Arixtra 5 mg/0.4 ml Enjeksiyonluk Çözelti Içeren Kullanıma Hazır Şırınga Kısa Ürün BilgisiKan ve Kan Yapıcı Organlar » Antitrombotikler » Antitrombotik İlaçlar » Diğer İlaçlar » Fondaparinuks Sodyum KISA ÜRÜN BİLGİSİ1. BEŞERİ TIBBİ ÜRÜNÜN ADIARIXTRA 5 mg/0.4 mL SC enjeksiyonluk çözelti içeren kullanıma hazır şırınga2. KALİTATİF VE KANTİTATİF BİLEŞİM Etkin madde:Fondaparinuks sodyum 5 mgYardımcı maddeler:Sodyum klorür 3 mgYardımcı maddeler için bölüm 6.1'e bakınız. 3. FARMASÖTİK FORMEnjeksiyonluk çözelti, kullanıma hazır şırınga.Berrak ve renksizden sarımsıya değişen renklerde olan bir çözeltidir. Çözeltinin pH'sı 5.0 ile 8.0 arasındadır. 4. KLİNİK ÖZELLİKLER4.1 Terapötik endikasyonlar Akut derin ven trombozu'nun (DVT) tedavisinde varfarin sodyum ile birlikte, Akut pulmoner emboli'nin (PE) tedavisinde varfarin sodyum ile birlikte endikedir. 4.2 Pozoloji ve uygulama şekli Pozoloji: Yetişkinlerde tavsiye edilen ARIXTRA dozu günde 1 kez subkütan enjeksiyon yoluyla uygulanan 7.5 mg/0.6 mL'dir (vücut ağırlığı 50 - 100 kg arasında olan hastalar için). Vücut ağırlığı 50 kg'ın altında olan hastalar için tavsiye edilen doz 5 mg/0.4 mL'dir. Vücut ağırlığı 100 kg'ın üzerinde olan hastalar için tavsiye edilen doz 10 mg/0.8 mL'dir. Uygulama sıklığı ve süresi:Tedaviye yeterli oral antikoagülasyon sağlanana kadar (Uluslararası normalize edilmiş oran 2- 3) en az 5 gün devam edilmelidir. Genellikle 72 saat içinde mümkün olduğunca çabuk eş zamanlı oral antikoagülan tedavisine başlanmalıdır. Klinik çalışmalarda ortalama uygulama süresi 7 gündür ve 10 günle sınırlıdır. Uygulama şekli:ARIXTRA, hasta uzanır durumdayken derin subkütan enjeksiyonla uygulanır. Uygulama sağ ve sol anteriolateral ile sağ ve sol posteriolateral karın duvarı arasında dönüşümlü olarak yapılmalıdır. Kullanıma hazır şırınga kullanılırken tıbbi ürün kaybını önlemek için enjeksiyondan önce şırınga içindeki hava kabarcığı dışarı çıkarılmamalıdır. İğne, baş parmak ve işaret parmağı arasında tutulan deri boğumuna dikey olarak tümüyle batırılmalıdır. Deri boğumunun tutulması enjeksiyon boyunca sürdürülmelidir(bkz. Bölüm 6.6 Beşeri tıbbi ürünlerden arta kalan maddelerin imhası ve diğer özel önlemler).Özel popülasyonlara ilişkin ek bilgilerBöbrek/Karaciğer yetmezliği:ARIXTRA orta düzeyde böbrek yetmezliği olan (kreatinin klerensi 30-50 mL/dak) hastalarda dikkatli şekilde kullanılmalıdır(bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri).ARIXTRA kullanan hastalarda periyodik olarak böbrek fonksiyonu değerlendirilmelidir. Tedavi sırasında şiddetli böbrek yetmezliği gelişen hastalarda ilaç derhal kesilmelidir. ARIXTRA kesildikten sonra antikoagülan etkiler böbrek fonksiyonu normal olan hastalarda 2-4 gün sürebilir (en az 3-5 yarı ömür süresi). ARIXTRA'nın antikoagülan etkileri böbrek yetmezliği olan hastalarda daha uzun sürebilir. ARIXTRA'nın farmakokinetik özellikleri, karaciğer yetmezliği olan hastalarda incelenmemiştir. Pediyatrik popülasyon:17 yaşın altındaki hastalarda ARIXTRA'nın güvenilirliği ve etkinliği araştırılmamıştır.Geriyatrik popülasyon:Doz ayarlaması gerekli değildir. 