farmakokinetik parametreler ağır Hemofili A'sı (faktör VIII düzeyi < %1 olan) olan kişilerde ölçülmüştür. Plazma numunelerinin analizi merkezi birlaboratuvarda kromojenik F VIII miktar tayini kullanılarak gerçekleştirilmiştir. 12 yaşındanbüyük ve daha önceden tedavi görmüş 18 hastada yapılan çaprazlamalı bir çalışmadan eldeedilen farmakokinetik parametreler aşağıdaki tabloda verilmektedir.
Emilim:Enjeksiyon sonrası plazmada ölçülen F VIII aktivitesi, beklenen plazma F VIII aktivitesinin %80-120'si kadar olur. Yapılan bir farmakokinetik çalışmada IMMUNATE enjeksiyonusonrası in vivo ortalama FVIII düzeylerinin %100 olduğu gösterilmiştir. Dağılım:Uygulanan faktör VIII, normal faktör VIII'le aynı şekilde dağılıma uğrar. Plazma FVIII aktivitesi iki fazlı eksponansiyel düşüş göstermektedir. Başlangıç fazında, intravasküler vediğer kompartmanlar arası dağılım (vücut sıvıları), 3-6 saatlik plazma eliminasyon yarılanmasüresine göre gerçekleşmekte; FVIII'in yaklaşık olarak % - % kadarı dolaşımda kalmaktadır.İkinci ve yavaş fazda ise (muhtemelen FVIII tüketimini yansıtan faz) yarılanma süresi 8-20saat arasında değişmektedir (ortalama 12 saat). Bu süre, biyolojik yarılanma süresi ileuyumludur. IMMUNATE ile gerçekleştirilen bu çalışmada, faktör VIII yarılanma sürelerimodele bağlı ve bağlı olmayan yöntemlerle belirlenmiştir. Her iki durumda da 11 saatlikortalama değerler bulunmuştur. Biyotransformasyon:
Plazmada IMMUNATE'den gelen faktör VIII, normal faktör VlII'le aynı şekilde tüketilerek biyotransformasyona uğrar. Aktive FVIII plazmada yer alan aktive protein C, aktive FIX veaktive FX ve plazmini içeren serin proteazlar tarafından parçalanarak inaktive olur. Eliminasyon:Faktör VIII normal yollardan tüketilir ve elimine edilir. LRP1 reseptörleri başta olmak üzere çeşitli hücre yüzey reseptörlerinin FVIII'in plazma klirensinde rol aldığı düşünülmektedirancak FVIII'in klirens mekanizmaları tam olarak açıklanamamıştır. 5.3. Klinik öncesi güvenlilik verileriIMMUNATE'in içerisinde yer alan insan kan koagülasyon faktörü VIII, insan plazmasının normal bileşenlerinden birisidir ve endojen faktör VIII gibi davranır. Klasik güvenlilik farmakolojisi, tekrarlayan doz toksisitesi, lokal tolerans ve immünojenite çalışmalarına dayanan klinik dışı veriler insanlarda spesifik bir zarar işaret etmemektedir. 6. FARMASÖTİK ÖZELLİKLERİ6.1. Yardımcı maddelerin listesiKuru toz içeren flakon: - İnsan albümini - Glisin - Lizin hidroklorür - Sodyum klorür - Trisodyum sitrat-2H2O - Kalsiyum klorür-2H2O Çözücü: - Steril Enjeksiyonluk Su 6.2. GeçimsizliklerBu tıbbi ürün Bölüm 6.6'da belirtilenler dışında başka bir ilaçla karıştırılmamalıdır. Yalnızca birlikte verilen infüzyon seti ile uygulanmalıdır; insan koagülasyon faktörü VIII infüzyon için kullanılan başka setlerin iç yüzeyi tarafından emilerek tedavide başarısızkalınabilir. 