75 yaşında ve üzeri hastalarda ARIXTRA dikkatli kullanılmalıdır(bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri).4.3 KontrendikasyonlarARIXTRA aşağıdaki durumlarda kontrendikedir:- Fondaparinuksa veya ilacın bileşenlerinden herhangi birine karşı aşırı duyarlılığı olanlarda. - Klinik yönden belirgin aktif kanaması olanlarda - Akut bakteriyel endokardit - Şiddetli böbrek yetmezliği olan hastalarda (kreatin klerensi <30 mL/dak) - ARIXTRA tedavisi sırasında in vitro pıhtılaşma testi pozitif sonuç vermiş olan trombositopeni hastalarında - Kalça kırığı, kalça replasman veya diz replasman ve abdominal cerrahi uygulanan 50 kg'ın altındaki ARIXTRA profilaktik tedavisi gören hastalarda Spinal/Epidural HematomaNöroaksiyel anestezi (epidural/spinal anesteziler) veya spinal ponksiyon kullanıldığında tromboembolik komplikasyonların önlenmesi için düşük moleküllü heparinler, heparinoidler veya ARIXTRA ile antikoagülasyon uygulanan veya uygulanması planlanan hastalarda uzun süreli veya kalıcı paralize neden olabilen epidural veya spinal hematom riski mevcuttur.Bu olayların riski analjezik uygulanması için kalıcı epidural kateter kullanımı veya nonsteroidal anti-inflamatuvar ilaçlar (NSAİİ'ler), antitrombositik ilaçlar veya diğer antikoagülanlar gibi hemostazı etkileyen ilaçların eşzamanlı kullanımı ile artmaktadır. Söz konusu riskin ayrıca travmatik veya tekrarlı epidural veya spinal ponksiyon ile arttığı düşünülmektedir.Hastalar nörolojik bozukluk belirti ve semptomları için sıklıkla izlenmelidir. Nörolojik bozukluk tespit edildiğinde acil tedavi gereklidir.Hekimler, tromboprofilaksi için antikoagülasyon uygulanan ve uygulanması planlanan hastalarda nöroaksiyel girişim öncesinde potansiyel yarar/risk oranını göz önünde bulundurmalıdır.ARIXTRA sadece subkütan uygulama için tasarlanmıştır.İntramüsküler yolla uygulamayınız.KanamaARIXTRA, konjenital ve kazanılmış kanama bozukluğu (örneğin: 50.000/mm3'ten az trombosit sayısı), aktif ülseratif gastrointestinal hastalığı ve yakın zamanda geçirilmiş intrakraniyal kanaması olanlarda, beyin, omurilik veya göz cerrahisinden hemen sonra ve aşağıda belirtilen özel hasta gruplarında dikkatli kullanılmalıdır. Kanama riskini artırma potansiyeli olan ajanlar fondaparinuksla birlikte uygulanmamalıdır. Dezirüdin, fibrinolitik ajanlar, GP IIb/IIIa reseptör antagonistleri, heparin, heparinoidler veya düşük molekül ağırlıklı heparin (LMWH) bu ajanlara dahildir. Venöz Trombo Emboli (VTE) tedavisi sırasında, bölüm 4.5'teki (Diğer tıbbi ürünlerle etkileşimler ve diğer etkileşim şekilleri)bilgilere uygun olarak vitamin K antagonist tedavisi de eş zamanlı uygulanmalıdır. Diğer antitrombositik ilaçlar (asetilsalisilik asit, dipiridamol, sülfinpirazon, tiklopidin veya klopidogrel) ve NSAiD'ler dikkatli kullanılmalıdır. Birlikte uygulama gerekli