6.3. Raf ömrü24 aydır. Sulandırılmış IMMUNATE çözeltisinin, oda sıcaklığında kullanılırken, yaklaşık olarak 3 saat kimyasal ve fiziksel stabilitesini koruduğu gösterilmiştir. Mikrobiyolojik nedenlerle,sulandırma işlemi mikrobiyal kontaminasyon riskini tamamen ortadan kaldıracak şekildekontrollü ve valide edilmiş aseptik koşullarda yapılmadığı sürece sulandırıldıktan hemen
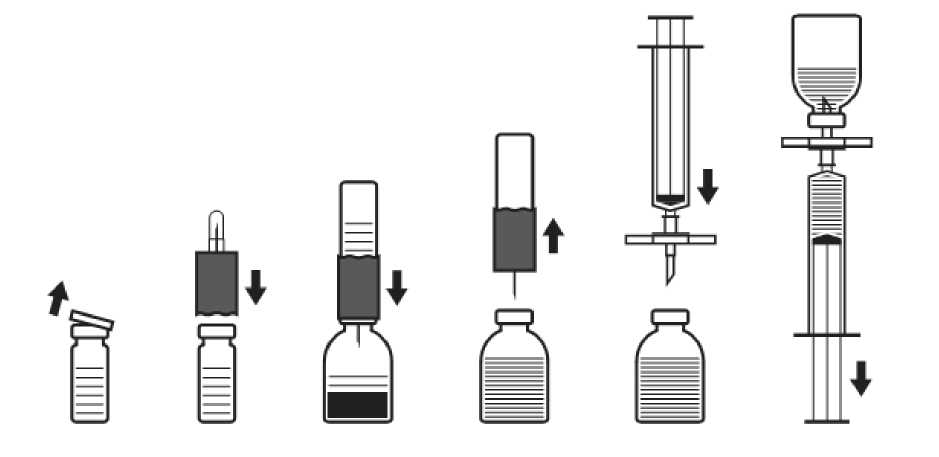
sonra kullanılması önerilir. Hemen kullanılmadığı durumlarda, kullanımda saklama süresi ve koşullarından kullanıcı sorumludur. Sulandırılan ürün tekrar buzdolabında saklanmamalıdır. Ürün raf ömrü içindeyken yalnızca bir dönem olmak üzere 25°C altı oda sıcaklığında 6 ay süreyle tutulabilir. Böyle saklanıyorsa lütfen ürünün ambalajı üzerine saklama süresinikaydediniz. Bu sürenin sonunda ürün tekrardan buzdolabında saklanmaz ancak hemenkullanılmalı veya atılmalıdır. 6.4. Saklamaya yönelik özel tedbirler2-8°C arasında saklanmalıdır ve transfer edilmelidir. Dondurulmamalıdır. Işıktan korumak amacıyla ambalajı içerisinde saklanmalıdır. Sulandırılarak kullanıma hazır hale getirilmiş ilacın saklanması için bkz. Bölüm 6.3. 6.5. Ambalajın niteliği ve içeriğiKutuda; klorobutil lastik tıpa ile kapatılmış liyofilize toz içeren Tip II cam flakon, 10 mL enjeksiyonluk su içeren Tip II cam flakon ve enjeksiyon ve sulandırma için kit (1 filtre iğnesi,1 transfer iğnesi, 1 tek kullanımlık şırınga, 1 tek kullanımlık iğne, 1 kanatlı infüzyon seti). 6.6. Beşeri tıbbi üründen arta kalan maddelerin imhası ve diğer özel önlemlerKullanılmamış olan ürünler ya da atık materyaller Tıbbi Atıkların Kontrolü Yönetmeliği ve Ambalaj Atıklarının Kontrolü Yönetmeliklerine uygun olarak imha edilmelidir. Son kullanma tarihi geçmiş veya kullanılmayan ilaçları çöpe atmayınız! Çevre, Şehircilik ve İklim Değişikliği Bakanlığınca belirlenen toplama sistemine veriniz. Uygulamadan öncesulandırma için yalnızca ambalajındaki seti kullanınız. Koruyucu içermediğindenIMMUNATE uygulamadan hemen önce sulandırılmalıdır. IMMUNATE infüzyonundan önce ve sonra, venöz uygulama cihazlarının içinden izotonik tuzlu su geçirilerek yıkanması önerilir. UYGULAMAYI YAPACAK DOKTOR İÇİN TALİMATLAR Kuru tozun sulandırılması:Aseptik teknik kullanılmalıdır! 1. Çözücü içeren (enjeksiyonluk steril su) kapalı flakonu oda sıcaklığına kadar ısıtınız(maksimum 37°C). 2. Kuru toz flakonunun ve çözücü flakonunun koruyucu kapaklarını çıkarınız (Şekil A) ve herikisinin de lastik tıpalarını dezenfekte ediniz. 