ise yakın takip şarttır.Spinal/Epidural anestezi ARIXTRA kullanımıyla eş zamanlı olarak uygulanan spinal/epidural anestezinin veya spinal ponksiyonun, uzun süreli veya kalıcı paraliz ile sonuçlanabileceği gözardı edilemez. Seyrek görülen bu olayların gelişme riski, postoperatif kalıcı epidural kateter kullanımı veya hemostazı etkileyen diğer tıbbi ürünlerin eş zamanlı kullanımıyla daha yüksek olabilir. Yaşlı hastalar Yaşlı popülasyon artmış kanama riski altındadır. Böbrek fonksiyonları genellikle yaşla azaldığı için, bu hastalarda eliminasyon azalabilir ve fondaparinuksa maruziyet artabilir (bkz.Bölüm 5.2 Farmakokinetik özellikler).(bkz. Bölüm 4.2 Pozoloji ve uygulama şekli).Düşük vücut ağırlığı Fondaparinuksun eliminasyonu vücut ağırlığı ile azalır. Vücut ağırlığı 50 kg'dan az olan hastalarda 5 mg/0.4 mL'lik doz dikkatli kullanılmalıdır (bkz. Bölüm 4.2 Pozoloji ve uygulama şekli).Böbrek yetmezliği Kreatin klerensi 50 mL/dk'dan az olan hastalar artmış kanama riski altındadır. Fondaparinuksun temel olarak böbrekler yoluyla atıldığı bilinmektedir. Orta düzeyde böbrek yetmezliği olan hastalarda ARIXTRA dikkatli kullanılmalıdır (bkz. Bölüm 4.2 Pozoloji ve uygulama şekli).Şiddetli karaciğer yetmezliği Şiddetli karaciğer yetmezliği olan hastalarda, pıhtılaşma faktörlerinin eksikliğine bağlı kanama riskinin artması nedeni ile ARIXTRA kullanımı dikkatle düşünülmelidir (bkz. Bölüm4.2 Pozoloji ve uygulama şekli).Trombositopeni DVT ve PE tedavisi klinik çalışmalarında ARIXTRA tedavisi uygulanan hastalarda %0.5 oranında orta şiddette trombositopeni meydana gelmiştir. DVT ve PE tedavisine yönelik klinik çalışmalarda ARIXTRA tedavi rejimi uygulanan hastalarda %0.04 oranında şiddetli trombositopeni meydana gelmiştir. Tüm trombositopeni vakaları, şiddet düzeyine bağlı olmaksızın yakından izlenmelidir. Platelet sayımı 100,000/mm3 düzeyinin altına düştüğü takdirde ARIXTRA tedavisi kesilmelidir. ARIXTRA her bir kullanıma hazır şırıngada 23 mg'dan daha az sodyum ihtiva eder. Sodyuma bağlı herhangi bir olumsuz etki beklenmez. 4.5 Diğer tıbbi ürünler ile etkileşimler ve diğer etkileşim şekilleriARIXTRA kanama riskini artırma ihtimali olan ilaçlarla birlikte uygulandığında kanama riski artar(bkz. Bölüm 4.4 Özel kullanım uyarıları ve önlemleri).ARIXTRA ile yapılan klinik çalışmalarda, oral antikoagülanlar (varfarin), ARIXTRA'nın farmakokinetiği ile etkileşime girmemiştir. 10 mg/0.8 mL'lik dozda kullanıldığı etkileşim çalışmalarında ARIXTRA varfarinin antikoagülasyon izleme (INR) aktivitesini etkilememiştir. Trombosit agregasyon inhibitörleri (asetilsalisilik asit), NSAİD'ler (piroksikam) ve digoksin ARIXTRA'nın farmakokinetiği ile etkileşime girmemiştir. 10 mg/0.8 mL dozunda kullanıldığı etkileşim çalışmalarında ARIXTRA, ne asetilsalisilik asit veya piroksikam tedavisi altında kanama zamanını ne de kararlı durumdaki digoksin farmakokinetiğini etkilememiştir. Özel popülasyonlara ilişkin ek bilgilerÖzel popülasyonlara ilişkin hiçbir klinik etkileşim çalışması yürütülmemi ştir.Pediyatrik popülasyon:Pediyatrik popülasyona ilişkin hiçbir klinik etkileşim çalışması yürütülmemi ştir.4.6 Gebelik ve laktasyonGenel tavsiye:Gebelik kategorisi B.