3. Transfer setinin dalgalı kenarını çözücü flakonunun üzerine yerleştiriniz ve bastırınız (Şekil B). 4. Transfer setinin koruyucu kapağını, açıkta kalan kısımlarına temas etmemeye dikkat ederekdiğer tarafından çıkarınız. 5. Çözücü flakonuna takılı haldeyken transfer setini ters çevirerek, iğnesini kuru toz içerenflakonun tıpasına batırınız (Şekil C). Çözücü, kuru toz içeren flakonun vakumu sayesindeemilecektir. 6. Yaklaşık 1 dakika kadar bekledikten sonra transfer setinin bağlı olduğu çözücü flakon ile kuru toz içeren flakonu birbirinden ayırınız (Şekil D). Preparat kolaylıkla çözündüğünden, flakonu sadece gerekiyorsa hafifçe sallayınız.FLAKONİÇERİĞİNİ ÇALKALAMAYINIZ. İÇERİĞİNİ ENJEKTÖRE ÇEKMENİN HEMEN ÖNCESİNE KADAR, KURU TOZ İÇEREN FLAKONU TERS ÇEVİRMEYİNİZ. 7. Sulandırılarak kullanıma hazır hale getirdikten sonra, uygulama öncesinde görsel olarakpartikül ve renk değişikliği açısından incelenmelidir. Çözelti berrak veya hafif opalesanolmalıdır. Sulandırma prosedürü tam olarak İzlense dahi, birkaç küçük partikül gözlegörülebilir. Ambalaj içeriğinde bulunan filtreli set, bu partikülleri uzaklaştıracaktır veambalaj etiketinde belirtilen potenste azalma olmayacaktır. Bulanık ya da partikül içerençözeltiler kullanılmamalıdır. Uygulama:Aseptik teknik kullanılmalıdır. 1. Tıpadan kopan lastik parçalarının ilaçla birlikte uygulanmasına engel olmak için(mikroemboli riski) ambalajdaki filtreli seti kullanınız. Çözünmüş preparatı çekmek içinfiltreli seti, ambalajdaki tek kullanımlık enjektöre takınız ve lastik tıpaya batırınız (Şekil E). 2. Enjektörü filtreli setten bir an için ayırınız. Kuru toz içeren flakona hava girecek ve oluşmuşköpük kaybolacaktır. Bundan sonra, filtreli set aracılığıyla çözeltiyi enjektöre çekiniz (ŞekilF). 3. Enjektörü filtreli setten ayırınız ve ambalajdaki kelebek infüzyon seti (ya da tek kullanımlıkiğne) ile çözeltiyi yavaş olarak (enjeksiyon hızı dakikada 2 mL'yi aşmamalıdır) intravenözolarak uygulayınız. Şek. A Şek. B Şek. C Şek. DŞek. EŞek. FHerhangi kullanılmamış tıbbi ürün veya atık materyal yerel gerekliliklere uygun olarak bertaraf edilmelidir. 7. RUHSAT SAHİBİTakeda İlaç Sağlık Sanayi Ticaret Limited Şirketi Levent-Şişli/İSTANBUL 8. RUHSAT NUMARASI(LARI)2016/116 9. İLK RUHSAT TARİHİ / RUHSAT YENİLEME TARİHİİlk ruhsat tarihi: 01.02.2016 Ruhsat yenileme tarihi: 10. KÜB'ÜN YENİLENME TARİHİ
|
İlaç BilgileriImmunate 1000/750 Iu Iv İnfüzyonluk Toz İçeren FlakonEtken Maddesi: İnsan Koagülasyon Faktörü Viii / Von Willebrand Faktörü Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||








Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2024 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.