Çocuk doğurma potansiyeli bulunan kadınlar / Doğum kontrolü (Kontrasepsiyon)Fondaparinuks için gebeliklerde maruz kalmaya ilişkin klinik veri mevcut değildir.Hayvanlar üzerinde yapılan çalışmalar, gebelik / embriyonal / fetal gelişim / doğum ya da doğum sonrası gelişim ile ilgili olarak doğrudan ya da dolaylı zararlı etkiler olduğunu göstermemektedir. Gebe kadınlara verilirken tedbirli olunmalıdır. Gebelik dönemiHayvan çalışmalarında maruziyet sınırlı olduğundan, gebelik, embriyo/fetal gelişim, doğum ve postnatal gelişim üzerindeki etkileri ile ilgili bilgiler yetersizdir.Gebe kadınlarda kullanımına ilişkin yeterli veri mevcut değildir. ARIXTRA açıkça gerekli olmadığı sürece hamile kadınlara reçete edilmemelidir. Laktasyon dönemiFondaparinuksun anne sütü ile atılıp atılmadığı bilinmemektedir. Hayvanlar üzerinde yapılan çalışmalar fondaparinuksun sütle atıldığını göstermektedir. Emzirmenin durdurulup durdurulmayacağına ya da ARIXTRA tedavisinin durdurulup durdurulmayacağına/tedaviden kaçınılıp kaçınılmayacağına ilişkin karar verirken, emzirmenin çocuk açısından faydası ve ARIXTRA tedavisinin emziren anne açısından faydası dikkate alınmalıdır.Üreme yeteneği/FertiliteSınırlı maruziyet nedeniyle hayvan çalışmaları, üreme toksisitesi üzerindeki etkileri ile ilgili bilgiler yetersizdir.4.7 Araç ve makine kullanımı üzerindeki etkilerAraç veya makine kullanımı üzerindeki etkisi ile ilgili herhangi bir çalışma yapılmamıştır.4.8 İstenmeyen etkilerARIXTRA'nın güvenilirliği venöz tromboemboli tedavisi gören 2.517 hasta üzerinde değerlendirilmiştir.Araştırıcı tarafından bildirilen ve ARIXTRA ile ilgili olma olasılığı olan advers olaylar ciddiyetlerindeki azalma sırasıyla, sıklık grupları çok yaygın (> 1/10); yaygın (>1/100, <1/10); yaygın olmayan (>1/1000, <1/100); seyrek (>1/10000, <1/1000), çok seyrek (<1/10000), bilinmiyor (eldeki verilerden hareketle tahmin edilemiyor) ve sistem organ sınıflarına göre aşağıda sıralanmıştır. Kan ve lenf sistemi hastalıklarıYaygın: Kanama (gastrointestinal, hamatüri, hematom, burun kanaması, hemoptiz,utero-vajinal kanama, hemartroz, oküler, purpura, morluk oluşumu) Yaygın olmayan: Anemi, trombositopeni Seyrek: Diğer kanamalar (hepatik, retroperitonel, intrakraniyal/intraserebral), trombositemi Metabolizma ve beslenme hastalıklarıSeyrek: Proteine bağlı olmayan (Npn)* nitrojen artışıSinir sistemi hastalıklarıYaygın olmayan: Baş ağrısıSeyrek: Baş dönmesi Gastrointestinal hastalıklarıYaygın olmayan: Bulantı, kusmaHepato-bilier hastalıklarıYaygın olmayan: Anormal karaciğer fonksiyonlarıDeri ve deri altı doku hastalıklarıSeyrek: Eritematöz döküntü, enjeksiyon yerinde reaksiyonGenel bozukluklar ve uygulama bölgesine ilişkin hastalıklarYaygın olmayan: Ağrı, ödemSeyrek: Alerjik reaksiyon *Üre, ürük asit, amino asit ve benzeri gibi protein olmayan nitrojenler için Npn standları.4.9 Doz aşımı ve tedavisiTavsiye edilenden yüksek ARIXTRA dozları kanama riskinde artışa yol açabilir.Doz aşımına bağlı kanama komplikasyonu görüldüğünde tedavi durdurulmalı ve birincil neden araştırılmalıdır. Cerrahi hemostaz, kan transfüzyonu, taze plazma transfüzyonu, plazmaferez gibi uygun bir tedaviye başlanması düşünülmelidir. 5. FARMAKOLOJİK ÖZELLİKLER5.1 Farmakodinamik özellikler:Farmakoterapötik grup: Antitrombotik, Antikoagülan ilaçlar ATC Kodu B01A X05Etki mekanizması Fondaparinuks aktif faktör X'in (Xa) sentetik ve selektif bir inhibitörüdür. Fondaparinuksun antitrombotik aktivitesi, Faktör Xa'nın antitrombin III (ATIII) aracılı selektif inhibisyonu ile sağlanır. ATIII'e selektif olarak bağlanan fondaparinuks, Faktör Xa'nın ATIII ile nötralizasyonunu artırır (yaklaşık 300 kat). Faktör Xa'nın nötralizasyonu, koagülasyon kaskadını bloke eder ve hem trombin oluşumunu hem de trombüs gelişimini önler. Fondaparinuks, trombini (aktif Faktör II) inaktive etmez ve trombositler üzerinde bilinen etkisi yoktur. Farmakodinamik etkiler 2.5 mg dozunda ARIXTRA, plazmada aktif parsiyel tromboplastin zamanı (aPTT), aktif pıhtılaşma zamanı (aPT) veya protrombin zamanı (PT)/uluslararası normalize edilmiş oran (INR) testleri gibi rutin koagülasyon testlerini, kanama zamanını veya fibrinolitik aktiviteyi etkilemez. Yüksek dozlarda aPTT'de orta düzeyde değişiklikler oluşabilir. 10 mg/0.8 mL dozunda kullanıldığı etkileşim çalışmalarında ARIXTRA varfarinin antikoagülasyon aktivitesini (INR) etkilememiştir. Fondaparinuks, heparinin indüklediği trombositopenisi olan hastaların serumlarıyla çapraz reaksiyona girmez. Klinik etkinlik ve güvenlilik Venöz Tromboemboli tedavisinde ARIXTRA klinik programı, ARIXTRA'nın derin ven trombozu (DVT) ve pulmoner emboli (PE) tedavisindeki etkinliğini göstermek için tasarlanmıştır. 4.875'ten fazla hasta üzerinde kontrollü faz II ve III klinik çalışmaları yapılmıştır. Derin ven torombozu'nun tedavisi Akut semptomatik DVT tanısı doğrulanmış hastalar üzerinde yapılan randomize, çift kör bir klinik çalışmada, günde 1 kez subkütan yolla uygulanan 5 mg/0.4 mL (vücut ağırlığı 50 kg'ın altında), 7.5 mg/0.6 mL (vücut ağırlığı 50 -100 kg arasında) veya 10 mg/0.8 mL (vücut ağırlığı 10 kg'ın üzerinde) ARIXTRA ile günde 2 kez subkütan yolla uygulanan 1 mg/kg enoksaparin karşılaştırılmıştır. Toplam 2.192 hasta tedavi edilmiştir; her iki grupta da hastalar en az 5 gün (ortalama 7 gün) tedavi görmüştür. Tedavi gruplarının ikisi de genellikle çalışma ilacının ilk uygulamasından sonraki 72 saat içinde kullanılmaya başlanan ve 90 ± 7 gün devam edilen vitamin K antagonisti almıştır; 2 - 3'lük INR sağlamak için düzenli doz ayarlamaları yapılmıştır. Primer etkinlik sonlanma noktası 97. güne kadar bildirilen doğrulanmış, semptomatik, reküran, fatal olmayan VTE ve fatal VTE'nin birleşimidir. Enoksaparin sodyumun % 4.1 'lik VTE oranına kıyasla ARIXTRA tedavisi % 3.9'luk VTE oranı ile ilişkilidir. Başlangıçtaki tedavi periyodu boyunca majör kanama enoksaparin alan hastalarda % 1.2 iken fondaparinuks alan hastalarda % 1.1 oranında gözlenmiştir. Pulmoner emboli tedavisi Akut semptomatik PE tanısı doğrulanan hastalar üzerinde yapılan randomize, açık etiket bir klinik çalışmada günde 1 kez subkütan yolla uygulanan 5 mg/0.4 mL (vücut ağırlığı 50 kg'ın altında), 7.5 mg/0.6 mL (vücut ağırlığı 50 - 100 kg arasında) veya 10 mg/ 0.8 mL (vücut ağırlığı 100 kg'dan yüksek) ARIXTRA ile anfraksiyone heparin IV bolus (5000 IU) uygulaması karşılaştırılmıştır; iv bolus uygulamasını takiben 1.5 - 2.5 kat aPTT kontrol değerini sürdürmek için ayarlanan sürekli IV infüzyonu yapılmıştır. Toplam 2.184 hasta tedavi edilmiştir; her iki tedavi grubundaki hastalar en az 5 gün (ortalama 7 gün) tedavi görmüştür. İki tedavi grubu da genellikle 2 - 3'lük INR sağlamak için düzenli doz ayarlamaları yapılan çalışma ilacının ilk dozunun uygulanmasını takiben 72 saat içinde başlanan ve 90 ± 7 gün devam ettirilen vitamin K antagonisti almıştır. Primer etkinlik sonlanma noktası, 97. güne kadar bildirilen doğrulanan semptomatik reküran fatal olmayan VTE ve fatal olan VTE'nin bileşimidir. Anfraksiyone heparin tedavisinin % 5.0'lik VTE oranına kıyasla ARIXTRA tedavisi % 3.8'lik VTE oranı ile ilişkilidir. Başlangıçtaki tedavi periyodu boyunca majör kanama, fraksiyonize olmayan heparin alan hastalarda % 1.1 iken fondaparinuks alan hastalarda % 1.3 oranında gözlenmiştir. 5.2 Farmakokinetik özellikler:Genel özelliklerFondaparinuks sodyum aktif faktör X(Xa)'nın sentetik ve selektif bir inhibitörüdür ve molekül ağırlığı 1728 g/mol'dür.Emilim: Subkütan uygulamayı takiben fondaparinuksun tamamı hızla absorbe olur (mutlak biyoyararlanım % 100). Sağlıklı genç bireylerde tek doz 2.5 mg ARIXTRA'nın subkütan uygulamasını takiben, doruk plazma konsantrasyonuna (ortalama C maksmaksksdeğerinin yarısı düzeyindeki plazma konsantrasyonlarına uygulamadan 25 dakika sonra ulaşılır.Fax. |
İlaç BilgileriArixtra 5 mg/0.4 ml Enjeksiyonluk Çözelti Içeren Kullanıma Hazır ŞırıngaEtken Maddesi: Fondaparinuks sodyum Atc Kodu: B01AX05 Kullanma talimatı ve kısa ürün bilgileri
Google Reklamları
|
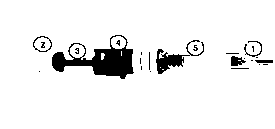
 1. Ellerinizi sabun ve su ile yıkayınız. Havlu ile kurulayınız.
1. Ellerinizi sabun ve su ile yıkayınız. Havlu ile kurulayınız. Şekil 1
Şekil 1 Şekil 2
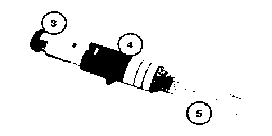
Şekil 2 Şekil 3
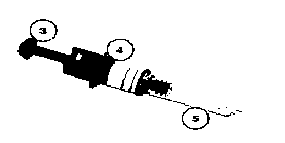
Şekil 3 Şekil 4
Şekil 4 Şekil 5
Şekil 5 Şekil 7
Şekil 7Ana Sayfa | Hakkımızda | İlaçlar | İlaç Ara | İlaç Firmaları | Gizlilik | Bize Ulaşın
Telif Hakkı 2008-2024 © İlaç Prospektüsü. Tüm Hakları Saklıdır.Uyarı: Sitemizde yayınladığımız ilaç bilgileri ile doktora danışmadan kesinlikle ilaç kullanmayınız!
Aksi halde doğabilecek sağlık sorunlarından ilacprospektusu.com sorumlu tutulamaz.